

Abstract
The effects of IQ on the promotion stage of DHPN‐induced lung carcinogenesis and contributions of oxidative stress were investigated in rats. Groups of 20 male 6‐week‐old F344 rats were given 0.1% DHPN in their drinking water for 2 weeks for initiation. From the age of 9 weeks, they were treated with 0, 150 and 300 p.p.m. of IQ in the diet for 27 weeks. Control rats were similarly fed 300 p.p.m. IQ or basal diet alone without the preceding initiation. IQ clearly (P < 0.01) enhanced the multiplicity of lung tumors in a dose‐dependent manner (DHPN alone, 3.63 ± 1.80; DHPN +150 p.p.m. IQ, 11.50 ± 5.04; DHPN +300 p.p.m. IQ, 18.83 ± 4.58 [no./rat]). In addition, the incidence of lung tumors in the 300 p.p.m. IQ alone group (25%) was significantly (P < 0.05) higher than that in the non‐treatment group (0%). In a second experiment, male rats were given IQ at doses of 0 and 300 p.p.m. in the diet for one week in order to analyze 8‐OHdG formation, levels of TBARS and BrdU‐LI in the lungs. There were no changes in 8‐OHdG or TBARS levels, but significant elevation of BrdU‐LI occurred in the IQ administration group. The overall data clearly indicate that IQ is a potent lung carcinogen in rats, in which oxidative stress may not be involved in lung carcinogenesis. (Cancer Sci 2006; 97: 368 –373)
Abbreviations:
- BrdU‐LI
bromodeoxyuridine labeling indices
- IQ
2‐amino‐3‐methylimidazo[4,5‐f]quinoline
- TBARS
thiobarbituric acid reactive substances.
IQ, one of the mutagenic and carcinogenic heterocyclic amines (HCAs), has been detected in cooked meat and fish( 1 , 2 ) and also in cigarette smoke condensate.( 3 ) It has been established to act as a complete carcinogen in the liver, colon, small intestine, Zymbal's gland, clitoral gland and skin of F344 rats( 4 ) and in the lungs of CDF1 mice.( 5 ) Of several carcinogenic HCAs, none have hitherto demonstrated lung carcinogenicity in rats( 6 ) although we recently suggested that IQ might be an exception.( 7 ) Thus, we incidentally found that incidences of alveolar hyperplasias, adenomas and adenocarcinomas in the lung were significantly increased in a group given 300 p.p.m. IQ for 27 weeks after initiation with diethylnitrosamine (DEN) and 1,2‐dimethylhydrazine (DMH), as compared with the initiation alone group.( 7 ) DEN is a genotoxic lung carcinogen and it was found to induce lung neoplastic lesions in approximately 50% of rats receiving a single dose (200 mg/kg body weight) in a long‐term (103 week) experiment.( 8 ) With regard to mechanisms of IQ carcinogenesis, metabolically activated IQ forms DNA adducts( 1 , 9 , 10 ) which have been detected in the lungs as well as in its established target organs in F344 rats.( 11 ) Therefore, previous results suggested that IQ enhanced lung carcinogenesis after initiation with DEN and that it is a complete lung carcinogen. The present study was conducted to test the hypothesis that IQ is indeed a lung carcinogen in rats, as well as in mice, using a suitable experimental condition and a common lung carcinogen, DHPN, as an initiator.
It was recently suggested that oxidative stress may participate in HCA carcinogenesis, based on the finding that induction of preneoplastic or neoplastic lesions was significantly inhibited when antioxidants were simultaneously administered( 12 , 13 ) and that superoxide anion radicals are generated during metabolism of HCAs by NADPH/cytochrome P‐450 reductase in vitro, as demonstrated using a spin‐trapping method.( 14 ) In addition, exposure to 2‐amino‐3,8‐dimethylimidazo[4,5‐f] quinoxaline (MelQx), another HCA, dose‐dependently raised 8‐hydroxydeoxyguanosine (8‐OHdG) levels in rat partially excised liver.( 15 ) Recently, we demonstrated that simultaneous treatment with NaNO2 enhanced the carcinogenic potential of IQ, especially in the colon and Zymbal's glands of rats with oxidative stress suggested to be partly responsible for the observed enhancement of colon carcinogenesis.( 7 ) Likewise, IQ treatment of Nrf2‐gene (transcriptional factor for inducible expression of a group of antioxidant enzymes) knockout mice increased liver tumor formation associated with increase in oxidative stress markers as compared with wild mice (Y. Kitamura et al., unpublished data).
