

Abstract
The breast cancer susceptibility gene (BRCA2) is localized mainly in the nucleus where it plays an important role in DNA damage repair. Some BRCA2 protein is also present in the centrosome. Here, we demonstrate that BRCA2 interacts with plectin, a cytoskeletal cross‐linker protein, and that this interaction controls the position of the centrosome. Phosphorylation of plectin by cyclin‐dependent kinase 1/cyclin B (CDK1/CycB) kinase has been reported to abolish its cross‐linking function during mitosis. Here, we induced phosphorylation of plectin in prepared fractions of HeLa cells by adding activated CDK1/CycB kinase. Consequently, there was significant dissociation of the centrosome from the nuclear membrane. Plectin has six homologous ankyrin‐like repeat domains (termed PLEC M1‐M6). Using a pull‐down assay, we found that GST–PLEC M1 and a GST‐C‐terminal region fusion protein (which comprised PLEC M6, along with an adjacent vimentin site) interacted with BRCA2. Since each PLEC module exhibits high homology to the others, the possibility of all six domains participating in this interaction was indicated. Moreover, when PLEC M1 was overexpressed in HeLa cells, it competed with endogenous plectin and inhibited the BRCA2–plectin interaction. This inhibitory effect resulted in dissociation of the centrosomes from the nucleus and increased the rate of micronuclei formation which may lead to carcinogenesis. In addition, when either BRCA2 or plectin was suppressed by the appropriate siRNA, a similar change in centrosomal positioning was observed. We suggest that the BRCA2–plectin interaction plays an important role in the regulation of centrosome localization and also that displacement of the centrosome may result in genomic instability and cancer development. (Cancer Sci 2009; 00: 000–000)
Mutations of breast cancer susceptibility gene (BRCA2), a tumor suppressor gene, are associated with an increased susceptibility to breast and ovarian carcinomas.( 1 , 2 ) The BRCA2 protein plays important roles in both transcriptional regulation and DNA damage repair mediated by homologous recombination.( 3 , 4 ) BRCA2 forms complexes with Rad51 that are important for the maintenance of genomic stability during DNA recombination and double‐strand break repair.( 5 , 6 ) Murine embryo fibroblasts that are homozygous for a targeted mutation of Brca2 (BRCA2Tr2014) show centrosome amplification, spontaneous accumulation of chromosomal abnormalities, and defective cytokinesis.( 7 , 8 ) Recently, we reported that BRCA2 interacts with γ‐tubulin (a component of the centrosome), has nuclear export and centrosome localization signals, and localizes to the centrosomes during the S and early M phases of the cell cycle.( 9 , 10 ) Moreover, suppression of BRCA2 using small interfering RNA (siRNA) causes abnormalities in the position or duplication of the centrosome.( 7 , 10 ) Nevertheless, despite these observations, the function of BRCA2 with respect to the centrosome remains uncertain.
Here, we demonstrate that plectin, a cytoskeletal cross‐linker protein widely expressed in a range of mammalian tissues and cell types,( 11 ) interacts with BRCA2. Plectin is a protein of exceptionally high‐molecular mass (>500 kDa), and electron microscopy of purified plectin molecules revealed an extended central rod and two flanking globular domains.( 12 ) Deficiency in the plectin gene causes epidermolysis bullosa simplex with muscular dystrophy, and more recently has been suggested to be implicated in tumorigenesis, such as pancreatic ductal adenocarcinoma and hepatocellular carcinoma.( 13 , 14 , 15 ) Moreover, it is known that plectin belongs to the plakin family and that has been shown to interact with microtubules, actin filaments, and intermediate filaments (IFs) such as vimentin, cytokeratin, and desmin, and plays a very important role in maintaining cell structure and mediating the mechanical integration of cells and tissues.( 11 , 16 , 17 , 18 , 19 ) It is known that ablation of plectin by siRNA in MCF7 epithelial cells had a negative effect on their migratory capacity,( 20 ) and that the distribution of vimentin structures is altered in mitotic plectin‐deficient fibroblasts.( 21 )
Plectin has been shown to be a direct target of mitotic cyclin‐dependent kinase 1/cyclin B (CDK1/CycB) kinase. Phosphorylation of plectin subunits by CDK1/CycB kinase at the onset of mitosis is correlated with disassembly of the plectin network.( 22 ) In view of the proposed role of plectin as a cytoplasmic cross‐linking element, specific regulation of its binding activities would seem to be particularly important during mitosis, when dramatic structural rearrangements of the cytoskeleton, including the IF networks, take place. Furthermore, plectin interacts with the nuclear membrane proteins lamin B and nesprin‐3.( 23 , 24 ) On the basis of the above information, we hypothesized that BRCA2‐plectin complexes might play a role in connecting the centrosome to the nuclear membrane. To test this hypothesis, we inhibited the interaction of BRCA2 with plectin and found that this inhibition altered localization of the centrosome and caused abnormalities in respect to nuclear replication. From these results, we propose that interaction between BRCA2 and plectin plays an important role in the positioning of the centrosome, and also contributes to maintaining genomic stability.
Materials and Methods
Cell lines
HeLa S3 cells were provided by the Riken Cell Bank (Tsukuba, Japan). COS7 and MCF7 (human breast cancer cell line) cells were purchased from ATCC (Manassas, VA, USA).
Antibodies
The following antibodies were used in this study: plectin (10F6)‐mouse monoclonal antibody (Bender MedSystems, Burlingame, CA, USA); γ‐tubulin (C‐11)‐mouse monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA); γ‐tubulin (T3559)‐rabbit polyclonal antibody (Sigma‐Aldrich, St. Louis, MO, USA); γ‐tubulin (C‐20)‐goat polyclonal antibody (Santa Cruz Biotechnology, San Diego, CA, USA); BRCA2 (Ab‐1)‐mouse monoclonal antibody (Calbiochem, San Diego, CA, USA); BRCA2 (Ab‐2)‐rabbit polyclonal antibody (Neomarkers, Fremont, CA, USA); FLAG (M2)‐mouse monoclonal antibody (Sigma‐Aldrich); HA (3F10)‐rat monoclonal antibody (Roche, Osaka, Japan); HA (12CA5)‐mouse monoclonal antibody (Roche); GST (Ab‐1)‐rabbit polyclonal antibody (Calbiochem); MCM7 (DCS‐141)‐mouse monoclonal antibody (Sigma‐Aldrich); vimentin (V9)‐mouse monoclonal antibody (Chemicon, Temecula, CA, USA); vinculin (FB‐11)‐mouse monoclonal antibody (Chemicon); lamin A/C (636)‐mouse monoclonal antibody (Santa Cruz Biotechnology); β‐actin (MAB1501R)‐mouse monoclonal antibody (Chemicon); and α‐tubulin‐rabbit polyclonal antibody (Abcam, Cambridge, MA, USA).
