

Abstract
Prostaglandin E2 is involved in colon carcinogenesis through its binding to the PGE2 receptor subtypes EP1, EP2, EP3 and EP4. We have demonstrated that administration of ONO‐8711, an EP1‐selective antagonist, suppresses development of AOM‐induced ACF in C57BL/6 mice and F344 rats. ONO‐8711 also reduced the numbers of intestinal polyps in Min mice. In the present study, we investigated the long‐term effects of ONO‐8711 on colon cancer development in rats treated with AOM. Male F344 rats were injected subcutaneously with AOM (15 mg/kg body weight) once a week for the first 2 weeks to develop colon cancer. Administration of 400 or 800 p.p.m. ONO‐8711 in their diets for 32 weeks reduced the incidence, multiplicity and volume of colon carcinomas. The incidence of colon adenocarcinomas in AOM‐treated rats was 97, 83 and 76% (P < 0.05) in the 0, 400 and 800 p.p.m. of ONO‐8711 groups, respectively. The multiplicity of adenocarcinomas was also decreased significantly, being 3.31 ± 0.33, 2.34 ± 0.27 (P < 0.05) and 2.06 ± 0.34 (P < 0.01) with 0, 400 and 800 p.p.m. of ONO‐8711, respectively. Moreover, treatment with 800 p.p.m. ONO‐8711 reduced the mean volume of adenocarcinomas to 49% (P < 0.05) of the value for the AOM treatment alone. Furthermore, the BrdU labeling index was decreased significantly in colon cancer cells by 800 p.p.m. ONO‐8711. These results confirm that EP1 is involved in colon carcinogenesis and that EP1‐selective antagonists might be promising candidates for colon cancer chemopreventive agents. (Cancer Sci 2005; 96: 260 –264)
Abbreviations:
- ABC
avidin biotin complex
- ACF
aberrant crypt foci
- AOM
azoxymethane
- Apc
adenomatous polyposis coli
- BrdU
5‐bromo‐2′‐deoxyuridine
- cAMP
cyclic AMP
- COX
cyclooxygenase
- FAP
familial adenomatous polyposis
- Gi
inhibitory G protein
- Gs
stimulatory G protein
- NSAID
non‐steroidal anti‐inflammatory drug
- PG
prostaglandin
- PhIP
2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine
- PKC
protein kinase C
- TX
thromboxane.
Colon cancer is one of the most frequent cancers in the world. (1) Epidemiological and experimental studies have indicated that aspirin and other NSAIDs can reduce the development of colon cancer, 1 , 2 , 3 , 4 , 5 the suggested mechanism being inhibition of COX activity, which catalyzes the synthesis of prostanoids. There are two isoforms of COX, the constitutively expressed COX‐1 and the inducible COX‐2. Both contribute to PGE2 production and colon carcinogenesis, and PGE2 levels are known to be elevated in human and rodent colon tumors. 6 , 7 , 8 , 9
Prostanoids such as PGD2, PGE2, PGF2, PGI2 and TXA2 exert their biological actions through binding to specific membrane receptors, which include DP for PGD2, FP for PGF2, IP for PGI2, TP for TXA2 and EP1 to EP4 for PGE2. 10 , 11 , 12 The PGE2 receptors are transmembrane G protein‐coupled receptors and it has been established that EP1 signals are transmitted by increased intracellular Ca2+ concentrations, with activation of phosphorylated PKC. (10) However, the species of G protein coupling to EP1 remains unidentified. EP2 and EP4 receptors couple to Gs and increase cAMP synthesis by adenylate cyclase, and the EP3 receptor couples to Gi and decreases cAMP synthesis by inhibition of adenylate cyclase. (13)
Our previous study using EP1–4, DP, FP, IP and TP receptor knockout mice showed that deficiency of either EP1 or EP4 receptors decreases formation of AOM‐induced colon ACF, putative preneoplastic lesions. 14 , 15 It has also been reported that the numbers of intestinal polyps in EP2‐deficient Apc Δ716 mice are lower than in their Apc Δ716 counterparts. (16) In contrast, enhancement of AOM‐induced colon cancer development has been observed in EP3 receptor knockout mice. (17) Thus, it is considered that EP1, EP2 and EP4 receptors are involved in enhancement of colon carcinogenesis, while the EP3 receptor acts against tumor development.
