

Abstract
Esophageal cancer is difficult to treat because of its rapid progression, and more effective therapeutic approaches are needed. The PPARγ is a nuclear receptor superfamily member that is expressed in many cancers. PPARγ expression is a feature of esophageal cancer cell lines, and in the present investigation, the PPARγ antagonists T0070907 and GW9662 could induce loss of invasion but could not induce growth reduction or apoptosis at low concentrations (<10 mM). A high concentration of antagonists (50 µM) inhibited cell growth and induced apoptosis, but these effects did not explain our result at the low concentration. Morphological change, decreased expression of the cell signaling pathway and inhibition of cancer cell invasion were observed in the low concentration. This suggested that PPARγ antagonists inhibited esophageal cancer cell invasion as well as cell adherence, most likely due to alteration in the FAK–MAPK pathway, and this was independent of apoptosis. These results suggested that PPARγ plays an important role in cancer cell invasion and that it might be a novel target for therapy of esophageal cancer. (Cancer Sci 2006; 97: 854–860)
Abbreviations:
- DMEM
Dulbecco's minimum essential medium
- ECM
extracellular matrix
- Erk
extracellular signal‐regulated kinase
- FAK
focal adhesion kinase
- FITC
fluorescein‐isothiocyanate
- MAPK
mitogen‐activated protein kinase
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- p‐Erk
phosphorylated extracellular signal‐regulated kinase
- p‐FAK
phosphorylated focal adhesion kinase
- PPARγ
peroxisome proliferator‐activated receptor gamma
- TZDs
thiazolizinediones.
Esophageal cancer is associated with a high mortality rate due to its typically late presentation and rapid progression. For tumors that are not amenable to surgical curative resection, chemotherapy and radiotherapy are commonly applied. But most patients continue to have a poor prognosis, along with an increased morbidity due to treatment‐related side‐effects.( 1 ) Clearly, new therapies for esophageal cancer are needed.
The nuclear transcription factor PPARγ has recently become a putative therapeutic cancer target for a variety of cancers.( 2 , 3 , 4 ) As PPARγ is mainly expressed in adipose tissue and activation plays a central role in adipocyte differentiation and insulin sensitivity,( 5 ) activating synthethic ligands, TZDs, are commonly used as oral antihyperglycemic agents in control of diabetes mellitus type 2. However, PPARγ is also overexpressed in many tumors, including examples in the esophagus, stomach, breast, lung and colon, suggesting that modulation of PPARγ function might impact on tumor survival.( 2 , 3 , 4 , 6 , 7 ) Initial efforts have focused on activation with the TZD ligands, as these have been shown to induce G1 cell cycle arrest in a variety of tumor cell lines.( 8 , 9 ) However, the results of clinical trials with TZDs have shown modest, if any, benefit.( 10 , 11 ) With esophageal cancers, PPARγ activation by TZDs in cell lines has been reported to inhibit in vitro cell growth and/or induce apoptosis.( 12 , 13 , 14 )
Several observations suggest that inhibition of PPARγ function might be beneficial in treating neoplasms.( 15 , 16 ) PPARγ is overexpressed in many cancer cell types, but loss‐of‐function mutations are rare,( 17 ) suggesting that the receptor is a tumor cell survival factor. The hypothesis that PPARγ function might contribute to carcinogenesis or cancer cell survival is also supported by the observation that in one murine model of colon cancer, PPARγ activation led to an increase in tumor formation.( 18 , 19 )
Little is known about inhibition of PPARγ function in esophageal cancer cells. In this study, we investigated the effect of PPARγ inhibition on esophageal cancer cell lines using PPARγ‐specific antagonists T0070907 and GW9662 in high (50 µM), low (<10 µM) and very low (<10 µM) concentrations. The PPARγ antagonist could prevent cell attachment to the ECM in high concentrations in our previous studies, however, the effect of PPARγ antagonists in low concentrations was not clear.( 20 , 21 ) A better understanding of the PPARγ function might lead to it being further utilized for cancer treatment.
Materials and methods
Reagents
The PPARγ‐specific antagonists T0070907 and GW9662 were purchased from Cayman Chemical (Ann Arbor, MI, USA) and Sigma Chemical (St Louis, MO, USA), respectively.