Plasmids and constructs
To obtain an expression vector coding for human plectin module 1 (PLEC M1) with an N‐terminal GST or HA tag, the gene was amplified by PCR and cloned into either pGEX 4T‐2 expression vector (Amersham Biosciences, Piscataway, NJ, USA) or pME18S HA. Furthermore, to obtain an expression vector coding for human PLEC M‐C with an N‐terminal GST tag, the gene was amplified by PCR and cloned into pGEX 4T‐1 expression vector (Amersham Biosciences). The gene was amplified by PCR using the following primers: GST‐PLEC M1, 5′‐CTGGAATTCTAGCCACAAAGACCCTGCCCAAT‐3′ and 5′‐TATCTCGAGCTAGCCTTATCCGTGAGTGGCA‐3′ (pGEX 4T‐2); HA‐PLEC M1, 5′‐CTCGAATTCTGCCACAAAGACCCTGCCCAAT‐3′ and 5′‐TATCTCGAGCTAGCCTTATCCGTGAGTGGCA‐3′ (pME18S HA); and GST‐PLEC M‐C, 5′‐ACCGGAATTCGAGAAGAAGCGGGAGCGGAA‐3′ and 5′‐AATCTCGAGCGCTGTAGTAGCCCTTGGTGG‐3′. The amplification was performed using 35 cycles of 94°C for 30 s, 65°C for 30 s, and 72°C for 1 min. The PLEC M1 gene was inserted into the EcoRI‐XhoI sites of pGEX 4T‐2 or pME18S HA and the PLEC M‐C gene was inserted into the EcoRI‐XhoI sites of pGEX 4T‐1. The plasmids were verified by DNA sequencing and protein expression, and their sizes confirmed by western blotting. Full‐length human BRCA2 cDNA, a gift from F.J. Couch (Mayo Clinic and Foundation, Rochester, MN, USA), was subcloned into the pME18S‐FL3 vector. The pME18S‐FL3 vector was kindly provided by Dr S. Sugano (University of Tokyo). In order to generate FLAG‐tagged BRCA2 protein, we fused the FLAG gene 3′ to the BRCA2 gene, such that the FLAG tag was fused to the carboxy terminus of the BRCA2 protein.
Glycerol density gradient centrifugation
HeLa S3 cell lysates were prepared by harvesting the cells in lysate buffer (20 mM Tris‐HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 1% NP40) containing protease inhibitors (completely EDTA free; Roche). The extract was centrifuged at 15 000 g for 30 min to remove cell debris. The lysates were layered on top of a 15–30% (w/v) glycerol gradient containing 20 mM Tris‐HCl (pH 8.0), 100 mM NaCl, and 1 mM EDTA. Centrifugation was performed in a Beckman SW28 rotor at 4°C for 12 h at 110 000g. Aliquots of 200 μL (total 15 fractions) were harvested from the top of the gradient. The distribution of proteins was visualized by running aliquots of each fraction on 5–20% denaturing polyacrylamide gels. The gels were either stained with silver staining or processed for western analysis using an anti‐BRCA2 (Ab‐1) antibody. Bovine serum albumin (68 kDa), aldolase (158 kDa), catalase (240 kDa), and thyroglobulin (669 kDa) were used for calibration.
Subcellular fractionation and treatment with CDK1/CycB kinase
Nuclear and cytosolic fractions were harvested from cell extracts using a CelLytic NuCLEAR Extraction Kit (Sigma‐Aldrich) according to the manufacturer’s instructions. Briefly, HeLa S3 cells at 80% to 90% confluence (150‐mm dishes) were washed twice in ice‐cold PBS and swollen in 1 mL of ice‐cold hypotonic buffer (100 mM HEPES [pH 7.9], containing 15 mM MgCl2, 100 mM KCl, protease complete cocktail [Roche], and phosphatase inhibitor cocktail 1 and 2) for 15 min. Cells were scraped from the dishes and centrifuged at 450g for 5 min at 4°C. The resultant supernatant was decanted off and the pellet resuspended in 400 μL of the hypotonic buffer. The cell resuspension was Dounce‐homogenized (50 strokes), and the resulting homogenate was centrifuged at 10 000g for 20 min at 4°C. The supernatant and pellet represent the cytosolic fraction and nuclear fraction, respectively. The nuclei were washed twice in 100 μL reaction buffer (25 mM Tris‐HCl [pH 7.5], 10 mM MgCl2, 2 mM DTT, and 5 mM β‐glycerophosphate containing protease and phosphatase inhibitor cocktails) and incubated in 200 μL reaction buffer containing 500 ng CDK1/CycB kinase (Cell Signaling) and 200 μM ATP at room temperature for 30 min. The reaction was terminated by the addition of stop buffer (50 mM EDTA [pH 8.0]). Some samples (100 μL) were fixed with 3.7% formaldehyde in PBS for 10 min on ice and immunostaining was performed as described previously.( 10 ) The remaining samples (100 μL) were centrifuged at 10 000g for 10 min and then solubilized in SDS‐PAGE sample buffer. Western blots were probed with antibodies against γ‐tubulin, vinculin, and MCM7.
GST pull‐down assay
pGEX 4T‐2, pGEX 4T‐2 PLEC M1, and pGEX 4T‐1 PLEC M‐C were transformed into Escherichia coli BL21 (DE3) PhysS bacteria, and GST, GST‐PLEC‐M1, and GST‐PLEC‐M‐C fusion proteins were purified using glutathione‐Sepharose 4B beads (Amersham Biosciences) according to the manufacturer’s instructions. Protein purity was determined by SDS‐PAGE followed by CBB staining. For the assay, COS7 cells (5 × 106) were transfected with pME18S‐FLAG or pME18S‐BRCA2‐FLAG using Lipofectamine 2000. After culturing for 24 h, the transfected cells were lysed with 200 μL lysis buffer (100 mM NaCl, 10 mM EDTA, 20 mM Tris‐HCl [pH 8.0], 1% Nonidet P‐40). For binding, GST or GST‐PLEC‐M1 or GST‐PLEC‐M‐C was mixed with 200 μL lysate containing a cocktail of protease inhibitors (Roche Diagnostics), and the mixture was rotated at 4°C for 30 min. The GST or GST‐PLEC‐M1 or GST‐PLEC‐M‐C were precipitated by anti‐GST antibody‐protein G‐Sepharose and washed five times with 0.2 mL lysis buffer. The Sepharose was then suspended in Laemmli buffer and proteins were resolved by SDS‐PAGE. After being transferred onto an Immobilon‐P membrane, BRCA2‐FLAG was visualized by western blotting using anti‐FLAG antibody.
GST–PLEC‐M1 competition assay
HeLa S3 cells (5 × 106) were lysed with 200 μL lysis buffer (100 mM NaCl, 10 mM EDTA, 20 mM Tris‐HCl [pH 8.0], 1% Nonidet P‐40). The cell lysate was mixed with 200 μL of a lysate of COS7 cells transfected with pME18S‐BRCA2‐FLAG, and GST or GST‐PLEC‐M1 (see “GST pull‐down assay” above) was added at the same molar ratio to a final volume of 400 μL. Following incubation at 4°C for 30 min, the samples were washed and proteins were detected by western blotting.
Immunofluorescence microscopy
Cell fixation and immunostaining were performed as described previously.( 10 ) FLAG‐tagged BRCA2 was detected directly following fixation, washing, and mounting. Anti‐FLAG (M2), anti‐BRCA2 (Ab‐2), vimentin (H‐84), γ‐tubulin (C‐20), anti‐γ‐tubulin (C‐11), and γ‐tubulin (T3559) antibodies were used at 1:200, 1:100, 1:100, 1:1000, 1:100, and 1:1000 dilutions, respectively, in the blocking solution. The cells were then incubated with Alexa Fluor 488‐ or 594‐conjugated secondary antibody (Molecular Probes, Eugene, OR, USA) diluted 1:200 in blocking solution. DNA was stained with Hoechst 33258, and coverslips were mounted on slides with ProLong Gold (Invitrogen, Carlsbad, CA, USA). Cells were examined using an Olympus Power BX51 fluorescence microscope (Olympus, Tokyo, Japan).
siRNA treatment
The target sequences of the plectin (PLEC1‐1 and 2) siRNAs were: PLEC1‐1, 5′‐GCACUCAUCUUGCGUGACA‐3′; and PLEC1‐2, 5′‐GAAGAGACACAGAUCGACAUU‐3′. Those of BRCA2 (BRCA2‐1 and 2) siRNAs were: BRCA2‐1, 5′‐GAAACGGACTTGCTATTTA‐3′; and BRCA2‐2, 5′‐GAAGAATGCAGGTTTAATA‐3′. As a control, we used the GL‐2 duplex to target the luciferase gene. Predesigned luciferase siRNA, plectin siRNA (1 and 2), and BRCA2 siRNAs (1 and 2) were purchased from Dharmacon (Lafayette, CO, USA). Transfections were performed in triplicate using 20 nmol siRNA and Oligofectamine reagent (Invitrogen) according to the manufacturer’s instructions. Suppression of plectin and BRCA2 was confirmed by immunoblotting protein extracts prepared from cells treated with siRNAs, using antibodies against plectin and BRCA2 (Ab‐1), respectively.