Epidemiological studies have shown that cigarette smoke is closely associated with development of human cancers in various organs, mainly in the lung.( 16 , 17 ) More than 20 carcinogens, which convincingly cause lung tumors in laboratory animals or humans exhibiting genotoxic effects, have been identified in cigarette smoke, including the tobacco‐specific nitrosamine 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone (NNK).( 18 , 19 ) Together with the specific DNA adduct formation,( 20 ) oxidative DNA damage was considered to be involved in nitrosamine NNK‐induced respiratory tract tumorigenesis in rodents.( 20 ) In addition, the amount of 8‐OHdG excreted in urine is higher in smokers as compared with non‐smokers.( 21 ) Accordingly, in the light of IQ detection in cigarette smoke,( 3 ) possible involvement of oxidative stress in its carcinogenesis was also examined in the present study.
To determine the effects of IQ in rats, we chose a two‐stage lung carcinogenesis model with DHPN initiation in male F344 rats. To investigate possible contributions of oxidative DNA damage and lipid peroxidation, 8‐OHdG formation in DNA and levels of thiobarbituric acid reactive substances (TBARS) were measured, respectively. Moreover, to assess influence on cell proliferation, bromodeoxyuridine (BrdU)‐positive cells were counted.
Materials and Methods
Chemicals
N‐bis(2‐hydroxypropyl)nitrosamine (DHPN) was purchased from Nacalai Tesque (Kyoto, Japan), IQ from Toronto Research Chemicals (North York, Canada), and BrdU and alkaline phosphatase from Sigma Chemical (St Louis, MO). Nuclease P1 was obtained from Yamasa Shoyu (Chiba, Japan). Monoclonal mouse anti‐BrdU, biotin‐labeled rabbit anti‐mouse immunoglobulin G, streptavidin‐biotin‐peroxidase complex horseradish peroxidase were purchased form DakoCytomation (Kyoto, Japan).
Animals
A total of 90 and 20 male F344/DuCrj SPF rats, at 5 weeks of age, were obtained from Charles River Japan (Tokyo, Japan) for experiments 1 and 2, respectively, and acclimatized for approximately 1 week before being assigned to four groups consisting of 20 animals each and a group consisting of 10 animals as the non‐treatment group (experiment 1), or two groups consisting of 10 animals each (experiment 2). They were housed in plastic cages (five rats/cage) with soft chips for bedding in a room with a barrier system and maintained under the following conditions: temperature (23 ± 2°C), relative humidity (60 ± 5%), ventilation frequency (18 times per hour) and a 12‐h illumination. Normal powdered diet (CRF‐1; Oriental Yeast, Tokyo, Japan) and tap water were available ad libitum. The protocols for these studies were approved by the Animal Care and Utilization Committee for the National Institute of Health Sciences, Japan.
Experimental design
The experimental protocol for experiment 1 is shown in Figure 1. Three groups of animals at the age of 6 weeks received water containing 0.1% DHPN during the first 2 weeks for initiation. From the age of 9 weeks, these initiated animals received basal diet containing 0, 150 or 300 p.p.m. IQ. The remaining two groups were treated with basal diet containing 300 p.p.m. IQ without initiation or basal diet alone. The duration of the post‐initiation period was set at 27 weeks. The highest dose level of IQ was selected according to the results of our previous study, in which lung tumors were observed at 300 p.p.m. in a DEN and DMH‐initiated two‐stage carcinogenesis model.( 7 )
Figure 1.
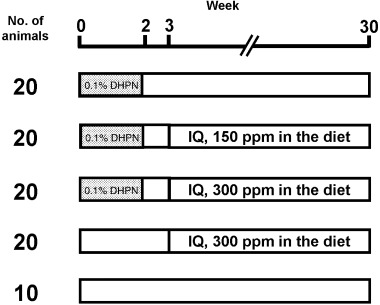
Experimental design of Experiment 1. Three groups of F344 male rats, at the age of 6 weeks, were given drinking water containing 0.1% DHPN during the first 2 weeks for initiation. From the age of 9 weeks, they received basal diet containing 0, 150 or 300 p.p.m. IQ. Non‐initiation groups were treated with basal diet containing 300 p.p.m. IQ or basal diet alone.
Body weights and food consumption were measured once a week until week 6 and thereafter once every three weeks. All surviving animals were killed under ether anesthesia at the end of week 30. At autopsy, the major organs/tissues were carefully examined and the liver, kidneys and lungs were weighed. All the major organs and tissues, including lungs, liver, kidneys, stomach, small intestine, colon, Zymbal's glands, thyroids, esophagus and any other grossly abnormal lesions were fixed and preserved in 10% phosphate‐buffered formalin solution. In the lung, all pulmonary lobes were injected with formalin solution from the trachea to ease histopathological examination. Organs of animals found dead or killed on becoming moribund were also fixed and preserved.