Cell culture
Human esophageal cancer cells (KYSE30, KYSE70, KYSE140) used in this study were obtained from the Human Science Foundation (Osaka, Japan). The histology of KYSE30 was well differentiated, KYSE70 poorly, and KYSE140 moderately. KYSE30 and KYSE70 were maintained in DMEM and KYSE140 in Ham's F12 supplemented with 2% fetal bovine serum. Cultures were maintained at 37°C, with an atmosphere of 5% CO2 and saturated humidity.
Western blot analysis
Adherent cells were washed in phosphate‐buffered saline, and cell extracts were prepared in Laemmli lysis buffer. Protein concentrations were measured using Bio‐Rad Protein Assay Reagent (Bio‐Rad, Richmond, CA, USA) following the manufacturer's suggested procedure. After electrophoresis of 20 µg aliquots using 10% sodium dodecylsulfate–polyacrylamide gel electrophoresis, proteins were transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA), blocked for 1 h in tris‐buffered saline with bovine serum albumin at room temperature, and incubated with primary monoclonal antibody for 1 h. The anti‐PPARγ antibody (E‐8) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti‐pFAK(pY397) from BD Biosciences (San Jose, CA, USA) and anti‐p‐Erk from Cellsignaling Technology (Beverly, MA, USA). After three washings, the membranes were incubated for 1 h at room temperature with secondary antibody, and immune complexes were visualized using the enhanced chemiluminescence detection kit (Amersham, London, UK) following the manufacturer's procedure.
Immunofluorescence and cell morphology
Cells (5 × 105 per well) were grown on collagen‐1 coated glass cover slips in six‐well flat bottom plates for 24 h. T0070907 and GW9662 were added and the cells were preincubated for 24 h. The cells were fixed with 4% formaldehyde followed by 100% ethanol at −20°C. Permeabilization was carried out with 0.1% Triton‐X, and non‐specific binding was blocked with 2% normal swine serum. Cells were incubated with antipaxillin monoclonal antibody (BD Biosciences) followed by FITC‐labeled secondary antibody. Alexa fluoro 594‐conjugated phalloidin (Molecular Probes, Eugene, OR, USA) was used to visualize F‐actin. The samples were then mounted with Vectashield (Vector Laboratories, Burlingame, CA, USA) and examined by confocal laser scanning microscopy (Carl Zeiss, Oberkochen, Germany). All experiments were done in triplicate.
Assessment of cell growth
Cell proliferation was measured by MTT assay.( 22 ) KYSE70 cells were plated in 96‐well plates at a concentration of 5 × 103 cells in 100 µL of DMEM. After 24 h incubation, the medium was changed with various concentrations of T0070907 and GW9662 added (1–50 µM), and the cells further incubated for 24–72 h. MTT solution (0.5%) was then added to each well. After the plates were incubated for 4 h, 20% sodium dodecylsulfate solution was incubated and absorbance at 595 nm was determined using a microplate reader (Model 550; Bio‐Rad). Control wells were treated with dimethylsulfoxide alone. Three independent experiments were carried out.
Apoptosis assay
To evaluate the apoptotic cell death, annexin V staining was carried out using an annexin V–FITC apoptosis detection kit I (Becton Dickinson, San Jose, CA, USA) according to the manufacturer's recommendations. Cells were subsequently analyzed by FACScan flow cytometry (Becton Dickinson).
Transwell invasion assays
In vitro cell invasion was assayed in BD BioCoat Matrigel invasion chambers (24 wells, 8 µm pore size; BD Biosciences). The top chamber was seeded with 5 × 104 KYSE70 cells in DMEM. The bottom chamber was filled with DMEM supplemented with 2% fetal bovine serum as a chemoattractant. Cells were preincubated with T0070907 and GW9662 (1–10 µM) in the top chamber, followed by incubation for 24 h in a humidified tissue culture incubator at 37°C under a 5% CO2 atmosphere. Noninvasive cells were removed from the upper surface of the membrane with a cotton swab, and cells on the lower surface of the membrane were fixed and stained with toluidine blue and mounted on glass slides. Five random fields/well were counted for quantitation of cell invasion. Triplicate wells were counted for each assay.
Statistical analysis
All results are expressed as means ± standard errors of the mean. Statistical comparisons were made using either Student's t‐test or Scheffe's method after anova. Differences were considered significant at P < 0.05.