In addition to genetic approaches, pharmacological research has been carried out to examine the roles of EP1 and EP4. Administration of ONO‐8711, 6‐[(2S,3S)‐3‐(4‐chloro‐2‐methylphenylsulfonylaminomethyl)‐bicyclo[2.2.2]octan‐2‐yl]‐5Z‐hexenoic acid, an EP1‐selective antagonist, reduced AOM‐induced ACF formation in C57BL/6 mice and F344 rats. 14 , 18 Furthermore, treatment with ONO‐8713, 4‐{2‐[N‐isobutyl‐N‐(2‐furylsulfonyl)amino]‐5‐trifluoromethylphenoxymethyl}cinnamic acid, another EP1 antagonist, and ONO‐AE2‐227, 2‐{2‐[2‐(1‐naphthyl)propanoylamino]phenyl}methylbenzoic acid, an EP4 antagonist, similarly suppressed AOM‐induced ACF formation in C57BL/6 mice. 15 , 19 Administration of ONO‐8711 and ONO‐AE2‐227 to Min mice, an animal model for human FAP, also reduced the number of intestinal polyps. 14 , 15 EP receptors transmit their signals by independent pathways, and combination treatment with ONO‐8711 and ONO‐AE2‐227 caused additional reduction in intestinal polyp formation in mice with a truncated adenomatous polyposis coli (Apc) gene at codon 1309 (Apc 1309 mice). (20)
Although evidence has thus accumulated that EP1 is involved in colon carcinogenesis, significant effects of EP1 antagonists on yields of actual colon cancers have yet to be reported. The present study was therefore designed to determine the suppressive influence of an EP1‐selective antagonist, ONO‐8711, on AOM‐induced colon tumor development in male F344 rats. Administration of ONO‐8711 reduced the colon cancer incidence, multiplicity and volume, along with cell proliferation in rat colon tumor cells. On the basis of these results, possible mechanisms of ONO‐8711 in colon cancer suppression are discussed.
Materials and Methods
Animals and chemicals. Male F344 rats, at 4 weeks of age, were purchased from Charles River Japan (Atsugi, Japan) and acclimated to laboratory conditions for 1 week. Two or three animals were housed per plastic cage, with sterilized softwood chips as bedding, in a barrier‐sustained animal room air‐conditioned at 24 ± 2°C and 55% humidity, on a 12 : 12 h light:dark cycle. AOM and BrdU were purchased from Sigma Chemical Co. (St Louis, MO, USA). The selective PGE receptor EP1 antagonist ONO‐8711, 6‐([2S,3S]‐3‐[4‐chloro‐2‐methylphenylsulfonylaminomethyl]‐bicyclo{2.2.2}octan‐2‐yl)‐5Z‐hexenoic acid, was synthesized chemically at Ono Pharmaceutical Co. (Osaka, Japan) and well mixed with powdered basal diet AIN‐76A (Dyets, Bethlehem, PA, USA) at concentrations of 400 and 800 p.p.m. The doses were selected based on the results of our previous study, in which 400 or 800 p.p.m. ONO‐8711 in the diet suppressed intestinal polyp formation in Apc 1309 mice and PhIP‐induced breast cancer development. 20 , 21
Animal experiments. The rats were divided into five groups as shown in Fig. 1. Groups 1–3 (36 animals per group) were treated subcutaneously with AOM in sterile saline at a dose of 15 mg/kg body weight once a week for 2 weeks. From the day of the first treatment with AOM, these animals were fed the following diets for 32 weeks: group 1, the basal diet; group 2, 400 p.p.m. ONO‐8711; group 3, 800 p.p.m. ONO‐8711. Groups 4 and 5, six animals per group, were the corresponding controls to groups 1 and 3, respectively, and were injected with saline without AOM followed by the basal diet or 800 p.p.m. of ONO‐8711. Food and water were available ad libitum. The animals were observed daily for clinical signs and mortality. Body weight and food consumption were measured weekly. At 37 weeks of age, all animals were sacrificed under anesthesia, 1 h after i.p. injection of BrdU in saline solution (50 mg/kg body weight) and complete autopsies were carried out. The liver, kidney and spleen were removed and weighed. Each intestinal tract was removed, opened longitudinally and fixed flat between sheets of filter paper in 10% neutral buffered formalin. The number, size and location of all intestinal tumors were determined. Estimation of tumor volume (V) was determined using the formula V = L × W × D × π/6, where L is length, W is width and D is depth of colon tumor. (22) Tissues were processed in paraffin, and sections were stained with hematoxylin and eosin for histopathological examination. Diagnosis of intestinal tumors was carried out according to the classification of Pozharisski. (23) The experimental protocol followed the guidelines for Animal Experiments in the National Cancer Center.