Methods of ‘Mass spectroscopy’ and ‘Immunoblot analysis and immunoprecipitation’ are described in Data S1.
Results
Identification of a BRCA2‐associated protein
In order to identify novel binding partners for BRCA2, a lysate derived from HeLa S3 cells was fractionated by glycerol density gradient centrifugation. Each fraction was analyzed by SDS‐PAGE and immunoblotted with an anti‐BRCA2 antibody. Although the molecular weight of the BRCA2 protein is 380 kDa (Fig. 1a, fraction 10), endogenous BRCA2 was observed at higher molecular weight fractions (Fig. 1a, fractions 11–14). This result raised the possibility that endogenous BRCA2 formed a complex with other proteins. Accordingly, we analyzed these fractions via SDS‐PAGE, followed by visualization of proteins via silver staining (Fig. 1b). Analysis of the 14th fraction revealed a discrete number of candidate BRCA2 interacting partners, not found in the other fractions, which were subsequently examined via tandem mass spectrometry. Following a database search, plectin was identified as a candidate for the BRCA2‐associated protein.
Figure 1.
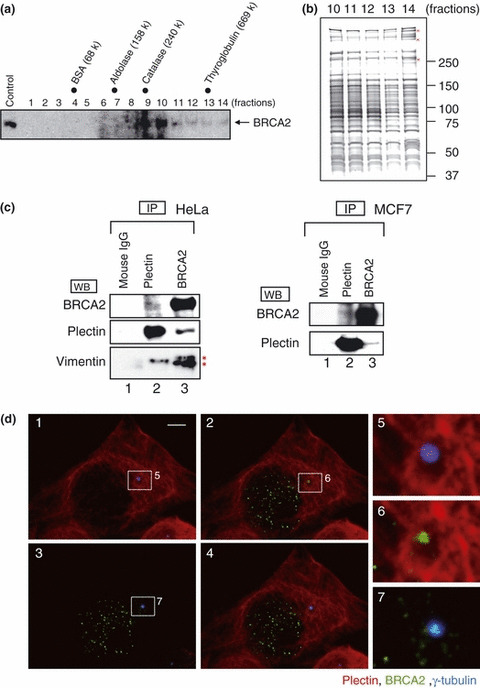
High molecular mass complex formation by the breast cancer susceptibility gene (BRCA2) and its association with plectin. (a) HeLa S3 cell extracts were separated by 15–30% glycerol gradient ultracentrifugation and the distribution of BRCA2 was examined by western blotting (lanes 1–14). Fraction numbers are indicated at the above of each panel. Molecular mass markers are indicated at the top of the figure. Arrows indicate the BRCA2 bands. (b) BRCA2‐associated proteins are present in a high molecular weight complex (lanes 11–14). Fractions (lanes 10–14) were separated by SDS‐PAGE and stained with silver‐ammoniac silver stain. Bands specifically present in lane 14 (red marks) were excised and the proteins identified by tandem mass spectrometry. (c) Coimmunoprecipitation of the endogenous BRCA2‐associated protein, plectin. A HeLa S3 extract (left panel) and MCF7 extract (right panel) were analyzed by immunoprecipitation, SDS‐PAGE, and western blotting using the indicated antibodies. Asterisks (*) indicate non‐specific bands originating from use of the anti‐BRCA2 antibody. (d) Immunostaining of BRCA2, γ‐tubulin, and plectin localization. HeLa S3 cells were incubated with anti‐BRCA2 antibody (Ab‐2; green), γ‐tubulin antibody (c‐20; blue), and plectin antibody (red). The boxed areas in 1, 2, and 3 are shown as magnified images in the row of images on the right (5–7). 1, γ‐tubulin (blue), plectin (red); 2, BRCA2 (green), plectin (red); 3, BRCA2 (green), γ‐tubulin (blue); 4, merged image. Scale bar, 5 μm.
In order to determine whether endogenous BRCA2 interacts with plectin, we performed co‐precipitation analysis. For this, BRCA2 was immunoprecipitated from HeLa cells, with analysis of anti‐BRCA2 immunoprecipitates via an anti‐plectin antibody demonstrating the presence of plectin in the immunoprecipitated complexes. Furthermore, analysis of anti‐plectin immunoprecipitates with the anti‐BRCA2 antibody demonstrated the presence of BRCA2 in the immunoprecipitated complexes. From these results, interaction of the two endogenous proteins BRCA2 and plectin was confirmed (Fig. 1c, left panel). No band was observed when precipitation was performed using mouse IgG. Analysis of anti‐plectin immunoprecipitates with an anti‐vimentin antibody demonstrated the presence of vimentin, indicating an interaction between plectin and vimentin. Moreover, in the analysis of anti‐BRCA2 immunoprecipitates with the anti‐vimentin antibody, vimentin was not detected in the immunoprecipitated complexes. The bands indicated by asterisks in Figure 1c, which arise from use of the anti‐BRCA2 antibody, were ascertained as non‐specific in nature. In summary, an interaction between BRCA2 and vimentin was not observed. These results suggest that the interaction with BRCA2 was not mediated indirectly by precipitating a larger cytoskeletal complex which contains vimentin. In addition, we investigated whether the BRCA2–plectin interaction could be observed in another cell line, namely MCF7 cells. MCF7 is a breast cancer cell line, with its BRCA2 status being confirmed as wild‐type. On the basis of immunoprecipitation analyses, interaction of BRCA2 and plectin was also observed in MCF7 cells (Fig. 1c, right panel).
We then conducted an immunostaining analysis of HeLa cells using an anti‐BRCA2 antibody in combination with an antibody against plectin and γ‐tubulin. BRCA2 localized to the centrosomal region, as represented by the anti‐γ‐tubulin antibody, as well as in the nucleus. Examination of plectin distribution identified a filamentous staining pattern throughout the cytoplasmic compartment (Fig. 1d).
Cyclin‐dependent kinase 1/cyclin B (CDK1/CycB) kinase treatment solubilizes plectin and results in centrosome displacement.