Results
Esophageal cancer expresses PPARγ protein
Human esophageal cancer tissues were stained using anti‐PPARγ‐specific antibody, and the expression was high in area of cancer invasion (Fig. 1a). To test whether inhibiting PPARγ activity affects esophageal cell growth or survival, three esophageal cell lines, KYSE30, KYSE70 and KYSE140, were examined. Western blot analysis revealed differential PPARγ protein expression in each. The expression level of PPARγ was very low in normal mucosa, low in KYSE30 cells (well differentiated), moderate in KYSE140 cells (moderately differentiated) and high in KYSE70 cells (poorly differentiated). As the degree of cell differentiation decreased from well differentiated to poorly differentiated, PPARγ protein expression increased. The expression of PPARγ was increased in esophageal cancer tissues compare with normal esophageal epithelial cells (Fig. 1b). The cell line with the highest expression of PPARγ, KYSE70, was used in subsequent investigations into the inhibitory effect of PPARγ activity.
Figure 1.
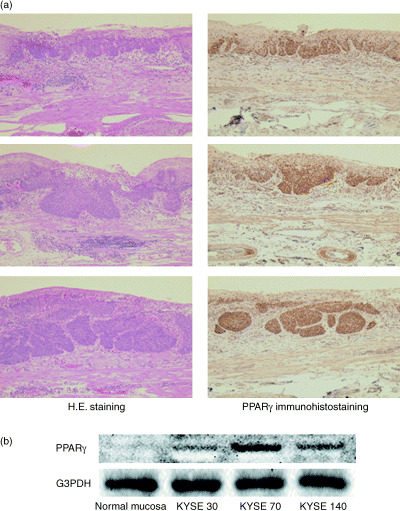
PPARγ expression in esophageal cancer. (a) Surgical resection of human esophageal cancer tissue stained using HE and antibody specific for PPARγ. The expression was high in area of cancer invasion. (b) The expression level of PPARγ was very low in normal mucosa, low in KYSE30 cells (well differentiated), moderate in KYSE140 cells (moderately differentiated) and high in KYSE70 cells (poorly differentiated). As the degree of cell differentiation decreased from well differentiated to poorly differentiated, PPARγ protein expression increased.
Treatment with PPARγ antagonists decreases cell adhesion to the ECM
Cells treated with 10 or 50 µM of T0070907 and GW9662 underwent morphological changes by 24 h, but those treated with 1 µM did not undergo any morphological change (Fig. 2). At this time point, most cells were still adherents to the plate. However, rather than becoming the normal elongated shape, they were rounded. This morphological change was not the result of apoptosis in cells treated with 10 µM T0070907 and GW9662, as cells at this time point were not positive for annexin V (Fig. 3). By 48 h, almost half of the cells treated with 50 µM T0070907 and GW9662 were nonadherent. In other words, 1 µM of antagonists induced no change, 10 µM induced morphological changes but did not inhibit cell adherence, and 50 µM induced morphological changes and inhibited cell adherence.
Figure 2.
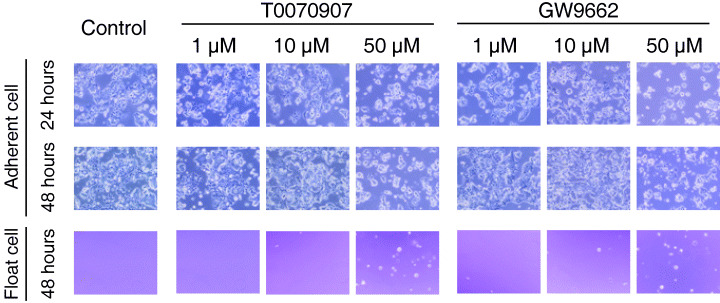
Morphological changes in esophageal cancer cell lines induced by PPARγ antagonists. KYSE70 cells were incubated with dimethylsulfoxide (control), and 1–50 µM T0070907 and GW9662. Cells treated with 10 and 50 µM of PPARγ antagonists underwent morphological changes by 24 h. By 48 h, 10 µM induced morphological changes but did not inhibit cell adherence; 50 µM induced morphological changes and inhibited cell adherence.
Figure 3.