In experiment 2, the two groups of 10 rats received basal diet alone or 300 p.p.m. IQ in the diet for one week from the age of 6 weeks. Five rats in each group were injected intraperitoneally with 100 mg/10 mL saline/kg of BrdU daily three times in total (the last 2 h before being killed). The animals were killed under ether anesthesia, autopsied and lungs were removed. For the BrdU‐treated animals, the lungs were inflated with 10% phosphate‐buffered formalin, injected through the trachea, for fixation of all pulmonary lobes. For the remaining five animals in each group, lungs were frozen immediately with liquid nitrogen and stored at −80°C until used for analysis of 8‐OHdG formation and lipid peroxidation.
Histopathology
All the preserved organs/tissues were embedded in paraffin, sectioned and stained with hematoxylin–eosin for routine histopathological examination. For the histopathological evaluation of lesions, rats who died or were killed on compassionate grounds were also included. Five lung lobes from each animal were examined.
Measurement of 8‐OHdG formation in DNA
The 8‐OHdG levels in lung DNA were determined by the method of Nakae et al.( 22 ) with approximately 0.3 g wet weight samples. Briefly, nuclear DNA was isolated with a DNA Extracter WB Kit (Wako Pure Chemical Industries, Osaka, Japan) and digested into deoxynucleotides by treatment with nuclease P1 and alkaline phosphatase. Levels of 8‐OHdG (8‐OHdG/105 deoxyguanosine) were then assessed by high‐performance liquid chromatography (column: Beckman Ultrasphere‐ODS, 5 µm, 4.6 × 250 mm; Beckman Coulter, Fullerton, CA) (elution, 10 mM NaH2PO4 containing 4% methanol) with an electrochemical detection system (Coulochem II; ESA, Bedford, MA) (guard cell, model 5020 [0.35 V]; analytical cell, model 5011 [electrode 1, 0.15 V; electrode 2, 0.3 V]).
Measurement of TBARS
Malondialdehyde (MDA; nmol/g) was assessed as an index of lipid peroxidation by the method of Uchiyama and Mihara.( 23 ) In brief, a 0.15 g portion of lung was homogenized with 1.35 mL of 1.15% KCl solution. In 0.05 mL of the homogenate, 0.2 mL of 8.1% sodium dodecyl sulphate and 3.0 mL of 0.4% thiobarbiturate in 10% acetic acid solution (pH 3.5) were added, followed by heating in a water bath at 95°C for 60 min. After cooling, 5.0 mL of n‐butanol and pyridine (15 : 1 v/v) and 1.0 mL distilled water were added to the sample, which was centrifuged at 1870 g for 10 min. The TBARS were measured by a Hitachi F‐2500 fluorescence spectrophotometer (Hitachi High‐Technologies, Tokyo, Japan) at 515 nm (excitation) and 553 nm (emission) in the butanol/pyridine phase.
Immunohistochemical staining for BrdU
Sections of lungs were treated with 0.5% hydrogen peroxide to block endogenous peroxidase activity, 4 mole HCl for denaturation of DNA and normal rabbit serum to block background staining. They were then treated with monoclonal mouse anti‐BrdU (1 : 100), biotin‐labeled rabbit anti‐mouse IgG (1 : 300) and strept avidin‐biotin‐peroxidase complex horseradish peroxidase. Peroxidase activity was visualized by treatment with diaminobenzidine tetrahydrochloride containing hydrogen peroxide and the nuclei were counterstained with hematoxylin.
Quantification of cell proliferation
In the lungs, at least 2000 alveolar type II cells were counted in the total of five lobes per animal. The BrdU‐LI were calculated as percentages of positive cells.
Statistical analysis
Incidences of lesions were analyzed by Fisher's exact probability test. Data for body weights, organ weights, lesion multiplicities, 8‐OHdG level, TBARS and BrdU‐LI were examined using analysis of variance. When positive results were obtained, Dunnett's multiple comparison test was applied to evaluate the statistical significance between pairs of groups. For DHPN‐initiated groups, simple linear regression analysis was conducted to confirm the IQ dose‐dependent effects on body weights, organ weights and lesion multiplicities.
Results
Experiment 1
Two animals in the DHPN +300 p.p.m. IQ group (week 25 and 28) and one animal in the DHPN alone group (week 28) were found dead. By the histopathological evaluation, these animals probably died of renal mesenchymal tumor and leukemia/malignant lymphoma in the DHPN +300 p.p.m. IQ group and renal mesenchymal tumor in the DHPN alone group. In lung, neoplastic lesions were not observed in the animal found dead at week 25, however, they were observed in the animals found dead at week 28 in both groups.
The results for final body and relative organ weights are shown in Table 1. In the groups given initiation, final body weights in the IQ‐treated groups were significantly lower than in the group given DHPN alone (P < 0.01) with dose dependence, and final body weights in the 300 p.p.m. IQ alone group were significantly lower than that in the non‐treatment group (P < 0.01). In initiated groups, relative organ weights of liver, kidneys and lungs were dose‐dependently and significantly increased by subsequent IQ administration. Relative organ weights in the 300 p.p.m. IQ alone group were also significantly increased as compared with those in the non‐treatment group (liver and kidneys, P < 0.01; lung, P < 0.05). In addition, relative lung weights in the DHPN alone group were significantly higher than that in the non‐treatment group (P < 0.01).