We previously reported that BRCA2 localizes around the centrosome and that it interacts with γ ‐tubulin, which is a component of the pericentriolar matrix.( 10 ) It has also been reported that plectin interacts with the nuclear membrane via either nesprin‐3 or lamin B proteins.( 23 , 24 ) The published data and the results of our analyses described above led us to speculate that the localization of the centrosome at the nucleus is mediated via the BRCA2‐plectin complex. Plectin is a versatile cross‐linking protein; phosphorylation of plectin by CDK1/CycB kinase causes the loss of its cross‐linking function during the M phase of the cell cycle, resulting in the detachment of plectin from cytoplasmic network structures.( 22 , 25 ) Our speculation on the possible role of the interaction between BRCA2 and plectin in the localization of the centrosome predicts that loss of the cross‐linking function will result in the detachment of the centrosome from the nucleus. In order to test this hypothesis, we prepared nuclear fractions in which the conformations of the nucleus, centrosome, and plectin were preserved. We then added activated CDK1/CycB kinase to these fractions and observed the position of the centrosomes. The nuclear and cytoplasmic fractions were confirmed by western blotting, employing antibodies against MCM7 (a nuclear protein) and vinculin (a cytoplasmic protein), respectively (Fig. 2a, left). In addition, untreated cells and nuclear fractions were immunostained with an antibody against lamin A/C (a outer nuclear membrane protein), confirming that the nuclear membrane had not been damaged seriously in the preparation of the nuclear fraction (Fig. 2a, right). Nuclear fractions were immunostained with anti‐γ‐tubulin and anti‐plectin antibodies, and the nuclear DNA counterstained with Hoechst 33258.
Figure 2.
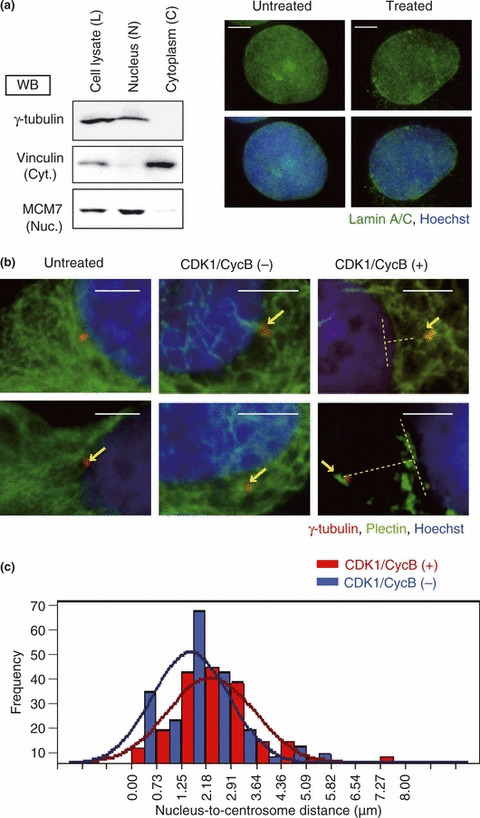
Cyclin‐dependent kinase 1/cyclin B (CDK1/CycB) kinase caused a change in cytoskeletal structure and centrosome localization. (a) HeLa cells were fractionated into nuclear and cytoplasmic compartments. Cell lysate (L), nuclear fraction (N), and cytoplasmic fraction (C) from HeLa cells were analyzed by immunoblotting with antibodies against γ‐tubulin (centrosomal marker), vinculin (cytosolic protein marker), and MCM7 (nuclear protein marker). Untreated cells (left row) and the nuclear fraction (right row) stained with antibodies against lamin A/C (green) and with Hoechst 33258 (blue) were analyzed by fluorescence microscopy. Scale bar, 5 μm. Upper row, lamin A/C; lower row, lamin A/C and Hoechst 33258. (b) The nuclear fraction was treated with CDK1/CycB kinase. The two images on the left show the untreated cells, the middle two images show the CDK1/CycB (−) fractions, and the right two images show the CDK1/CycB (+) fraction. Samples were stained with antibodies against plectin (green), γ‐tubulin (red), and Hoechst 33258 (blue). Arrows indicate localization of the centrosome and dotted lines indicate the measured distance between the centrosome and nucleus. Scale bar, 5 μm. (c) Minimum distance between the nuclear edge and centrosome was measured using Adobe Photoshop CS3 software (Adobe, San Jose, CA, USA). Centrosomes overlying the nucleus were excluded from the analysis. Distances were measured in the CDK1/CycB kinase positive (+) and CDK1/CycB (−) groups (n = 157 and 180, respectively, P = 0.046).
Next, untreated cells (Fig. 2b, untreated) and nuclear fractions (Fig. 2b, CDK1/CycB [−] and [+]) were immunostained with an antibody against plectin and γ‐tubulin. In the case of CDK1/CycB kinase‐free, plectin was located peripherally around the nucleus, and the centrosome was located close to the nucleus, as in untreated cells (Fig. 2b, CDK1/CycB [−]). However, following the addition of CDK1/CycB kinase, plectin became diffusely distributed throughout the cytoplasm, and there was a tendency for the centrosome to become detached from the nucleus (Fig. 2b, CDK1/CycB [+]). In order to examine this detachment, we measured the distance between the centrosome and the nucleus. This analysis revealed that in the CDK1/CycB kinase‐treated group, there was a greater variation in the distance between the centrosome and the nucleus than in the CDK1/CycB (−) group (F‐test, P = 0.046; Fig. 2c). These results support our suggestion that the centrosome is located near the nucleus as a consequence of the cross‐linking effects of plectin.
GST–PLEC M1 interacts with BRCA2 and inhibits the interaction between BRCA2 and plectin
As described above, we hypothesized that the centrosome would become detached from the nucleus if interaction between BRCA2 and plectin was inhibited. To test this prediction, we investigated the site of interaction between these two proteins. Plectin has six repeat domains (PLEC module [M] 1–6, Fig. 3a) in its C terminus, each having a hairpin‐helix‐loop‐helix (β2α2) structure. It has been reported that the secondary elements of the PLEC modules and the ankyrin repeats of the cyclin‐dependent kinase inhibitor p18ink4c show strikingly similar compositions.( 26 ) Since the ankyrin repeat domain has been demonstrated to participate in intramolecular and intermolecular protein–protein interactions, we postulated that the PLEC M1 domain might be involved in the interaction with BRCA2. Accordingly, we examined whether a GST–PLEC M1 fusion protein would bind BRCA2‐FLAG protein. In a GST pull‐down assay in which GST–PLEC M1 was added to a lysate of COS7 cells in which BRCA2‐FLAG had been overexpressed, we confirmed the interaction of these two proteins (Fig. 3b, lane 4).
Figure 3.
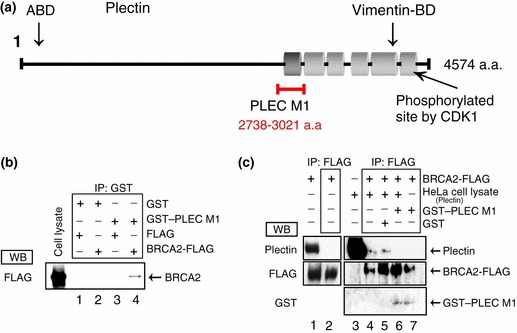
PLEC M1 plays an important role in the breast cancer susceptibility gene (BRCA2)–plectin interaction. (a) Structurally, plectin consists of a ∼200‐nm central coiled‐coil rod flanked by globular N‐ and C‐terminal domains. The actin‐binding domain (ABD) is located close to the amino terminus. The structure of the C‐terminal domain is dominated by six highly homologous repeat domains comprising an intermediate filament (vimentin) binding domain and a cyclin‐dependent kinase phosphorylation site. Among these domains, we focused on PLEC M1 (2738–3021 amino acids). (b) GST–PLEC M1 and GST were incubated with BRCA2‐FLAG or FLAG, and a pull‐down assay was then performed. Lane 1, GST incubated with FLAG; lane 2, GST incubated with BRCA2‐FLAG; lane 3, GST–PLEC M1 incubated with FLAG; lane 4, GST–PLEC M1 incubated with BRCA2‐FLAG. (c) HeLa S3 cell lysate was incubated with BRCA2‐FLAG. BRCA2‐FLAG was overexpressed transiently in COS7 cells. Cell lysates of these COS7 cells were subjected to immunoprecipitation with anti‐FLAG antibody followed by western blotting with the corresponding antibodies (left panel). GST–PLEC M1 or GST was added and samples were subjected to immunoprecipitation with anti‐FLAG antibody followed by western blotting with the corresponding antibodies (right panel). GST–PLEC M1 and GST were purified and checked for quantity on a CBB‐stained SDS‐PAGE gel (not shown).