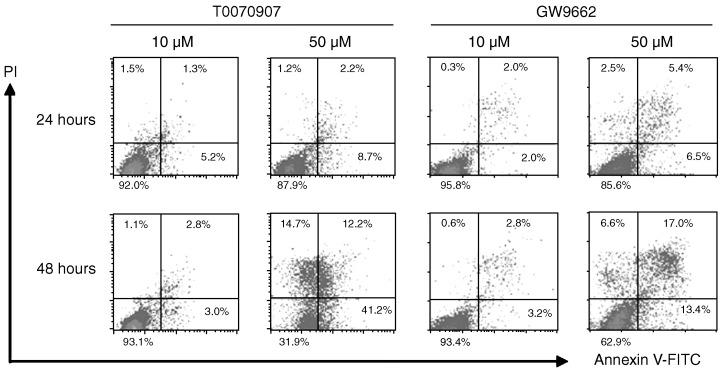
PPARγ antagonists induced apoptosis in KYSE70 esophageal cancer cells at 48 h. Cells were incubated with 10 and 50 µM T0070907 and GW9662, followed by flow cytometry analysis using annexin V and propidium iodide double staining. Apoptotic cells were observed in 50 µM antagonists at the 48 h time point, but not in 10 µM.
PPARγ antagonists induce change of actin organization
Actin fibers play an important role in maintaining the cytoskeletal structure, and paxillin is functionally important in transducing intracellular messages that are associated with growth factor signaling and cell–ECM interactions.( 23 ) To determine whether the observed cell rounding was associated with alterations in cytoskeletal function, these proteins were examined by confocal microscopy. Before antagonist treatment, actin fibers and paxillin were visible (Fig. 4). After 12 h of treatment in 10 and 50 µM PPARγ antagonists, the cells began to lose their actin fibers, and by 24 h almost all the KYSE70 cells had changed to a round shape with loss of actin fibers. Similar results were obtained with KYSE30 and KYSE140 cells (data not shown).
Figure 4.
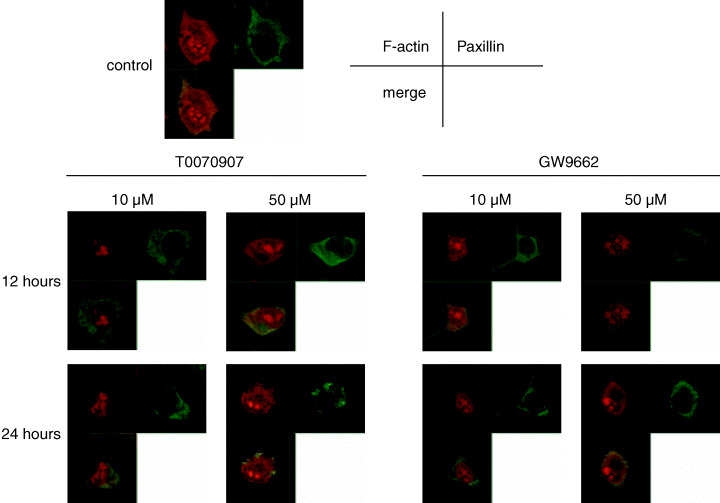
Before PPARγ antagonist treatment of KYSE70 esophageal cancer cells, actin fibers and paxillin were visible. After 12 h of treatment with 10 and 50 µM T0070907 and GW9662, the cells began to lose their actin fibers, and by 24 h almost all of the cells had changed to a round shape with complete loss of actin fibers.
Effect of PPARγ antagonists on cell growth
In order to compare the effects of PPARγ antagonism on esophageal cancer cell growth, KYSE70 cells were incubated with T0070907 and GW9662. At 24, 48 and 72 h, the number of cells was measured using MTT assay (Fig. 5). The results were similar in all cell lines, therefore we used KYSE70 cells because they had the highest expression of PPARγ. The compounds did not reduce the growth ratio at concentrations below 10 µM, but did at 50 µM.
Figure 5.