Figure 1.
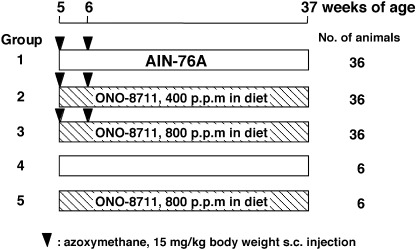
Experimental protocol. The animals were male F344 rats at 5 weeks of age at the commencement. They were treated subcutaneously with AOM (15 mg/kg body weight) once a week for 2 weeks and from the day of the first treatment were fed a basal diet or a diet containing 400 or 800 p.p.m. ONO‐8711 for 32 weeks.
BrdU labeling index. Two serial sections, including colon adenocarcinoma tissues and normal colonic epithelium, were prepared. One section was used for determination of BrdU labeling indexes and the other for apoptotic indexes. Cell proliferation in the colon carcinomas and normal colonic epithelium were examined by immunohistochemical detection of BrdU incorporation using the ABC method (Vectastain Elite ABC kit, Vector Laboratories, Burlingame, CA, USA) and a mouse monoclonal antibody to BrdU (1 : 300; Dako, Kyoto, Japan). For determination of BrdU labeling indexes, three representative fields in each section of a colon carcinoma and surrounding normal‐appearing tissue were selected under light‐microscope examination at a magnification of ×400. In each section, one carcinoma per rat was used for counting. The number of BrdU‐positive nuclei in a minimum of 1500 cells was counted and the result was expressed as a percentage value.
Apoptotic index. Sections were stained with the aid of an ApopTag peroxidase in situ apoptosis detection kit (Chemicon International, Temecula, CA, USA) according to the manufacturer's instructions. For analysis of apoptosis, well‐defined and darkly stained apoptotic cells and bodies in the colon carcinomas were counted in the same way as for BrdU‐positive cells. The percentage of positive cells (apoptotic index) was determined by calculating the positive cell number : total cell number × 100.
Statistical analysis. Data are presented as mean ± SE values. Data for body weight, organ weight, tumor multiplicity, tumor volume, BrdU labeling and apoptotic index were compared by Student's t‐test or Welch's t‐test. Data for tumor incidences were analyzed by Fisher's exact probability test. Differences were considered to be statistically significant with P < 0.05 (two‐tailed).
Results
Administration of ONO‐8711 at doses of 400 and 800 p.p.m. in the diet for 32 weeks did not affect food intake or clinical signs of AOM‐treated animals. The mean body weights at 37 weeks of age were 355.0 ± 3.3 g in the AOM control group, 350.5 ± 3.0 g in the AOM + 400 p.p.m. ONO‐8711 group, 340.4 ± 3.8 g in the AOM + 800 p.p.m. ONO‐8711 group, 354.3 ± 5.9 g in the saline control group and 342.7 ± 10.5 g in the saline + 800 p.p.m. ONO‐8711 group. In AOM‐ and saline‐treated rats fed the diet containing ONO‐8711, there were no gross changes in body weight or any organ weight that would point to any toxicity of ONO‐8711.