We then went on to examine whether GST–PLEC M1 would inhibit the binding of BRCA2‐FLAG to plectin. First of all, we examined whether endogenous plectin from COS7 cells, in which BRCA2‐FLAG was overexpressed, interacted with BRCA2‐FLAG protein. In COS7 lysates, both plectin and BRCA2 was detected (Fig. 3c, lane 1), but analysis of anti‐FLAG immunoprecipitates with an anti‐plectin antibody demonstrated the absence of plectin (Fig. 3c, lane 2). So, it was demonstrated that the endogenous plectin from COS7 cells did not interact with overexpressed BRCA2‐FLAG protein.
Next, we performed immunoprecipitation studies, using an anti‐FLAG antibody, in which we added a HeLa cell lysate containing plectin to a lysate of COS7 cells in which the BRCA2‐FLAG protein had been overexpressed. Subsequent immunoblotting with the anti‐plectin antibody revealed the presence of plectin (Fig. 3c, lane 4). Since plectin was not detected in the absence of the HeLa cell lysate (Fig. 3c, lane 2), we conclude that the BRCA2‐FLAG combined with plectin originating from the HeLa cells rather than plectin of COS7 cell origin. However, when we added GST–PLEC M1 and plectin (HeLa cell lysate) to a cell lysate containing BRCA2‐FLAG and performed immunoprecipitation with the anti‐FLAG antibody, we were unable to detect plectin (Fig. 3c, lane 6). Since plectin was detected when GST was added (Fig. 3c, lane 5), we suggest that GST–PLEC M1 inhibited the binding of BRCA2‐FLAG and plectin. On the basis of these results, we propose that BRCA2 and plectin interact via the PLEC M1 domain.
Plectin module 1 (PLEC M1) also inhibits endogenous plectin–BRCA2 association
Next, we examined whether PLEC M1 would inhibit the interaction of endogenous BRCA2 and plectin. Immunoprecipitation was performed using an anti‐BRCA2 antibody and a lysate of HeLa cells in which HA‐PLEC M1 had been overexpressed. Immunoblotting with the anti‐plectin antibody failed to detect endogenous plectin (Fig. 4a, lane 4). Plectin was, however, detected in both HA‐transfected and non‐transfected cells (Fig. 4a, lanes 5 and 6). Therefore, HA‐PLEC M1 inhibited the interaction between endogenous BRCA2 and endogenous plectin in HeLa cells. We then overexpressed HA‐PLEC M1 in HeLa cells and used an immunofluorescence assay to detect any changes in centrosomal localization. The assay revealed that in the cells in which HA‐PLEC M1 had been introduced, the centrosome had become detached from the nucleus (Fig. 4b, left upper panel). Conversely, in the HA‐transfected cells, the centrosome was located adjacent to the nucleus (Fig. 4b, left lower panel). We then attempted to determine the distances between the nuclei and centrosomes in these different groups of cells. In the HA‐PLEC M1‐transfected group, we observed a larger dispersion in nucleus–centrosome distances than in the HA‐transfected group, with this difference shown to be statistically significant (F‐test, P = 0.018; Fig. 4b, right panel).
Figure 4.
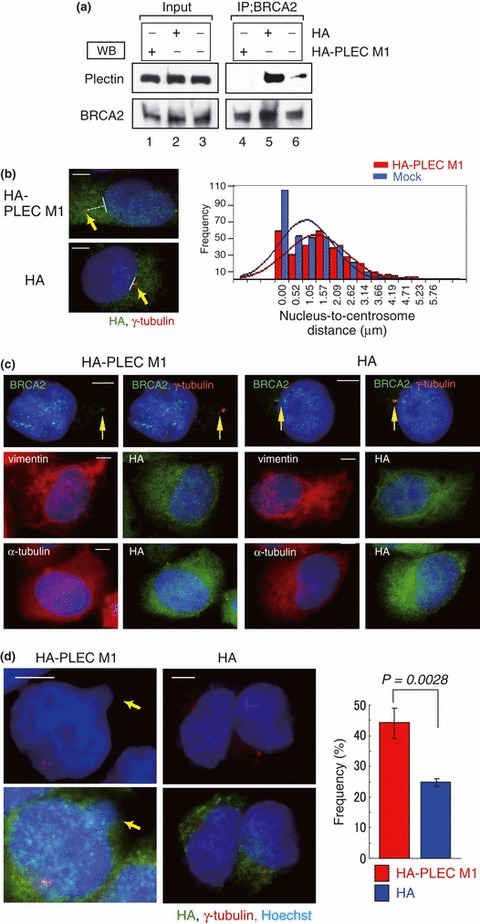
Subcellular localization of the centrosome in HeLa S3 cells transfected or untransfected with HA‐PLEC M1. (a) HA‐PLEC M1 was expressed transiently in HeLa S3 cells (lanes 1 and 4). HA‐transfected cells (lanes 2 and 5). Untransfected cells (lanes 3 and 6). Western blotting of cell lysates (input, left panel) and immunoprecipitation with anti‐breast cancer susceptibility gene (BRCA2) antibody (right panel) were performed using anti‐plectin or anti‐BRCA2 antibodies. Endogenous BRCA2 interacted with endogenous plectin in HeLa S3 cells (lane 5); however, endogenous BRCA2 did not interact with endogenous plectin in HA‐PLEC M1‐transfected cells (lane 4). (b) Indirect immunofluorescent staining with antibodies against HA (green) and γ‐tubulin (red) and with Hoechst 33258 (blue). In the HA‐PLEC M1‐transfected cells (left upper panel), displacement of centrosomes was observed. In the HA‐transfected cells (left lower panel), no comparable change in centrosome location was observed. Arrows indicate centrosomes. Bar, 5 μm. Minimum distance between the nuclear edge and centrosome was measured using Adobe Photoshop CS3 software. Distances were measured in HA‐PLEC M1‐transfected cells and HA‐transfected cells (n = 293 and 348, respectively; P = 0.018) (right panel). (c) Indirect immunofluorescent staining with antibodies against BRCA2 (green) and γ‐tubulin (red) (upper), with antibodies against HA (green) and vimentin (red) (middle), with antibodies against HA (green) and α‐tubulin (red) (lower). Nucleus stained with Hoechst 33258 (blue). (d) The numbers of abnormal nuclei were counted in HA‐PLEC M1‐transfected cells or HA‐transfected cells. The panels on the left show the HA‐PLEC M1‐transfected cells, with the panels on the right showing the HA‐transfected cells. Arrow indicates micronuclei, with the margin of the nucleus was indicated by a dotted line. Experiments were performed in triplicate; the histogram shows the mean frequencies (± SD) of abnormal nuclei in three independent experiments. The mean frequency of abnormal nuclei was 44.3% for HA‐PLEC M1‐transfected cells (red, n = 287) and 25% for HA‐transfected cells (blue, n = 288).