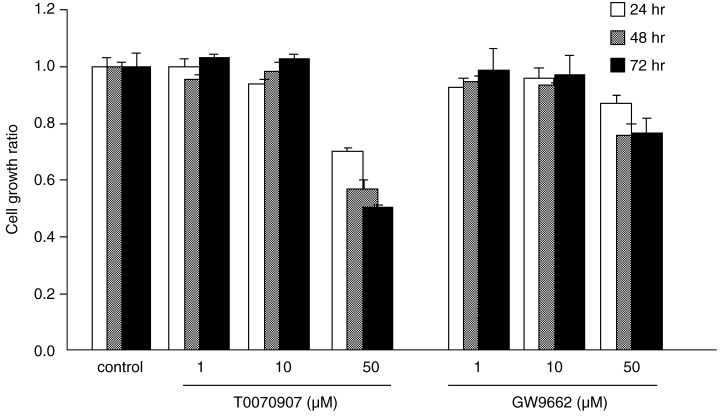
The PPARγ antagonists T0070907 and GW9662 prevented cell growth in KYSE70 esophageal cancer cells. MTT assay showed the cell growth ratio was significantly reduced in cells treated with 50 µM PPARγ antagonists, and this effect was time‐dependent. PPARγ antagonists at concentrations below 10 µM did not reduce the cell growth ratio.
Effect of PPARγ antagonists on apoptotic cell death
The results of propidium iodide–annexin V–FITC staining showed that the PPARγ‐specific antagonists both induced apoptosis in KYSE70 cells by 48 h at a concentration of 50 µM (Fig. 3), but did not induce apoptosis at 10 µM.
PPARγ antagonism affects the ability of esophageal cancer cell invasion
The PPARγ antagonists had the potential to decrease cell invasiveness. In transwell invasion assays, the number of invasive KYSE70 cells significantly decreased with antagonist treatment below 10 µM, and this effect dose‐dependent (Fig. 6). At this concentration (10 µM), the PPARγ antagonists did not induce apoptosis, suggesting that the effect of invasion reduction was independent of apoptosis.
Figure 6.
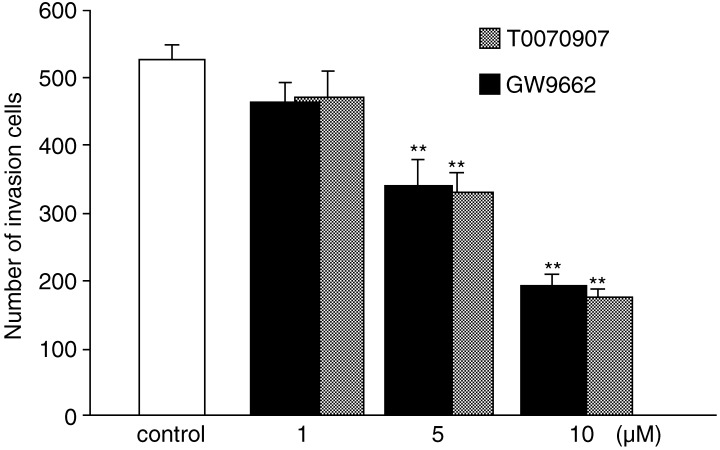
KYSE70 esophageal cancer cells were incubated with PPARγ antagonists for 24 h during a transwell invasion assay. The number of invasive cells was decreased with antagonist treatment below 10 µM, and this effect was dose‐dependent. Error bars represent standard errors of the mean for three replicates. *P < 0.05, **P < 0.01.
PPARγ antagonism inhibits the phosphorylation of FAK and Erk
To determine possible mechanisms of the cell growth inhibition by PPARγ antagonism, important adhesion and survival cell signaling pathways were investigated. PPARγ antagonists altered FAK (Tyr397) and Erk phosphorylation. KYSE70 cells were incubated with 10 µM of each antagonist.
The results of Western blot analysis revealed decreased expression of p‐FAK and p‐Erk by the treatment with the antagonists (Fig. 7). p‐FAK decreased after 9 h and p‐Erk decreased after 12 h.
Figure 7.
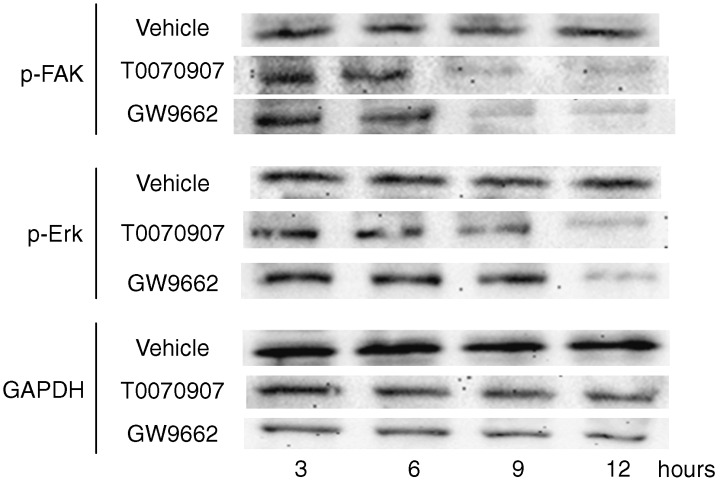
Expression of FAK (Tyr397) and Erk phosphorylation was altered in KYSE70 esophageal cancer cells incubated with 10 µM PPARγ antagonists. p‐FAK was decreased at 9 h and p‐Erk was decreased at 12 h.