Data for incidences and multiplicities of intestinal tumors are summarized in Table 1. More were well‐differentiated tubular adenocarcinomas and the main sites of development were the distal and middle colons. The incidence of colon adenocarcinomas in AOM‐treated rats was 97, 83 and 76% (P < 0.05) in the 0, 400 and 800 p.p.m. ONO‐8711‐treated groups, respectively (Table 1). The multiplicity of colon adenocarcinomas also decreased dose‐dependently, from 3.31 ± 0.33 for the AOM‐treated control group to 2.34 ± 0.27 (P < 0.05), and to 2.06 ± 0.34 (P < 0.01) by 400 and 800 p.p.m. ONO‐8711 treatments, respectively (Table 1). The incidence and multiplicity of colon adenomas in the groups treated with 800 p.p.m. ONO‐8711 were increased slightly, but not significantly. The percentage of well‐differentiated adenocarcinomas was also increased slightly in the 800 p.p.m. group (Table 2). On the other hand, suppression of adenocarninoma volume was observed by ONO‐8711 administration (Fig. 2). The reduction of adenocarcinomas was from 26.3 ± 5.8 mm3 for the AOM control group to 14.1 ± 4.5 mm3 at 400 p.p.m. and to 13.0 ± 3.1 mm3 (P < 0.05) at 800 p.p.m. The mean volumes of colon adenomas in the 400 and 800 p.p.m. ONO‐8711‐treated groups also tended to be reduced, but they were not big enough to evaluate a significant difference from that in the control group (0.48 ± 0.26 mm3 vs 0.30 ± 0.13 mm3 and 0.14 ± 0.04 mm3 in the 400 and 800 p.p.m. groups, respectively). In the small intestine, the incidence and multiplicity of adenocarcinomas were not significantly lower in rats treated with ONO‐8711, and no dose‐dependency was observed: the incidences were 29, 9 and 18% in the 0, 400 and 800 p.p.m. of ONO‐8711‐treated groups, respectively, and the multiplicities were 0.29 ± 0.08, 0.09 ± 0.05 (P < 0.05) and 0.21 ± 0.08 (Table 1).
Table 1.
Effects of ONO‐8711 treatment on the incidences and multiplicities of AOM‐induced intestinal tumors in rats
| Treatment | Effective no. of animals | No. of animals with tumors (%) | No. of tumors per rat (mean ± SE) | ||||
|---|---|---|---|---|---|---|---|
| Small intestine (carcinoma) | Large intestine | Small intestine (carcinoma) | Large intestine | ||||
| Adenoma | Carcinoma | Adenoma | Carcinoma | ||||
| AOM alone | 35 | 10 (29) | 15 (43) | 34 (97) | 0.29 ± 0.08 | 0.66 ± 0.15 | 3.31 ± 0.33 |
| AOM + ONO‐8711, 400 p.p.m. | 35 | 3 (9) | 16 (46) | 29 (83) | 0.09 ± 0.05* | 0.66 ± 0.15 | 2.34 ± 0.27* |
| AOM + ONO‐8711, 800 p.p.m. | 33 | 6 (18) | 16 (48) | 25 (76)* | 0.21 ± 0.08 | 0.85 ± 0.19 | 2.06 ± 0.34** |
| Saline alone | 6 | 0 | 0 | 0 | 0 | 0 | 0 |
| ONO‐8711, 800 p.p.m. | 6 | 0 | 0 | 0 | 0 | 0 | 0 |
Significantly different from the control value at P < 0.05.
Significantly different from the control value at P < 0.01.
Table 2.
Effects of ONO‐8711 on histological types of AOM‐induced colon carcinomas
| Treatment | Effective no. of animals | Total no. of carcinomas (%) | No. of carcinomas diagnosed | |||
|---|---|---|---|---|---|---|
| Well‐ differentiated (%) | Moderately‐ differentiated (%) | Signet‐ring cell (%) | Mucinous (%) | |||
| AOM alone | 35 | 116 (100) | 108 (93) | 3 (3) | 1 (1) | 4 (3) |
| AOM + ONO‐8711, 400 p.p.m. | 35 | 82 (100) | 77 (94) | 2 (2) | 3 (4) | 0 (0) |
| AOM + ONO‐8711, 800 p.p.m. | 33 | 68 (100) | 65 (96) | 0 (0) | 2 (3) | 1 (1) |
Figure 2.
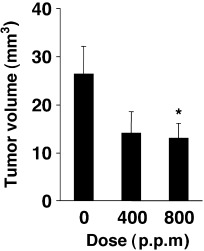
Effects of ONO‐8711 on the mean volumes of adenocarcinomas of the AOM‐treated rat colon. Bars show AOM alone (control group), AOM + 400 p.p.m. ONO‐8711 and AOM + 800 p.p.m. ONO‐8711. *Significantly different from the control value at P < 0.05.