Table 1.
Final body and relative organ weights of rats treated with DHPN and/or IQ
| Treatment | No. of rats | Body weight (g) | Liver (%) | Kidneys (%) | Lungs (%) |
|---|---|---|---|---|---|
| 0.1% DHPN | 19 | 345 ± 16 | 2.61 ± 0.14 | 0.58 ± 0.04 | 0.46 ± 0.04** |
| 0.1% DHPN/150 p.p.m. IQ | 20 | 322 ± 12**** | 3.16 ± 0.16**** | 0.62 ± 0.09*** | 0.54 ± 0.07*** |
| 0.1% DHPN/300 p.p.m. IQ | 18 | 303 ± 13**** | 3.60 ± 0.32**** | 0.65 ± 0.06**** | 0.71 ± 0.23**** |
| 300 p.p.m. IQ | 20 | 314 ± 11** | 3.37 ± 0.17** | 0.64 ± 0.03** | 0.39 ± 0.03* |
| Non‐treatment | 10 | 351 ± 17 | 2.57 ± 0.11 | 0.55 ± 0.02 | 0.36 ± 0.03 |
P < 0.05;
P < 0.01, significantly different from non‐treatment group:
P < 0.05;
P < 0.01, significantly different from DHPN alone. For DHPN‐initiated groups, IQ dose‐dependency was observed in all parameters (P < 0.01).
Data for final incidence and multiplicity of lung tumors are summarized in Table 2. In the initiated groups, incidences were 100% in all three groups and multiplicities showed significant increase (P < 0.01) in a clear dose‐dependent manner with IQ administration (DHPN alone, 3.63 ± 1.80; DHPN +150 p.p.m. IQ, 11.50 ± 5.04; DHPN +300 p.p.m. IQ, 18.83 ± 4.58). Notably, the incidence of lung adenomas in the 300 p.p.m. IQ alone group was also significantly increased (25%) as compared with the non‐treatment group (0%) (P < 0.05).
Table 2.
Incidence and multiplicity of lung tumors in rats treated with DHPN and/or IQ
| Treatment | No. of rats | Adenoma | Carcinoma | Total | |||
|---|---|---|---|---|---|---|---|
| Incidence (%) | Multiplicity (no./rat) | Incidence (%) | Multiplicity (no./rat) | Incidence (%) | Multiplicity (no./rat) | ||
| 0.1% DHPN | 19 | 18 (95)** | 2.84 ± 1.57** | 11 (58)** | 0.79 ± 0.85** | 19 (100)** | 3.63 ± 1.80** |
| 0.1% DHPN/150 p.p.m. IQ | 20 | 20 (100) | 8.30 ± 3.93*** | 19 (95)*** | 3.20 ± 2.24*** | 20 (100) | 11.50 ± 5.04*** |
| 0.1% DHPN/300 p.p.m. IQ | 18 | 18 (100) | 11.72 ± 3.80*** | 18 (100)*** | 7.11 ± 3.01*** | 18 (100) | 18.83 ± 4.58*** |
| 300 p.p.m. IQ | 20 | 5 (25)* | 0.25 ± 0.44 | 0 (0) | 0 | 5 (25)* | 0.25 ± 0.44 |
| Non‐treatment | 10 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
P < 0.05;
P < 0.01, significantly different from non‐treatment group:
P < 0.01, significantly different from DHPN alone. For DHPN‐initiated groups, IQ dose‐dependency was observed in all multiplicity data (P < 0.01).
Incidences of other preneoplastic and neoplastic lesions are summarized in Table 3. In the thyroids, incidences of follicular cell hyperplasias, adenomas and adenocarcinomas tended to increase with IQ administration among the initiated groups, though without statistical significance. In the other organs, incidences of lesions in the liver, kidney, Zymbal's glands and colon showed no statistically significant variation among the groups.
Table 3.