Plectin is a cytoplasmic protein that acts as a link between various cytoskeletal systems, and the interaction with vimentin intermediate filaments is well known. To confirm the influence of overexpressed HA‐PLEC M1 to the other cytoskeletal proteins, we performed immunostaining analysis with anti‐vimentin and anti‐α‐tubulin antibodies. No significant change was detected in respect to the configuration of vimentin and α‐tubulin structure in HA‐PLEC M1 overexpressed cells (Fig. 4c). In addition, nuclear abnormalities, such as micronuclei and multinuclei, were observed more frequently in the HA‐PLEC M1‐transfected HeLa cells than in the HA‐transfected cells (Fig. 4d). On the basis of these results, we suggest that the interaction between endogenous plectin and BRCA2 is inhibited by the overexpression of HA‐PLEC M1 and that this inhibition gives rise to a change in centrosome localization and induction of nuclear abnormalities.
Suppression of plectin causes change centrosomal localization and abnormal duplication of the nucleus
We examined the effect of silencing the plectin gene on centrosomal localization. Two types of siRNA (PLEC1‐1, 2) were used separately to silence the plectin gene, and their effects were examined by western blotting 48 h after transfection. In HeLa cells transfected with a plectin siRNA, the expression of plectin was considerably reduced compared to control cells transfected with luciferase siRNA (Fig. 5a, left). This suppression of plectin expression by the plectin siRNAs was confirmed by immunostaining (Fig. 5a, right).
Figure 5.
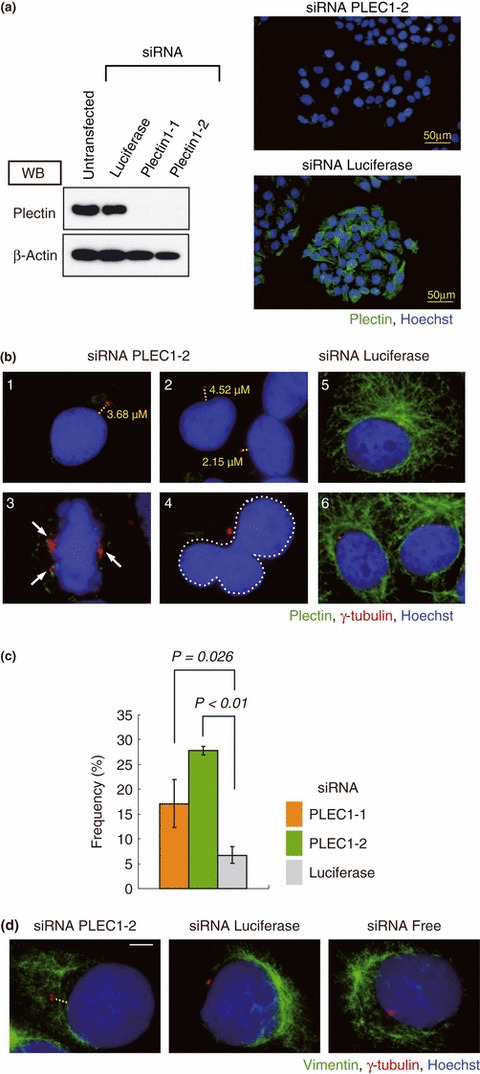
Suppression of plectin causes serious damage to the cell. (a) HeLa cells were transfected with control siRNA or with plectin siRNA (PLEC1‐1, ‐2), and 48 h after transfection, cells were harvested and subjected to immunoblotting with antibodies against plectin and β‐actin. Non‐transfected cells were also subjected to the same immunoblotting process. PLEC1‐1‐ and 1‐2‐transfected cells exhibited markedly reduced plectin expression; consequently, these siRNAs were selected for use in all the subsequent experiments. Immunofluorescence microscopy of PLEC1‐2 siRNA (right upper panel) and luciferase siRNA (right lower panel) transfected cells is shown. Bars, 50 μm. (b) Immunostaining with antibodies against plectin (green), γ‐tubulin (red), and with Hoechst 33258 (blue). PLEC 1‐2 siRNA‐transfected cells (1–4) and control siRNA (5 and 6). Measured distances between centrosome and nucleus are indicated. Arrows indicate centrosomes. The margin of the aberrant nucleus is indicated by a dotted line in picture 4. (c) The histogram shows the mean frequencies (± SD) of nuclear anomalies and centrosomal displacement in three independent experiments. Two people independently counted cells that displayed a nuclear abnormality. The mean values for PLEC1‐1, PLEC1‐2, and luciferase transfected cells were 17.1%, 27.9%, and 6.7%, respectively. (d) Immunostaining with antibodies against vimentin (green), γ‐tubulin (red), and with Hoechst 33258 (blue). PLEC 1‐2 siRNA‐transfected cells (left), luciferase siRNA‐transfected cells (middle), and siRNA free cells (right).
We then performed an immunofluorescence assay of HeLa cells transfected with PLEC1‐2 siRNA (1, 2, 3, 4, 5) or luciferase siRNA (5, 6). In addition to detachment of the centrosome from the nucleus (1, 2, 5), silencing of plectin also resulted in abnormalities in respect to centrosome replication (3, 5) or in aberrant nuclei (4, 5). Similar results were observed in cells transfected with PLEC1‐1 siRNA. The frequency of aberrant cells was significantly lower in cells transfected with luciferase siRNA (Fig. 5c). Moreover, to examine the influence on vimentin structure of plectin ablation by siRNA, immunostaining assay was performed with an anti‐vimentin antibody. In the plectin siRNA transfected cells, luciferase siRNA transfected cells, and untransfected cells (siRNA Free), no significant change was observed in respect to vimentin structure around the periphery of the nucleus (Fig. 5d). Transfection of HeLa cells with BRCA2 siRNA also caused similar nuclear and centrosomal abnormalities (Fig. S1). On the basis of these results, we suggest that, in addition to playing important roles in the positioning of the centrosome, both plectin and BRCA2 have an effect on nuclear duplication.
Figure 6.
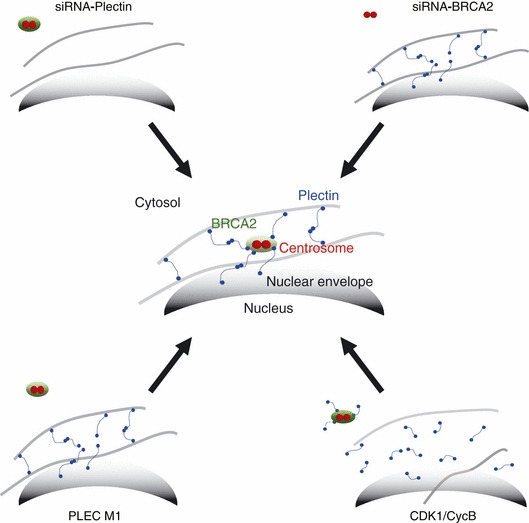
Model for centrosome localization at the nucleus. Plectin (blue) and the breast cancer susceptibility gene (BRCA2) (green) interact directly, and the complexes thus formed attach to intermediate filaments (gray lines). The cytoskeletal intermediate filaments are connected to the nuclear membrane by plectin and nesprin‐3. The centrosomes localize around the nucleus by attaching to the cytoskeletal protein through the plectin–BRCA2 complex. Thus, the centrosome is indirectly anchored to the nucleus.