Data for BrdU‐positive cells (BrdU labeling indexes) and apoptotic cells (apoptotic indexes) in the normal colon epithelium and colon carcinomas are summarized in Table 3. The mean BrdU labeling index for carcinomas in the AOM‐treated control group was higher than in the other groups. Administration of AOM + 400 and 800 p.p.m. ONO‐8711 reduced the BrdU labeling index to 59 and 54% (P < 0.05) of the AOM‐treated control value, respectively. On the other hand, AOM + 400 and 800 p.p.m. ONO‐8711 treatment did not show any effect on BrdU labeling indexes in normal mucosa of rats. The apoptotic indexes for carcinomas in the groups of AOM + 400 and 800 p.p.m. ONO‐8711 treatment were 103 and 118% of the AOM control value, respectively (Table 3).
Table 3.
Effects of ONO‐8711 on cell proliferation and apoptosis in the colon
| Treatment | No. of rats tested | BrdU labeling index (mean ± SE) | Apoptotic index (mean ± SE) | |
|---|---|---|---|---|
| Normal mucosa | Carcinoma | Carcinoma | ||
| AOM alone | 4 | 3.5 ± 0.1 | 9.0 ± 1.2 | 0.39 ± 0.06 |
| AOM + ONO‐8711, 400 p.p.m. | 4 | 3.8 ± 0.1 | 5.3 ± 1.1 | 0.40 ± 0.16 |
| AOM + ONO‐8711, 800 p.p.m. | 4 | 3.3 ± 0.2 | 4.9 ± 0.8* | 0.46 ± 0.05 |
Significantly different from the control value at P < 0.05.
Discussion
The present study demonstrated administration of the EP1‐selective antagonist ONO‐8711 to significantly reduce the incidence and multiplicity of AOM‐induced rat colon carcinomas, without any toxic effects in terms of body and organ weights. A decrease in the mean volume of adenocarcinomas was observed, and the BrdU labeling index for colon carcinoma was also reduced by treatment with ONO‐8711. These results support our previous short‐term experiment that treatment with 800 p.p.m. ONO‐8711 in the diet reduced the proliferative activity of the colon ACF in rats. (18)
In our previous study, 800 p.p.m. ONO‐8711 administration inhibited ACF development (31% reduction), (18) and a 38% reduction was observed in the number of tumors in the present study. The suppressive effect of 800 p.p.m. ONO‐8711 was observed not only in the number of ACF, but also in their growth, (18) as mentioned. For instance, the numbers of ACF with one crypt, two crypts, three crypts and greater than four crypts were decreased by 11, 32, 51 and 55%, respectively. (18) This is in line with the decreased carcinoma volume and BrdU labeling index in the colon carcinoma in the present study. Our previous report (18) also shows that the BrdU labeling index in the normal colonic epithelium in AOM‐treated rats given 800 p.p.m. ONO‐8711 for 5 weeks from 5 weeks of age were almost the same as those in the AOM‐treated control rats. The present study confirmed the previous findings. Although the incidence and multiplicity of colon adenomas and the percentages of well‐differentiated adenocarcinomas in the 800 p.p.m. ONO‐8711 group were slightly increased, it may not be stating it too strongly that ONO‐8711 could block progression of colon carcinogenesis. Substances that prevent early stages of carcinogenesis or delay the carcinogenic process are considered to be good candidates as chemopreventive agents. Therefore, EP1 antagonists, including ONO‐8711, deserve consideration in this respect.
The tumor suppressive mechanisms of ONO‐8711 need to be revealed in more detail. EP1 signals are transmitted by increased intracellular Ca2+ concentrations, with activation of PKC. (10) As noted above, the physiological activities of the EP2 and EP4 receptors and EP3 receptor through activation and inhibition of cAMP synthesis being consistent with their respective effects on colon tumor development. (13) In contrast, the effect of the EP1‐selective antagonist on PKC activity is uncertain. It has been reported decreased PKCα and δ and increased PKCβII expression levels were observed in colon tumors compared with surrounding normal epithelial cells. (24) PKCα and PKCδ inhibit cell proliferation and promote differentiation of many cell types in vitro, 25 , 26 , 27 , 28 and PKCβII is linked to enhancement of cell proliferation and suppression of apoptosis. (29) In our preliminary experiments, the expression levels of phosphorylated PKCα and δ in the colon tumors were not affected by 800 p.p.m. ONO‐8711 treatment (data not shown). However, the roles of PKC in colon cancer formation appear complex, and further studies are needed to investigate the relationship between EP1 receptor signaling and downstream targets, like PKC isozymes, in colon carcinogenesis.