Incidence of preneoplastic and neoplastic lesions in other organs of rats treated with DHPN and/or IQ
| Group | 0.1% DHPN | 0.1% DHPN/ 150 p.p.m. IQ | 0.1% DHPN/ 300 p.p.m. IQ | 300 p.p.m. IQ | Non‐ treatment |
|---|---|---|---|---|---|
| No. of animals | 19 | 20 | 18 | 20 | 10 |
| Organs/findings | |||||
| Thyroid | |||||
| Follicular cell hyperplasia | 1 (5%) | 2 (10%) | 3 (17%) | 0 | 0 |
| Follicular cell adenoma | 0 | 1 (5%) | 3 (17%) | 0 | 0 |
| Follicular cell carcinoma | 2 (11%) | 6 (30%) | 2 (11%) | 0 | 0 |
| Liver | |||||
| Hepatocellular adenoma | 0 | 0 | 1 (6%) | 0 | 0 |
| Hepatocellular carcinoma | 0 | 0 | 1 (6%) | 0 | 0 |
| Kidney | |||||
| Adenoma | 1 (5%) | 0 | 0 | 0 | 0 |
| Adenocarcinoma | 0 | 1 (5%) | 0 | 0 | 0 |
| Transitional cell hyperplasia | 0 | 1 (5%) | 0 | 1 (5%) | 0 |
| Renal pelvic carcinoma | 0 | 3 (15%) | 0 | 0 | 0 |
| Renal mesenchymal tumor | 0 | 0 | 1 (6%) | 0 | 0 |
| Zymbal's gland | |||||
| Carcinoma | 0 | 1 (5%) | 2 (11%) | 1 (5%) | 0 |
| Colon | |||||
| Adenocarcinoma | 0 | 1 (5%) | 1 (6%) | 0 | 0 |
No significant difference was observed in any of these groups.
Experiment 2
The results for 8‐OHdG, TBARS and BrdU‐LI in lungs are shown in Table 4. Neither 8‐OHdG nor TBARS levels showed any significant changes with IQ administration. Although results of toxicity such as hypertrophy, necrosis and hyperplasia in alveolar epithelial cells were not observed, BrdU‐LI showed a significant increase with IQ administration (P < 0.05).
Table 4.
8‐OHdG, TBARS and BrdU‐LI levels in lung of rats treated with or without IQ
| Treatment | No. of rats | 8‐OHdG levels (8‐OHdG/105 dG) | TBARS levels (nmol MDA/g lung) | BrdU labeling index (BrdU positive cells/total cells (%)) |
|---|---|---|---|---|
| Control | 5 | 0.89 ± 0.20 | 31.9 ± 4.4 | 10.9 ± 7.7 |
| IQ (300 p.p.m) | 5 | 1.01 ± 0.55 | 28.6 ± 9.3 | 26.5 ± 8.2* |
P < 0.05, significantly different from the control values.
Discussion
In the present experiment, IQ administration in the post‐initiation stage dose‐dependently enhanced lung carcinogenesis in rats pretreated with DHPN. In addition, the fact that IQ administration alone without initiation induced lung tumors is clearly indicative of complete carcinogenic potential in the rat lung. HCAs like IQ have genotoxic effects in many in vitro and in vivo assays and they form DNA adducts in their target organs.( 1 , 9 , 24 , 25 ) Schut et al. has reported that IQ‐DNA adduct levels in the lung to be second highest among target and non‐target organs examined in F344 rats.( 11 ) Thus, it is possible that genotoxic effects are responsible for the positive results with IQ in the present DHPN two‐stage carcinogenesis model. In CDF1 mice, the lung was previously reported as a target of IQ( 5 ) but the lungs did not demonstrate lesions in one rat study.( 4 ) Also, no other HCAs have so far shown carcinogenicity in the rat lung.( 6 ) Therefore, this first report of potent lung carcinogenicity is particularly important. As a possible mechanism of IQ carcinogenesis, in addition to genotoxicity, a relevance of oxidative stress was expected from the literature( 7 , 12 , 13 , 14 ) but representative oxidative stress markers such as 8‐OHdG and TBARS were not increased in the lung in the present experiment. It should be stressed, however, that Møller et al. recently presented evidence that oxidative stress biomarkers are unaltered by IQ treatment in Big Blue rats, even if dose dependent DNA adduct formation and DNA strand breaks were observed in both liver and colon.( 26 ) In addition, our previous experiment demonstrating that IQ administration to F344 rats for one week did not enhance any oxidative stress markers including 8‐OHdG, TBARS and acrolein‐modified protein in both liver and colon( 7 ) clearly concurs with the present results.
Our present data showing that IQ significantly increased cell proliferation in alveolar epithelium monitored in terms of the BrdU‐LI strongly suggest that IQ is able to exert a promotional effect on DHPN lung carcinogenesis in rat. It has been considered that pulmonary type I cell damage by toxic insult is followed by proliferation of type II epithelial cells which eventually transform into new type I cells.( 27 ) However, indicators of lung toxicity such as hypertrophy, necrosis and hyperplasia in alveolar epithelial cells by IQ administration were not observed in the same preparations stained with hematoxylin–eosin. Although IQ exposure was reported to cause cytotoxicity as well as mutagenicity in mammalian cells in vitro ( 28 , 29 ) lack of cytotoxicity in alveolar epithelium in vivo strongly indicates that the increase in BrdU‐LI was due to the primary effect of this chemical. Whatever the case, the increased type II epithelial cell proliferation observed in the present study might not be related to oxidative stress, but might be due to some direct action of IQ.
