

Abstract
RelA‐associated inhibitor (RAI) was initially identified as a protein that interacts with the p65 subunit (RelA) of nuclear factor‐κB. It was recently found to interact with the p53 tumor suppressor protein. RAI is a structural homolog of the p53‐binding protein 2 and IκB family proteins, and is known to inhibit the DNA‐binding activities of p65 and p53. In the present study, we have attempted to predict the 3‐dimensional structure of RAI in complex with p53 using computational chemistry. In order to evaluate the predicted structure model, we created a series of RAI mutants in which the amino acid residues involved in the interaction with p53 were mutated, and examined their activities in blocking p53‐mediated bax gene expression. Our observations support the validity of the predicted 3‐dimensional model of the p53–RAI protein complex. Based on the p53–RAI complex model, we have demonstrated the biological importance of the R248 and R273 residues of p53, and the D775 and E795 residues of RAI, in the protein–protein interaction between p53 and RAI and the biological actions of these proteins. These findings will further clarify the biological actions of RAI in carcinogenesis and can be used for the development of a novel strategy in blocking the actions of RAI. The possible biological implications of RAI are also discussed. (Cancer Sci 2008; 99: 615–622)
Abbreviations:
- 53BP
p53‐binding protein
- AP
activator protein
- DTT
dithiothreitol
- EDTA
ethylene diamine tetra‐acetic acid
- iASPP
inhibitory member of the ankyrin repeats‐, SH3 domain‐, and Pro‐rich region‐containing proteins
- NF‐κB
nuclear factor‐κB
- PCR
polymerase chain reaction
- RAI
RelA‐associated inhibitor
- TK
thymidine kinase
RelA‐associated inhibitor is a novel inhibitor of the transcription factor NF‐κB and has been identified as an interacting protein of the RelA subunit of NF‐κB by yeast two‐hybrid screening.( 1 ) RAI shares similar structural configurations with the closely related protein 53BP2 in that they both contain a Pro‐rich domain, ankyrin repeats, and an SH3 domain.( 1 , 2 , 3 ) However, in contrast to 53BP2, RAI exists primarily in the nucleus and inhibits the DNA‐binding activity of NF‐κB.( 4 ) In addition, the rai gene was rediscovered as a p53‐binding protein that inhibits the actions of p53, thus named as iASPP.( 5 , 6 )
Nuclear factor‐κB is a heterodimeric protein complex consisting of the p65 and p50 subunits and is known to stimulate the generation of cancer cells by blocking apoptosis and promoting cell cycling.( 7 ) Thus, RAI is considered to play a role in inhibiting carcinogenesis as a tumor suppressor protein. In spite of the extensive search for mutations in the rai gene, no evidence has been found that rai is mutated in various human malignancies.( 8 ) However, expression of the rai gene was found to be downmodulated in some acute leukemia cells that were refractory to chemotherapy.( 8 ) Although the molecular details are not known, it was found that Nkipl (NF‐κB interacting protein 1), a gene newly identified by positional cloning that is considered to be responsible for cardiomyopathy and abnormal skin formation with curly hair because of the gene knockdown phenotype, is identical to the rai gene.( 9 )
There are three isoforms of RAI proteins, consisting of 351 (39 kDa), 407 (50 kDa), and 828 (95 kDa) amino acids, which share the same C‐terminal regions containing a Pro‐rich domain, ankyrin repeats, and a SH3 domain.( 1 , 4 , 5 , 10 ) RAI inhibits the DNA‐binding activities of not only NF‐κB but also p53 and AP1.( 1 , 4 , 5 ) With regard to the inhibitory action of RAI against p53, RAI can exert an inhibitory action on the p53‐mediated transactivation of the bax gene but not that of the p21 gene;( 5 ) thus, it is suggested that binding of RAI might give the p53 protein additional promoter specificity. The RAI protein is therefore considered a unique transcriptional regulator and it is hoped that analysis of the molecular interaction between RAI and its molecular partners (such as p53) should provide useful information in developing a novel cancer therapy with p53 as a target.