Discussion
Currently, there is very little information about the inhibition of PPARγ in cancer cells, including esophageal cancer cells. Our previous studies using hepatocellular carcinoma and tongue cancer cells have demonstrated that PPARγ antagonists (high concentration, 50 µM) could prevent cell attachment to ECM, leading to loss of adhesion‐induced apoptosis.( 20 , 21 ) Tongue and esophageal cancer are similar in that they are both squamous cell carcinomas, but the treatment approaches are different. However, esophageal cancer is clinically very important because its mortality rate is very high.
In this study, we demonstrated using esophageal cancer cells that a very low concentration (<10 µM) of PPARγ antagonists could induce the inhibition of invasive properties, but not induce growth reduction or apoptosis. MTT assay (Fig. 5) showed that a low and very low concentration of PPARγ antagonists did not inhibit the growth ratio, even after 72 h. Similarly, a low concentration (10 µM) of PPARγ antagonists did not induce apoptosis (Fig. 3). However, a low concentration of PPARγ antagonists could inhibit the cancer cell invasion in the transwell migration assays (Fig. 6). These results suggested that the mechanism by which the PPARγ antagonists inhibited the cancer cell invasion at low concentrations was different from the mechanism by which the high concentration induced apoptosis and cell growth reduction. Therefore, our results suggested that PPARγ might play an important role in cancer cell invasion.
Several reports have clearly demonstrated that PPARγ agonist ligands, the TZDs, could inhibit cell growth and apoptosis of adenocarcinomas, as well as esophageal cancer tissues.( 14 , 24 , 25 , 26 ) The PPARγ antagonists T0070907 and GW9662 could induce a very similar inhibition of cell growth at a high concentration (50 µM) (Fig. 4). Although it appears paradoxical that both over‐activation and inhibition of PPARγ activity could lead to reduced cell growth, this might be a result of different mechanisms. In the TZD setting, a well‐recognized G0–G1 cell cycle arrest occurs. In contrast, with PPARγ antagonists, apoptosis appeared to follow loss of adhesion, which was not observed using TZDs. It is suggested that both PPARγ antagonists and TZDs should be considered important candidates for further development as anticancer agents.
PPARγ antagonists were found to first affect cell morphology, with almost all cells changing their cytoskeletal structure, involving both actin fibers and paxillin, within the first 12 h. After adopting a rounded shape, the cells then began detaching from the ECM by 24 h. At this time point, the cells were clearly not apoptotic, as determined by flow cytometric analysis in low concentration (10 µM). This suggested there were different mechanisms dependent on PPARγ activity.
FAK, a 125 kDa non‐receptor tyrosine kinase, is an important regulator of cell survival, invasion, migration, and cell cycle progression.( 27 , 28 , 29 ) This overexpression of FAK has been observed in a number of human malignant cells, and this might play an important role in determining cellular invasion and metastasis.( 30 , 31 ) FAK is functionally important in transducing intracellular messages associated with growth factor signaling, cell–ECM interactions, modifying the cytoskeleton and activating MAPK cascades, including Erk. In the present study, the inhibition of phosphorylation of FAK in KYSE70 cells treated with antagonists (10 µM) was observed at 9 h, followed by a reduction in Erk phosphorylation at 12 h. Inhibition of Erk phosphorylation occurred after the inhibition of FAK phosphorylation by PPARγ antagonists, suggesting that PPARγ might play an important role in the MAPK pathway.