In an informative report concerning human risk, IQ induced hepatocellular tumors but failed to induce lung tumors in cynomolgus monkey primates.( 30 ) In these animals, N‐hydroxylation, subsequent adduct formation and probable carcinogenesis of IQ appears to be largely dependent on hepatic CYP3A4 and/or CYP2C9/10( 31 ) and not CYP1A2 having high specificity and catalytic activity for IQ,( 32 ) which is not expressed in liver of this species.( 31 ) Accordingly, our results cannot be simply extrapolated to the human case. However, care must be taken because genotoxic IQ is contained in cigarette smoke, though a small amount (estimated to be 0.26 ng per cigarette),( 3 ) combined with other many genotoxic, carcinogenic, and cocarcinogenic substances, as well as in cooked meat (estimated to be 20 ng per kilogram).( 1 , 2 , 33 ) The major pathway of HCA activation involves phase I hepatic CYP1A1/2‐mediated N‐hydroxylation followed by phase II (mainly N‐acetyltransferase and sulfotransferase) esterification of the N‐hydroxylamines to reactive ester derivatives that covalently bind C8 or N 2 positions of guanine in DNA.( 32 ) Cigarette smoke is known to be a lung CYP1A1 inducer in smokers( 34 , 35 ) and in rats.( 36 , 37 ) In addition, it has been reported in a case‐control study that there are significant associations among CYP1A1,( 38 , 39 ) N‐acetyltransferase( 40 , 41 ) and sulfotransferase( 42 ) polymorphisms and increased risk of lung cancer. Therefore, IQ might be classified as one of the important lung carcinogens in cigarette smoke.
In conclusion, our data indicate that IQ exerts strong promoting effects on DHPN‐lung carcinogenesis and may act as a complete carcinogen in the rat lung. As oxidative stress may not be a major factor for IQ carcinogenesis in the rat lung, further studies appear warranted to elucidate the underlying mechanisms. In addition, further experiments to evaluate rat lung carcinogenesis of other IQ‐type HCAs are now a high priority.
Acknowledgments
We appreciate the expert technical assistance provided by Machiko Maeda, Ayano Ogose and Ayako Kaneko. This work was supported in part by Health and Labour Science Research Grants from the Ministry of Health, Labour and Welfare of Japan.
References
- 1. Sugimura T. Overview of carcinogenic heterocyclic amines. Mutat Res 1997; 376: 211–9. [DOI] [PubMed] [Google Scholar]
- 2. Skog KI, Johansson MA, Jagerstad MI. Carcinogenic heterocyclic amines in model systems and cooked foods: a review on formation, occurrence and intake. Food Chem Toxicol 1998; 36: 879–96. [DOI] [PubMed] [Google Scholar]
- 3. Yamashita M, Wakabayashi K, Nagao M et al. Detection of 2‐amino‐3‐methylimidazo[4,5‐f]quinoline in cigarette smoke condensate. Jpn J Cancer Res 1986; 77: 419–22. [PubMed] [Google Scholar]
- 4. Takayama S, Nakatsuru Y, Masuda M, Ohgaki H, Sato S, Sugimura T. Demonstration of carcinogenicity in F344 rats of 2‐amino‐3‐methyl‐imidazo[4,5‐f]quinoline from broiled sardine, fried beef and beef extract. Jpn J Cancer Res (Gann) 1984; 75: 467–70. [PubMed] [Google Scholar]
- 5. Ohgaki H, Kusama K, Matsukura N et al. Carcinogenicity in mice of a mutagenic compound, 2‐amino‐3‐methylimidazo[4,5‐f]quinoline, from broiled sardine, cooked beef and beef extract. Carcinogenesis 1984; 5: 921–4. [DOI] [PubMed] [Google Scholar]
- 6. Nagao M. A new approach to risk estimation of food‐borne carcinogens – heterocyclic amines – based on molecular information. Mutat Res 1999; 431: 3–12. [DOI] [PubMed] [Google Scholar]
- 7. Kitamura Y, Umemura T, Okazaki K et al. Enhancing effects of simultaneous treatment with sodium nitrite on 2‐amino‐3‐methylimidazo[4,5‐f]quinoline‐induced rat liver, colon and Zymbal's gland carcinogenesis after initiation with diethylnitrosamine and 1,2‐dimethylhydrazine. Int J Cancer 2006; 118: 2399–404. [DOI] [PubMed] [Google Scholar]