In conclusion, the present study demonstrated that the EP1‐selective antagonist ONO‐8711 has the potential to suppress AOM‐induced development of colon cancers in rats. ONO‐8711 may be a promising candidate chemopreventive agent for colon cancer.
Acknowledgments
This work was supported by Grants‐in‐Aid for Cancer Research, for the Third‐Term Comprehensive 10‐Year Strategy for Cancer Control, and for the Research on Advanced Medical Technology from the Ministry of Health, Labor and Welfare of Japan, and a Grant‐in‐Aid for the Sagawa Foundation for Promotion of Cancer Research.
References
- 1. Stewart BW, Kleihues P, eds. World Cancer Report. Lyon: IARC Press, 2003; 12–17. [Google Scholar]
- 2. Thun MJ, Namboodiri MM, Heath CW Jr. Aspirin use and reduced risk of fatal colon cancer. N Engl J Med 1991; 325: 1593–6. [DOI] [PubMed] [Google Scholar]
- 3. Rao CV, Rivenson A, Simi B, Zang E, Kelloff G, Steele V, Reddy BS. Chemoprevention of colon carcinogenesis by sulindac, a nonsteroidal anti‐inflammatory agent. Cancer Res 1995; 55: 1464–7. [PubMed] [Google Scholar]
- 4. Smalley WE, DuBois RN. Colorectal cancer and nonsteroidal anti‐inflammatory drugs. Adv Pharmacol 1997; 39: 1–20. [DOI] [PubMed] [Google Scholar]
- 5. Chan AT. Aspirin, non‐steroidal anti‐inflammatory drugs and colorectal neoplasia: future challenges in chemoprevention. Cancer Causes Control 2003; 14: 413–18. [DOI] [PubMed] [Google Scholar]
- 6. Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol 1998; 38: 97–120. [DOI] [PubMed] [Google Scholar]
- 7. Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O, Langenbach R. Genetic disruption of Ptgs‐1, as well as Ptgs‐2, reduces intestinal tumorigenesis in Min mice. Cancer Res 2000; 60: 4705–8. [PubMed] [Google Scholar]
- 8. Rigas B, Goldman IS, Levine L. Altered eicosanoid levels in human colon cancer. J Lab Clin Med 1993; 122: 518–23. [PubMed] [Google Scholar]
- 9. Pugh S, Thomas GA. Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2 . Gut 1994; 35: 675–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev 1994; 46: 205–29. [PubMed] [Google Scholar]
- 11. Suda M, Tanaka K, Natsui K, Usui T, Tanaka I, Fukushima M, Shigeno C, Konishi J, Narumiya S, Ichikawa A, Nakao N. Prostaglandin E receptor subtypes in mouse osteoblastic cell line. Endocrinology 1996; 137: 1698–705. [DOI] [PubMed] [Google Scholar]
- 12. Ushikubi F, Hirata M, Narumiya S. Molecular biology of prostanoid receptors; an overview. J Lipid Mediat Cell Signal 1995; 12: 343–59. [DOI] [PubMed] [Google Scholar]
- 13. Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev 1999; 79: 1193–226. [DOI] [PubMed] [Google Scholar]
- 14. Watanabe K, Kawamori T, Nakatsugi S, Ohta T, Ohuchida S, Yamamoto H, Maruyama T, Kondo K, Ushikubi F, Narumiya S, Sugimura T, Wakabayashi K. Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Res 1999; 59: 5093–6. [PubMed] [Google Scholar]
- 15. Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, Tani K, Kobayashi M, Maruyama T, Kobayashi K, Ohuchida S, Sugimoto Y, Narumiya S, Sugimura T, Wakabayashi K. Involvement of prostaglandin E receptor subtype EP4 in colon carcinogenesis. Cancer Res 2002; 62: 28–32. [PubMed] [Google Scholar]
- 16. Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc Δ716 knockout mice. Nature Med 2001; 7: 1048–51. [DOI] [PubMed] [Google Scholar]