High concentration (50 µM) PPARγ antagonists induced apoptosis and cell detachment, and reduced cell growth, but a low concentration (10 µM) could reduce cell invasion and alter the MAPK signaling pathway. These results suggest that the effects of a low concentration of antagonists were independent of the effects of apoptosis, detachment and cell growth inhibition. One study has reported that the difference in effect depends on PPARγ concentration,( 32 ) but further investigation is necessary.
In summary, PPARγ antagonists inhibited esophageal cancer cell invasion as well as cell adherence to ECM, most likely due to alteration in the FAK–MAPK pathway, and this was independent of apoptosis. Our results suggest that PPARγ plays important roles in cancer cell invasion, therefore it might be a novel target for esophageal cancer therapy.
Acknowledgments
This work was supported by a Grants‐in‐Aid for the Third Term Comprehensive Control Research for Cancer from the Ministry of Health, Labour, and Welfare of Japan, and the Ministry of Education, Science, Sports, Culture, Japan.
References
- 1. Blazeby JM, Alderson D, Farndon JR. Quality of life in patients with oesophageal cancer. Recent Results Cancer Res 2000; 155: 193–204. [DOI] [PubMed] [Google Scholar]
- 2. Takashima T, Fujiwara Y, Higuchi K et al. PPAR‐gamma ligands inhibit growth of human esophageal adenocarcinoma cells through induction of apoptosis, cell cycle arrest and reduction of ornithine decarboxylase activity. Int J Oncol 2001; 19: 465–71. [PubMed] [Google Scholar]
- 3. Chang TH, Szabo E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator‐activated receptor gamma in non‐small cell lung cancer. Cancer Res 2000; 60: 1129–38. [PubMed] [Google Scholar]
- 4. DuBois RN, Gupta R, Brockman J, Reddy BS, Krakow SL, Lazar MA. The nuclear eicosanoid receptor, PPARgamma, is aberrantly expressed in colonic cancers. Carcinogenesis 1998; 19: 49–53. [DOI] [PubMed] [Google Scholar]
- 5. Desvergne B, Wahli W. Peroxisome proliferator‐activated receptors: nuclear control of metabolism. Endocr Rev 1999; 20: 649–88. [DOI] [PubMed] [Google Scholar]
- 6. Mueller E, Sarraf P, Tontonoz P et al. Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell 1998; 1: 465–70. [DOI] [PubMed] [Google Scholar]
- 7. Sato H, Ishihara S, Kawashima K et al. Expression of peroxisome proliferator‐activated receptor (PPAR) gamma in gastric cancer and inhibitory effects of PPARgamma agonists. Br J Cancer 2000; 83: 1394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guan YF, Zhang YH, Breyer RM, Davis L, Breyer MD. Expression of peroxisome proliferator‐activated receptor gamma (PPARgamma) in human transitional bladder cancer and its role in inducing cell death. Neoplasia 1999; 1: 330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kitamura S, Miyazaki Y, Shinomura Y, Kondo S, Kanayama S, Matsuzawa Y. Peroxisome proliferator‐activated receptor gamma induces growth arrest and differentiation markers of human colon cancer cells. Jpn J Cancer Res 1999; 90: 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Debrock G, Vanhentenrijk V, Sciot R, Debiec‐Rychter M, Oyen R, Van Oosterom A. A phase II trial with rosiglitazone in liposarcoma patients. Br J Cancer 2003; 89: 1409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kulke MH, Demetri GD, Sharpless NE et al. A phase II study of troglitazone, an activator of the PPARgamma receptor, in patients with chemotherapy‐resistant metastatic colorectal cancer. Cancer J 2002; 8: 395–9. [DOI] [PubMed] [Google Scholar]
- 12. Hashimoto Y, Shimada Y, Itami A et al. Growth inhibition through activation of peroxisome proliferator‐activated receptor gamma in human oesophageal squamous cell carcinoma. Eur J Cancer 2003; 39: 2239–46. [DOI] [PubMed] [Google Scholar]