In the present study, we have applied a structure‐based bioinformatic approach in predicting the 3D structure of the p53–RAI protein complex. In order to examine the predicted 3D structure, we compared the biological activities of mutant RAI proteins, in which the contact amino acid residues were mutated, developed by site‐directed mutagenesis.
Materials and Methods
Isolation of a full‐length RAI clone. Human full‐length RAI cDNA was obtained by PCR using oligonucleotide primers, 5′‐gga att cca tgg aca gcg agg cat tcc aga gcg‐3′ (forward) and 5′‐cgg gat ccc gct aga ctt tac tcc ttt gag gct tc‐3′ (reverse), and a full‐length human cDNA library prepared from EST clone BC032298 as a template. The PCR was carried out using the Expand High Fidelity system (Roche) and the PCR products were cloned into the pFLAG‐CMV2 vector (Sigma), thus creating the RAI‐expression plasmid pFLAG‐CMV2‐RAI. Nucleotide sequencing was carried out using the ABI PRISM dye terminator cycle sequencing ready reaction kit with Applied Biosystems 313 automated DNA sequencer (ABI). Nucleotide sequence of RAI was determined using the forward CMV30 primer and six internal sequencing primers: 5′‐caa ccc tac acc cct aca g‐3′ (forward1), 5′‐aga gca gcc tgg atg gac‐3′ (forward2), 5′‐ctc tct ggg acg cca gcg ccg agt tc‐3′ (forward3), 5‐ctc aga tcc ctc agt gat ggg gac ag‐3′ (reverse1), 5′‐gaa ctc ggc gct ggc gtc cca gag ag‐3′ (reverse2), and 5′‐cgg gat ccc gct aga ctt tac tcc ttt gag gct tc‐3′ (reverse3) corresponding to the various portions of RAI cDNA.
Plasmids. Eukaryotic expression plasmids for p53 (pC53‐SN3)( 11 ) and p65 (pcDNA‐p65)( 4 , 12 ) were as reported previously. pFLAG‐CMV2‐RAI, expressing RAI in human cells, and its mutants were created in the present study and the details are described below. The luciferase expression vectors under the control of the bax‐ and p21‐promoters,( 13 , 14 ) and pGL3‐κB‐luc,( 4 , 12 , 15 ) were as reported previously. The Renilla luciferase expression vector under the control of the constitutively active TK promoter, pTK‐RL, was used as an internal control.( 1 , 4 , 15 )
Site‐directed mutagenesis. RAI mutants were created using a commercial site‐directed mutagenesis kit (QuickChange site‐directed mutagenesis kit; Stratagene) as reported previously.( 15 ) RAI‐m1, containing the F773A substitution, was created by mutagenesis primers including 5′‐cta cag cgc cga ggc cgg gga cga gct g‐3′ (forward) and 5′‐cag ctc gtc ccc ggc ctc ggc gct gta‐3′ (reverse). The mutated sequences are underlined. Similarly, mutagenesis primers for other RAI mutants were as follows: RAI‐m2 containing the D775A substitution, 5′‐gcc gag ttc ggg gcc gag ctg tcc ttc‐3′ (forward) and 5′‐gaa gga cag ctc ggc ccc gaa ctc ggc‐3′ (reverse); RAI‐m3 with the D775R substitution, 5′‐gcc gag ttc ggg cgc gag ctg tcc ttc‐3′ (forward) and 5′‐gaa gga cag ctc gcg ccc gaa ctc ggc‐3′ (reverse); RAI‐m4 with the Y769A substitution, 5′‐ctc tct ggg acg cca gcg ccg agt tc‐3′ (forward) and 5′‐gaa ctc ggc gct ggc gtc cca gag ag‐3′ (reverse); RAI‐m5 with the E795A substitution, 5′‐cgg gcc gga ggc gac cga ctg gtg‐3′ (forward) and 5′‐cac cag tcg gtc gcc tcc ggc ccg‐3′ (reverse); RAI‐m6 with the E795R substitution, 5′‐cgg gcc gga gcg gac cga ctg gtg‐3′ (forward) and 5′‐cac cag tcg gtc cgc tcc ggc ccg‐3′ (reverse); and RAI‐m7 with double substitution of Y769A and D775A. The primers used in creating RAI‐m2 and RAI‐m4 DNA were used to create the double mutant. The nucleotide sequencing was carried out at least twice to confirm the site‐directed mutagenesis.