- 17. Shoji Y, Takahashi M, Kitamura T, Watanabe K, Kawamori T, Maruyama T, Sugimoto Y, Negishi M, Narumiya S, Sugimura T, , Wakabayashi K . Downregulation of prostaglandin E receptor subtype EP3 during colon cancer development. Gut 2004; 53: 1151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kawamori T, Uchiya N, Kitamura T, Ohuchida S, Yamamoto H, Maruyama T, Sugimura T, Wakabayashi K. Evaluation of a selective prostaglandin E receptor EP1 antagonist for potential properties in colon carcinogenesis. Anticancer Res 2001; 21: 3865–9. [PubMed] [Google Scholar]
- 19. Watanabe K, Kawamori T, Nakatsugi S et al. Inhibitory effect of a prostaglandin E receptor subtype EP1 selective antagonist, ONO‐8713, on development of azoxymethane‐induced aberrant crypt foci in mice. Cancer Lett 2000; 156: 57–61. [DOI] [PubMed] [Google Scholar]
- 20. Kitamura T, Itoh M, Noda T, Tani K, Kobayashi M, Maruyama T, Kobayashi K, Ohuchida S, Sugimura T, Wakabayashi K. Combined effects of prostaglandin E receptor subtype EP1 and subtype EP4 antagonists on intestinal tumorigenesis in adenomatous polyposis coli gene knockout mice. Cancer Sci 2000; 94: 618–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kawamori T, Uchiya N, Nakatsugi S, Watanabe K, Ohuchida S, Yamamoto H, Maruyama T, Kondo K, Sugimura T, Wakabayashi K. Chemopreventive effects of ONO‐8711, a selective prostaglandin E receptor EP1 antagonist, on breast cancer development. Carcinogenesis 2001; 22: 2001–4. [DOI] [PubMed] [Google Scholar]
- 22. Rao CV, Indranie C, Simi B, Manning PT, Connor JR, Reddy BS. Chemopreventive properties of a selective inducible nitric oxide synthase inhibitor in colon carcinogenesis, administered alone or in combination with celecoxib, a selective cyclooxygenase‐2 inhibitor. Cancer Res 2002; 62: 165–70. [PubMed] [Google Scholar]
- 23. Pozharisski KM. Tumors of the intestines. In: Turusov VS, ed. Pathology of Tumours in Laboratory Animals. Lyon: IARC Scientific Publications, 1990; 119–40. [PubMed] [Google Scholar]
- 24. Verstovsek G, Byrd A, Frey MR, Petrelli NJ, Black JD. Colonocyte differentiation is associated with increased expression and altered distribution of protein kinase C isozymes. Gastroenterology 1998; 115: 75–85. [DOI] [PubMed] [Google Scholar]
- 25. Diaz‐Meco MT, Berra E, Municio MM, Sanz L, Lozano J, Dominguez I, Diaz‐Golpe V, Lain de Lera MT, Alcami J, Paya CV et al. A dominant negative protein kinase C ζ subspecies blocks NF‐κB activation. Mol Cell Biol 1993; 13: 4770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frey MR, Saxon ML, Zhao X, Rollins A, Evans SS, Black JD. Protein kinase C isozyme‐mediated cell cycle arrest involves induction of p21waf1/cip1 and p27kip1 and hypophosphorylation of the retinoblastoma protein in intestinal epithelial cells. J Biol Chem 1997; 272: 9424–35. [DOI] [PubMed] [Google Scholar]
- 27. Borner C, Ueffing M, Jaken S, Parker PJ, Weinstein IB. Two closely related isoforms of protein kinase C produce reciprocal effects on the growth of rat fibroblasts. Possible molecular mechanisms. J Biol Chem 1995; 270: 78–86. [DOI] [PubMed] [Google Scholar]
- 28. Scaglione‐Sewell B, Abraham C, Bissonnette M, Skarosi SF, Hart J, Davidson NO, Wali RK, Davis BH, Sitrin M, Brasitus TA. Decreased PKCα expression increases cellular proliferation, decreases differentiation, and enhances the transformed phenotype of CaCo‐2 cells. Cancer Res 1998; 58: 1074–81. [PubMed] [Google Scholar]
- 29. Davidson LA, Brown RE, Chang WC, Morris JS, Wang N, Carroll RJ, Turner ND, Lupton JR, Chapkin RS. Morphodensitometric analysis of protein kinase C βII expression in rat colon: modulation by diet and relation to in situ cell proliferation and apoptosis. Carcinogenesis 2000; 21: 1513–19. [PubMed] [Google Scholar]