- 13. Fujii D, Yoshida K, Tanabe K, Hihara J, Toge T. The ligands of peroxisome proliferator‐activated receptor (PPAR) gamma inhibit growth of human esophageal carcinoma cells through induction of apoptosis and cell cycle arrest. Anticancer Res 2004; 24: 1409–16. [PubMed] [Google Scholar]
- 14. Rumi MA, Sato H, Ishihara S, Ortega C, Kadowaki Y, Kinoshita Y. Growth inhibition of esophageal squamous carcinoma cells by peroxisome proliferator‐activated receptor‐gamma ligands. J Lab Clin Med 2002; 140: 17–26. [DOI] [PubMed] [Google Scholar]
- 15. Martelli ML, Iuliano R, Le Pera I et al. Inhibitory effects of peroxisome poliferator‐activated receptor gamma on thyroid carcinoma cell growth. J Clin Endocrinol Metab 2002; 87: 4728–35. [DOI] [PubMed] [Google Scholar]
- 16. Panigrahy D, Shen LQ, Kieran MW, Kaipainen A. Therapeutic potential of thiazolidinediones as anticancer agents. Expert Opin Invest Drugs 2003; 12: 1925–37. [DOI] [PubMed] [Google Scholar]
- 17. Posch MG, Zang C, Mueller W et al. Somatic mutations in peroxisome proliferator‐activated receptor‐gamma are rare events in human cancer cells. Med Sci Monit 2004; 10: BR250–254. [PubMed] [Google Scholar]
- 18. Lefebvre AM, Chen I, Desreumaux P et al. Activation of the peroxisome proliferator‐activated receptor gamma promotes the development of colon tumors in C57BL/6J‐APCMin/+ mice. Nat Med 1998; 4: 1053–7. [DOI] [PubMed] [Google Scholar]
- 19. Saez E, Tontonoz P, Nelson MC et al. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat Med 1998; 4: 1058–61. [DOI] [PubMed] [Google Scholar]
- 20. Schaefer KL, Wada K, Takahashi H et al. Peroxisome proliferator‐activated receptor gamma inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Res 2005; 65: 2251–9. [DOI] [PubMed] [Google Scholar]
- 21. Masuda T, Wada K, Nakajima A et al. Critical role of peroxisome proliferator‐activated receptor gamma on anoikis and invasion of squamous cell carcinoma. Clin Cancer Res 2005; 11: 4012–21. [DOI] [PubMed] [Google Scholar]
- 22. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth 1983; 65: 55–63. [DOI] [PubMed] [Google Scholar]
- 23. Giancotti FG, Ruoslahti E. Integrin signaling. Science 1999; 285: 1028–32. [DOI] [PubMed] [Google Scholar]
- 24. Eibl G, Wente MN, Reber HA, Hines OJ. Peroxisome proliferator‐activated receptor gamma induces pancreatic cancer cell apoptosis. Biochem Biophys Res Commun 2001; 287: 522–9. [DOI] [PubMed] [Google Scholar]
- 25. Ohta K, Endo T, Haraguchi K, Hershman JM, Onaya T. Ligands for peroxisome proliferator‐activated receptor gamma inhibit growth and induce apoptosis of human papillary thyroid carcinoma cells. J Clin Endocrinol Metab 2001; 86: 2170–7. [DOI] [PubMed] [Google Scholar]
- 26. Shimada T, Kojima K, Yoshiura K, Hiraishi H, Terano A. Characteristics of the peroxisome proliferator activated receptor gamma (PPARgamma) ligand induced apoptosis in colon cancer cells. Gut 2002; 50: 658–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen HC, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan JL. Interaction of focal adhesion kinase with cytoskeletal protein talin. J Biol Chem 1995; 270: 16995–9. [DOI] [PubMed] [Google Scholar]
- 28. Sieg DJ, Hauck CR, Ilic D et al. FAK integrates growth‐factor and integrin signals to promote cell migration. Nat Cell Biol 2000; 2: 249–56. [DOI] [PubMed] [Google Scholar]
- 29. Schaller MD. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim Biophys Acta 2001; 1540: 1–21. [DOI] [PubMed] [Google Scholar]
- 30. Agochiya M, Brunton VG, Owens DW et al. Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene 1999; 18: 5646–53. [DOI] [PubMed] [Google Scholar]
- 31. Owens LV, Xu L, Dent GA et al. Focal adhesion kinase as a marker of invasive potential in differentiated human thyroid cancer. Ann Surg Oncol 1996; 3: 100–5. [DOI] [PubMed] [Google Scholar]
- 32. Yamauchi T, Waki H, Kamon J et al. Inhibition of RXR and PPARgamma ameliorates diet‐induced obesity and type 2 diabetes. J Clin Invest 2001; 108: 1001–13. [DOI] [PMC free article] [PubMed] [Google Scholar]