Cell culture and luciferase assay. A transient luciferase assay was carried out to evaluate the effects of RAI and its mutants as reported previously.( 1 , 4 , 15 ) Human embryonic kidney 293 cells and Saos‐2 cells derived from osteosarcoma were cultured as reported.( 15 , 16 ) Briefly, 293 cells were grown at 37°C in culture in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum. Saos2 cells were maintained in Macoy's 5A medium with 10% fetal bovine serum. Cells were transfected with various plasmids using Fugene‐6 transfection reagent (Roche) in 12‐well plates with 1.5 µL Fugene‐6 transfection reagent per mL culture medium and a total of 0.5 µg plasmid DNA as described previously.( 17 , 18 ) The control plasmid pUC19 was used to equalize the amount of DNA for each transfection. Fugene‐6–DNA complexes were allowed to form liposomes for 45 min at room temperature in serum‐free medium before being added to the cells. After 24 h of transfection, cells were incubated for an additional 24 h by changing to a complete culture media and then harvested. The luciferase activity was measured using the luciferase assay system (Promega) and the relative light units emitted from the cells expressing luciferase were determined with a TD‐20/20 luminometer (Promega). Transfection efficiency was monitored by the Renilla luciferase activity derived from the pRL‐TK plasmid containing the TK promoter cotransfected as an internal control. All luciferase activities shown in transient transfection assays were corrected by the internal control activity of Renilla luciferase by pRL‐TK. These assays were carried out in triplicate. The results were presented as the fold increase in luciferase activity (mean ± SD) relative to the control in three independent transfections.
Western blot assay. In order to examine the expression of RAI protein in cultured Saos2 and 293 cells transfected with pFLAG‐CMV2‐RAI, cells were cultured for 48 h and the cell extracts were prepared by treatment with lysis buffer (50 mM Tris‐HCl [pH 8.0], 5 mM EDTA, 1.0% Nonidet P‐40, 10% glycerol, 1 mM DTT, and 1× complete protease inhibitors) containing 100 mM NaCl, followed by western blotting. The cell extracts were resolved by 8% sodium dodecylsulfate–polyacrylamide gel electrophoresesis and transferred to nitrocellulose membranes (Hybond‐C; Amersham). The membranes were incubated with mouse monoclonal anti‐FLAG M2 antibody (Sigma) at a dilution of 1/1000. Anti‐mouse IgG antibody was used as a secondary antibody at a dilution of 1/2000.