- 8. Shivapurkar N, Hoover KL, Poirier LA. Effect of methionine and choline on liver tumor promotion by phenobarbital and DDT in diethylnitrosamine‐initiated rats. Carcinogenesis 1986; 7: 547–50. [DOI] [PubMed] [Google Scholar]
- 9. Alexander J, Wallin H. Metabolic fate of heterocyclic amines from cooked food. In: Hayatsu H, ed. Mutagens in Food: Detection and Prevention. Boca Raton: CRC Press, 1991; 143–52. [Google Scholar]
- 10. Schut HA, Snyderwine EG, Zu HX, Thorgeirsson SS. Similar patterns of DNA adduct formation of 2‐amino‐3‐methylimidazo[4,5‐f]quinoline in the Fischer 344 rat, CDF1 mouse, cynomolgus monkey and Salmonella typhimurium . Carcinogenesis 1991; 12: 931–4. [DOI] [PubMed] [Google Scholar]
- 11. Schut HA, Herzog CR, Cummings DA. Accumulation of DNA adducts of 2‐amino‐3‐methylimidazo[4,5‐f]quinoline (IQ) in tissues and white blood cells of the Fischer‐344 rat after multiple oral dosing. Carcinogenesis 1994; 15: 1467–70. [DOI] [PubMed] [Google Scholar]
- 12. Hirose M, Futakuchi M, Tanaka H et al. Prevention by antioxidants of heterocyclic amine‐induced carcinogenesis in a rat medium‐term liver bioassay: results of extended and combination treatment experiments. Eur J Cancer Prev 1998; 7: 61–7. [PubMed] [Google Scholar]
- 13. Hirose M, Takahashi S, Ogawa K, Futakuchi M, Shirai T. Phenolics: blocking agents for heterocyclic amine‐induced carcinogenesis. Food Chem Toxicol 1999; 37: 985–92. [DOI] [PubMed] [Google Scholar]
- 14. Sato K, Akaike T, Kojima Y, Ando M, Nagao M, Maeda H. Evidence of direct generation of oxygen free radicals from heterocyclic amines by NADPH/cytochrome P‐450 reductase in vitro . Jpn J Cancer Res 1992; 83: 1204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kato T, Hasegawa R, Nakae D et al. Dose‐dependent induction of 8‐hydroxyguanine and preneoplastic foci in rat liver by a food‐derived carcinogen, 2‐amino‐3,8‐dimethylimidazo[4,5‐f]quinoxaline, at low dose levels. Jpn J Cancer Res 1996; 87: 127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doll R. Strategy for detection of cancer hazards to man. Nature 1977; 265: 589–96. [DOI] [PubMed] [Google Scholar]
- 17. Thun MJ, Henley SJ, Calle EE. Tobacco use and cancer: an epidemiologic perspective for geneticists. Oncogene 2002; 21: 7307–25. [DOI] [PubMed] [Google Scholar]
- 18. Hecht SS, Chen CB, Ornaf RM, Hoffmann D, Tso TC. Chemical studies on tobacco smoke LVI. Tobacco specific nitrosamines: origins, carcinogenicity and metabolism. IARC Sci Publications 1978; 19: 395–413. [PubMed] [Google Scholar]
- 19. Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 1999; 91: 1194–210. [DOI] [PubMed] [Google Scholar]
- 20. Hecht SS. DNA adduct formation from tobacco‐specific N‐nitrosamines. Mutat Res 1999; 424: 127–42. [DOI] [PubMed] [Google Scholar]
- 21. Irie M, Tamae K, Iwamoto‐Tanaka N, Kasai H. Occupational and lifestyle factors and urinary 8‐hydroxydeoxyguanosine. Cancer Sci 2005; 96: 600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakae D, Mizumoto Y, Kobayashi E, Noguchi O, Konishi Y. Improved genomic/nuclear DNA extraction for 8‐hydroxydeoxyguanosine analysis of small amounts of rat liver tissue. Cancer Lett 1995; 97: 233–9. [DOI] [PubMed] [Google Scholar]
- 23. Uchiyama M, Mihara M. Determination of malonaldehyde precursor in tissue by the thiobarbituric acid test. Anal Biochem 1978; 86: 271–8. [DOI] [PubMed] [Google Scholar]
- 24. Snyderwine EG, Davis CD, Nouso K, Roller PP, Schut HA. 32P‐postlabeling analysis of IQ, MeIQx and PhIP adducts formed in vitro in DNA and polynucleotides and found in vivo in hepatic DNA from IQ‐, MeIQx‐ and PhIP‐treated monkeys. Carcinogenesis 1993; 14: 1389–95. [DOI] [PubMed] [Google Scholar]
- 25. Schut HA, Herzog CR. Formation of DNA adducts of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP) in male Fischer‐344 rats. Cancer Lett 1992; 67: 117–24. [DOI] [PubMed] [Google Scholar]