Discussion
In this study, we identified plectin as a protein that interacts with the breast cancer susceptibility protein BRCA2. Plectin has six highly conserved tandem repeats (PLEC module [M] 1–6) at its C terminus; each repeat module has five tandem repeats that consist of 38 amino acids.( 26 ) These modules have a composition of secondary elements which are similar to the ankyrin repeat structures which participate in protein interactions.( 27 ) Furthermore, the IF‐binding site of plectin has been located between PLEC M5 and M6 ( 18 ); in addition, the carboxy‐terminal M6 contains a unique cell cycle kinase cdk1 phosphorylation site.( 25 )
So, reflecting on the possibility that BRCA2 interacts with any of the plectin modules, we took notice of PLEC M1 (2738–3021 a.a) and the C‐terminal region (4150–4503 a.a) (which comprised PLEC M6, along with an adjacent vimentin site; hereafter called PLEC M‐C), and examined whether BRCA2 and PLEC M1 or PLEC M‐C would interact. Our pull‐down assay showed that BRCA2‐FLAG and GST–PLEC M1 indeed interact (Fig. 3b), with PLEC M‐C also interacting with BRCA2‐FLAG (Fig. S2). From these results, given that each module has high homology to each of the others, the possibility that BRCA2 may interact with all of these six modules was suggested. How BRCA2 associates with these six modules is a question to be tackled in the future. We further demonstrated that overexpression of HA‐PLEC M1 inhibited the interaction between endogenous plectin and BRCA2 in HeLa S3 cells, resulting in displacement of the centrosomes from the perinuclear region. Our observations indicate the possibility that the centrosome exists at the perinuclear region through a plectin/BRCA2 interaction.
In comparison to luciferase siRNA transfection, displacement of the centrosome was caused by knockdown of plectin or BRCA2 expression using siRNA. Since plectin interacts with vimentin and plays an essential role in cytoskeleton network organization, it was considered that when expression of plectin was silenced by siRNA, collapse of the cytoskeleton network including vimentin would happen, and consequently, detachment of the centrosome would be observed. However, immunostaining of cells in which plectin expression is suppressed by siRNA with an anti‐vimentin antibody revealed that the vimentin structure at the periphery around the nucleus was not different from that of normal control cells. Taken together, it was considered that displacement of the centrosome caused by ablation of plectin with siRNA transfection is due to the impossibility of plectin–BRCA2 interaction.
Currently, it is assumed that in mammalian cells the positioning of the centrosome at the cell center is mediated predominantly by a pulling mechanism involving cortical dynein; that is, cortical dynein is thought to pull on those microtubules which are nucleated from the centrosome.( 28 , 29 ) However, this leaves unanswered the question of why centrosomes remain in the nuclear fraction when cell membranes are destroyed. We previously reported that BRCA2 interacts with γ‐tubulin, a component of the pericentriolar matrix of the centrosome.( 10 ) In addition to its interaction with cytoskeletal proteins such as actin and IFs, plectin has also been reported to interact with the nuclear membrane proteins lamin B and nesprin‐3.( 23 , 24 , 30 ) With regard to the positioning of the centrosome, a recent study demonstrated that the ZYG‐12 protein of Caenorhabditis elegans is essential for attachment of the centrosome to the nucleus.( 31 , 32 )
In human dermal fibroblasts, it is shown that emerin and microtubules both are necessary for the association of the centrosome with the nuclear envelope.( 33 ) From our results, we inferred the mechanism by which this interaction connects the centrosome and nucleus. We accordingly propose a model in which the centrosome is localized close to the nuclear membrane via the BRCA2–plectin interaction (Fig. 6).
Furthermore, although the centrosome starts moving at the M phase, the mechanism that determines the timing of this event has yet to be clarified. Plectin is phosphorylated by CDK1/CycB kinase during the M phase and thereby becomes unable to bridge the IFs.( 22 , 25 ) We suggest that phosphorylation of plectin is the signal that initiates centrosome movement. When CDK1/CycB kinase was added to a nuclear fraction containing centrosomes prepared from HeLa cells, the distribution of plectin observed surrounding the nuclei became diffuse and the centrosomes were separated from the nuclei. These observations suggest that the centrosome starts to move in response to the phosphorylation of plectin by CDK1/CycB kinase during the M phase.
Recently, it was reported that abnormalities of the centrosome are involved in oncogenesis. In Drosophila, larval brain cells with extra centrosomes can generate metastatic tumors when transplanted into the abdomens of wild‐type hosts.( 34 ) Similar tumor development has also been observed in larval brain tissues of Drosophila that carry a mutated form of the DSas‐4 protein that is required for centriole replication.( 35 ) In addition, centrosome amplification is an early event in the development of breast cancer, and amplification of centrosome size and number correlate with chromosome instability.( 36 )
To date, however, the effects of centrosomal displacement on the cell have remained poorly understood. In the present study, abnormalities in nuclear morphology and division were observed in HeLa cells in which PLEC M1 had been transiently overexpressed. Since no abnormalities in centrosome duplication were observed, the possibility is suggested that inhibition of the interaction between BRCA2 and plectin is implicated in the observed centrosomal displacement. In C. elegans, Hook protein ZYG‐12 is indispensable for attachment of the centrosome and nucleus. Mutations in the zyg‐12 gene of C. elegans perturb the attachment of the centrosome to the nucleus in interphase cells, and following the aberrant positioning of the centrosome, some chromosomes fail to congregate on the metaphase plate in zyg‐12 mutant embryos.( 31 ) Gönczy indicates the possibility that attachment of the centrosome and nucleus may be important in preventing genome instability.( 32 ) This infers the possibility that detachment of the centrosome from the nuclear membrane leads to genomic instability, and consequently contributes to carcinogenesis. Micronuclei are also extensively used to evaluate genotoxic effects and genomic instability.( 37 ) It was suggested that the formation of nuclear anomalies, such as micronuclei, are events commonly seen in the early stages of carcinogenesis.( 38 ) We showed that transient overexpression of HA‐PLEC M1, and depletion of BRCA2 and plectin by siRNA in HeLa cells, caused aberrant position of centrosomes, and resulted in micronucleus formation. These considerations give mechanistic support to a possible causal association between abnormal centrosomal positioning, micronuclei formation, and carcinogenesis.
Previously, we reported that disruption of the nuclear export sequence results in deregulation of BRCA2 export.( 9 ) According to the Breast Cancer Information Core (BIC), which was established at the National Human Genome Research Institute (Bethesda, MD, USA) to coordinate the information related to BRCA1 and BRCA2 research, 12 examples of mutations in this Nuclear export signal (NES) sequence have been reported as unverified changes. In this paper, we demonstrated that disruption of the BRCA2–plectin interaction leads to abnormal positioning of the centrsome and nuclear abnormalities, such as micronuclei or multinuclei. Then, we also considered that the germline mutations in the plectin binding region of the BRCA2 gene may have the same influence. In this study, we could not detect the plectin binding domain of BRCA2, but we want to clarify it in the future. However, from these results, it is conceivable that BRCA2 has an important role at the centrosome. Therefore, it was inferred that germline mutations of these domains in BRCA2, which include a nuclear export sequence and a plectin binding domain, have an influence on breast tumorigenesis.
Taken together, the data reported here suggest that BRCA2–plectin complexes may participate in both the formation of the microenvironment surrounding the centrosome and in nuclear duplication (nuclear morphological aberration and abnormal nuclear division). Our observations suggest a novel role for BRCA2 in the localization of the centrosome, which may be possibly linked to the normal duplication of the nucleus or genomic stability.
Finally, this paper is the first report to describe the BRCA2–plectin interaction and its role in centrosomal regulation of the perinuclear region.
Supporting information
Fig. S1. Suppression of BRCA2 by siRNA causes a change in centrosomal localization.
Fig. S2. PLEC M‐C also interacts with the breast cancer susceptibility gene (BRCA2).
Data S1. A part of “Material and Methods”.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We thank all members of the Department of Molecular Diagnosis of The Cancer Institute, Japanese Foundation for Cancer Research, for helpful discussions and encouragement. This work was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Wooster R, Bignell G, Lancaster J et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378: 789–92. [DOI] [PubMed] [Google Scholar]
- 2. Venkitaraman A. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002; 108: 171–82. [DOI] [PubMed] [Google Scholar]
- 3. Pellegrini L, Venkitaraman A. Emerging functions of BRCA2 in DNA recombination. Trends Biochem Sci 2004; 29: 310–6. [DOI] [PubMed] [Google Scholar]
- 4. Esashi F, Christ N, Gannon J et al. CDK‐dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005; 434: 598–604. [DOI] [PubMed] [Google Scholar]
- 5. Wong A, Pero R, Ormonde P, Tavtigian S, Bartel P. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem 1997; 272: 31941–4. [DOI] [PubMed] [Google Scholar]
- 6. Yang H, Li Q, Fan J, Holloman W, Pavletich N. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA‐ssDNA junction. Nature 2005; 433: 653–7. [DOI] [PubMed] [Google Scholar]
- 7. Tutt A, Gabriel A, Bertwistle D et al. Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr Biol 1999; 9: 1107–10. [DOI] [PubMed] [Google Scholar]
- 8. Daniels M, Wang Y, Lee M, Venkitaraman A. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science 2004; 306: 876–9. [DOI] [PubMed] [Google Scholar]
- 9. Han X, Saito H, Miki Y, Nakanishi A. A CRM1‐mediated nuclear export signal governs cytoplasmic localization of BRCA2 and is essential for centrosomal localization of BRCA2. Oncogene 2008; 27: 2969–77. [DOI] [PubMed] [Google Scholar]
- 10. Nakanishi A, Han X, Saito H et al. Interference with BRCA2, which localizes to the centrosome during S and early M phase, leads to abnormal nuclear division. Biochem Biophys Res Commun 2007; 355: 34–40. [DOI] [PubMed] [Google Scholar]
- 11. Foisner R, Bohn W, Mannweiler K, Wiche G. Distribution and ultrastructure of plectin arrays in subclones of rat glioma C6 cells differing in intermediate filament protein (vimentin) expression. J Struct Biol 1995; 115: 304–17. [DOI] [PubMed] [Google Scholar]
- 12. Foisner R, Wiche G. Structure and hydrodynamic properties of plectin molecules. J Mol Biol 1987; 198: 515–31. [DOI] [PubMed] [Google Scholar]
- 13. McMillan J, Akiyama M, Rouan F et al. Plectin defects in epidermolysis bullosa simplex with muscular dystrophy. Muscle Nerve 2007; 35: 24–35. [DOI] [PubMed] [Google Scholar]
- 14. Cheng C, Liu Y, Ho C et al. The influence of plectin deficiency on stability of cytokeratin18 in hepatocellular carcinoma. J Mol Histol 2008; 39: 209–16. [DOI] [PubMed] [Google Scholar]
- 15. Kelly K, Bardeesy N, Anbazhagan R et al. Targeted nanoparticles for imaging incipient pancreatic ductal adenocarcinoma. PLoS Med 2008; 5: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Svitkina T, Verkhovsky A, Borisy G. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol 1996; 135: 991–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrä K, Nikolic B, Stöcher M, Drenckhahn D, Wiche G. Not just scaffolding: plectin regulates actin dynamics in cultured cells. Genes Dev 1998; 12: 3442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nikolic B, Mac Nulty E, Mir B, Wiche G. Basic amino acid residue cluster within nuclear targeting sequence motif is essential for cytoplasmic plectin‐vimentin network junctions. J Cell Biol 1996; 134: 1455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wiche G. Role of plectin in cytoskeleton organization and dynamics. J Cell Sci 1998; 111(Pt 17): 2477–86. [DOI] [PubMed] [Google Scholar]
- 20. Boczonadi V, McInroy L, Määttä A. Cytolinker cross‐talk: periplakin N‐terminus interacts with plectin to regulate keratin organisation and epithelial migration. Exp Cell Res 2007; 313: 3579–91. [DOI] [PubMed] [Google Scholar]
- 21. Spurny R, Gregor M, Castañón M, Wiche G. Plectin deficiency affects precursor formation and dynamics of vimentin networks. Exp Cell Res 2008; 314: 3570–80. [DOI] [PubMed] [Google Scholar]
- 22. Foisner R, Malecz N, Dressel N, Stadler C, Wiche G. M‐phase‐specific phosphorylation and structural rearrangement of the cytoplasmic cross‐linking protein plectin involve p34cdc2 kinase. Mol Biol Cell 1996; 7: 273–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foisner R, Traub P, Wiche G. Protein kinase A‐ and protein kinase C‐regulated interaction of plectin with lamin B and vimentin. Proc Natl Acad Sci U S A 1991; 88: 3812–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilhelmsen K, Litjens S, Kuikman I et al. Nesprin‐3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J Cell Biol 2005; 171: 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Malecz N, Foisner R, Stadler C, Wiche G. Identification of plectin as a substrate of p34cdc2 kinase and mapping of a single phosphorylation site. J Biol Chem 1996; 271: 8203–8. [DOI] [PubMed] [Google Scholar]
- 26. Janda L, Damborský J, Rezniczek G, Wiche G. Plectin repeats and modules: strategic cysteines and their presumed impact on cytolinker functions. BioEssays 2001; 23: 1064–9. [DOI] [PubMed] [Google Scholar]
- 27. Mosavi L, Cammett T, Desrosiers D, Peng Z. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci 2004; 13: 1435–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vallee R, Stehman S. How dynein helps the cell find its center: a servomechanical model. Trends Cell Biol 2005; 15: 288–94. [DOI] [PubMed] [Google Scholar]
- 29. Burakov A, Nadezhdina E, Slepchenko B, Rodionov V. Centrosome positioning in interphase cells. J Cell Biol 2003; 162: 963–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Foisner R, Feldman B, Sander L, Wiche G. Monoclonal antibody mapping of structural and functional plectin epitopes. J Cell Biol 1991; 112: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Malone C, Misner L, Le Bot N et al. The C. elegans hook protein, ZYG‐12, mediates the essential attachment between the centrosome and nucleus. Cell 2003; 115: 825–36. [DOI] [PubMed] [Google Scholar]
- 32. Gönczy P. Centrosomes: hooked on the nucleus. Curr Biol 2004; 14: R268–70. [DOI] [PubMed] [Google Scholar]
- 33. Salpingidou G, Smertenko A, Hausmanowa‐Petrucewicz I, Hussey P, Hutchison C. A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J Cell Biol 2007; 178: 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Basto R, Brunk K, Vinadogrova T et al. Centrosome amplification can initiate tumorigenesis in flies. Cell 2008; 133: 1032–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Castellanos E, Dominguez P, Gonzalez C. Centrosome dysfunction in drosophila neural stem cells causes tumors that are not due to genome instability. Curr Biol 2008; 18: 1209–14. [DOI] [PubMed] [Google Scholar]
- 36. Lingle W, Barrett S, Negron V et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci U S A 2002; 99: 1978–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fenech M. Chromosomal biomarkers of genomic instability relevant to cancer. Drug Discov Today 2002; 7: 1128–37. [DOI] [PubMed] [Google Scholar]
- 38. Bonassi S, Znaor A, Ceppi M et al. An increased micronucleus frequency in peripheral blood lymphocytes predicts the risk of cancer in humans. Carcinogenesis 2007; 28: 625–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Suppression of BRCA2 by siRNA causes a change in centrosomal localization.
Fig. S2. PLEC M‐C also interacts with the breast cancer susceptibility gene (BRCA2).
Data S1. A part of “Material and Methods”.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item