- 26. Møller P, Wallin H, Vogel U et al. Mutagenicity of 2‐amino‐3‐methylimidazo[4,5‐f]quinoline in colon and liver of Big Blue rats: role of DNA adducts, strand breaks, DNA repair and oxidative stress. Carcinogenesis 2002; 23: 1379–85. [DOI] [PubMed] [Google Scholar]
- 27. Witschi HP. Cell damage and cell renewal in the lung. In: Sipes IG, McQueen CA, Gandolfi JA, eds. Comprehensive Toxicology, Vol. 8 of Roth RA, ed. Toxicology of the Respiratory System. Oxford: Elsevier, 1997; 231–48. [Google Scholar]
- 28. Felton JS, Bjeldanes LF, Hatch FT. Mutagens in cooked foods – metabolism and genetic toxicity. Adv Exp Med Biol 1984; 177: 555–66. [DOI] [PubMed] [Google Scholar]
- 29. Fan L, Schut HA, Snyderwine EG. Cytotoxicity, DNA adduct formation and DNA repair induced by 2‐hydroxyamino‐3‐methylimidazo [4,5‐f]quinoline and 2‐hydroxyamino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine in cultured human mammary epithelial cells. Carcinogenesis 1995; 16: 775–9. [DOI] [PubMed] [Google Scholar]
- 30. Adamson RH, Thorgeirsson UP, Snyderwine EG et al. Carcinogenicity of 2‐amino‐3‐methylimidazo[4,5‐f]quinoline in nonhuman primates: induction of tumors in three macaques. Jpn J Cancer Res 1990; 81: 10–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Snyderwine EG, Turesky RJ, Turteltaub KW et al. Metabolism of food‐derived heterocyclic amines in nonhuman primates. Mutat Res 1997; 376: 203–10. [DOI] [PubMed] [Google Scholar]
- 32. Schut HA, Snyderwine EG. DNA adducts of heterocyclic amine food mutagens: implications for mutagenesis and carcinogenesis. Carcinogenesis 1999; 20: 353–68. [DOI] [PubMed] [Google Scholar]
- 33. Felton JS, Knize MG, Shen NH, Andresen BD, Bjeldanes LF, Hatch FT. Identification of the mutagens in cooked beef. Environ Health Perspect 1986; 67: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bartsch H, Castegnaro M, Rojas M, Camus AM, Alexandrov K, Lang M. Expression of pulmonary cytochrome P4501A1 and carcinogen DNA adduct formation in high risk subjects for tobacco‐related lung cancer. Toxicol Lett 1992; 64–65: 477–83. [DOI] [PubMed] [Google Scholar]
- 35. Kim JH, Sherman ME, Curriero FC, Guengerich FP, Strickland PT, Sutter TR. Expression of cytochromes P450 1A1 and 1B1 in human lung from smokers, non‐smokers, and ex‐smokers. Toxicol Appl Pharmacol 2004; 199: 210–9. [DOI] [PubMed] [Google Scholar]
- 36. Wardlaw SA, Nikula KJ, Kracko DA, Finch GL, Thornton‐Manning JR, Dahl AR. Effect of cigarette smoke on CYP1A1, CYP1A2 and CYP2B1/2 of nasal mucosa in F344 rats. Carcinogenesis 1998; 19: 655–62. [DOI] [PubMed] [Google Scholar]
- 37. Lee CZ, Royce FH, Denison MS, Pinkerton KE. Effect of in utero and postnatal exposure to environmental tobacco smoke on the developmental expression of pulmonary cytochrome P450 monooxygenases. J Biochem Mol Toxicol 2000; 14: 121–30. [DOI] [PubMed] [Google Scholar]
- 38. Taioli E, Gaspari L, Benhamou S et al. Polymorphisms in CYP1A1, GSTM1, GSTT1 and lung cancer below the age of 45 years. Int J Epidemiol 2003; 32: 60–3. [DOI] [PubMed] [Google Scholar]
- 39. Song N, Tan W, Xing D, Lin D. CYP 1A1 polymorphism and risk of lung cancer in relation to tobacco smoking: a case‐control study in China. Carcinogenesis 2001; 22: 11–6. [DOI] [PubMed] [Google Scholar]
- 40. Wikman H, Thiel S, Jager B et al. Relevance of N‐acetyltransferase 1 and 2 (NAT1, NAT2) genetic polymorphisms in non‐small cell lung cancer susceptibility. Pharmacogenetics 2001; 11: 157–68. [DOI] [PubMed] [Google Scholar]
- 41. Zhou W, Liu G, Thurston SW et al. Genetic polymorphisms in N‐acetyltransferase‐2 and microsomal epoxide hydrolase, cumulative cigarette smoking, and lung cancer. Cancer Epidemiol Biomarkers Prev 2002; 11: 15–21. [PubMed] [Google Scholar]
- 42. Liang G, Miao X, Zhou Y, Tan W, Lin D. A functional polymorphism in the SULT1A1 gene (G638A) is associated with risk of lung cancer in relation to tobacco smoking. Carcinogenesis 2004; 25: 773–8. [DOI] [PubMed] [Google Scholar]