Computer‐assisted molecular docking and 3D analyses of proteins. The atomic coordinates of the 3D structures of the p53–DNA (1tsr) and p53–53BP2 (1ycs) complexes, determined by X‐ray crystallography, were obtained from the Protein Data Bank (http://www.rcsb.org/pdb/home). Because the 3D structure of RAI has not been determined, it was predicted from the homologous protein 53BP2, with 52% homology with RAI by homology modeling using integrated molecular analysis software, Molecular Operating Environment (Chemical Computing Group) and confirmed by another web‐based software program, 3D‐Jigsaw (http://www.bmm.icnet.uk/servers/3djigsaw/). Another software program, DS Visualizer (Accelrys) was used to visualize the protein structures. Because the predicted protein structures by homology modeling often suffer from artificial bias such as unified distribution of dihedral angles between adjacent amino acids, we applied the superimpose method. We have predicted the 3D structure of RAI by homology modeling and substituted the moiety of 53BP2 with the predicted RAI coordinates, and the superimpose method was applied subsequently to predict the molecular complex between p53 and RAI. Prediction of the protein complex was carried out using GreenPepper software (a subset application of SYBYL; Tripos), newly developed by Kondo et al.( 19 ) and based on a computer algorithm of BiGGER,( 20 ) with modification by applying fast Fourier transformation and also applying the method of Glaser et al.,( 21 ) which considers flexibility of amino acid side chains present on the protein surface. In practice, we carried out molecular docking simulations in silico by carrying out approximately 1010 independent calculations with a special shift of 1 Å between two contacted protein molecules and an angle shift of 15 degrees each alongside the contact surface of these two proteins. The extent of protein contact was expressed as the contact scores, with the assumption that the overlapping rigid core was ‘0’, that of the overlapping flexible core was ‘<5’, and that of the flexible surface was ‘>200’. The structure with the highest score was adapted as the most probable 3D protein‐complex structure.
Results
Isolation of the full‐length RAI clone. In order to obtain the full‐length cDNA clone encoding all of the RAI amino acids, we subcloned the full‐length EST clone BC032298 into the pCMV2‐FLAG vector at its multicloning site after digestion with EcoRI and BamHI, thus expressing full‐length RAI protein in fusion with the FLAG tag at the N‐terminus. The pCMV2‐FALG‐RAI plasmid was thus created and transfected into Saos2 and 293 cells by liposome‐mediated transfection. As shown in Fig. 1, when cell lysates of the transfected cells were analyzed by western blotting, the recombinant RAI protein was detected in Saos2 and 293 cells as a fusion protein with the FLAG epitope. The anti‐FLAG tag antibody detected a specific band migrating at approximately 95 kDa in two independent experiments. Other reacted bands corresponding to 77 and 23 kDa, detected in Saos2 cells, were considered non‐specific because they were detected in cells transfected with either empty vector or pFLAG‐CMV2‐RAI. Thus, it is concluded that the 95‐kDa protein is the major form of RAI.
Figure 1.
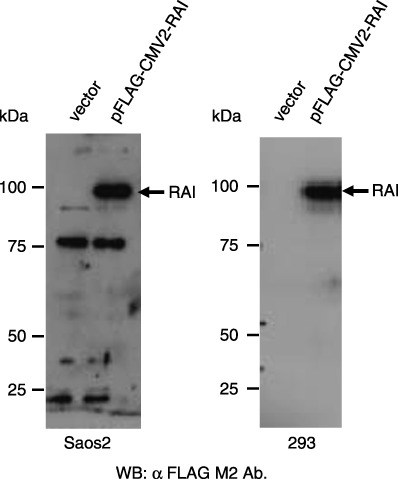
Western blot detection of RelA‐associated inhibitor (RAI) protein in transfected cells. Saos2 (right panel) and 293 cells (left panel) were transfected with the construct expressing FLAG‐tagged RAI or empty vector. Protein extracts were analyzed by western blotting with anti‐FLAG tag antibody. The positions of the standard protein markers are indicated from the left in kDa.
Inhibition of p53‐ and p65‐mediated gene expression by RAI. We then examined the effects of RAI on p53‐mediated gene expression by transfecting pFLAG‐CMV2‐RAI together with pC53‐SN3, expressing wild‐type p53, and the luciferase reporter plasmids under the control of the promoters from bax (bax‐luc), p21 (p21‐luc), or a promoter regulated by NF‐κB (κB‐luc) in 293 cells wherein the endogenous p53 is defective. As demonstrated in Fig. 2, although gene expression from the p53‐driven bax or p21 genes and that from κB‐luc driven by p65 were augmented in a dose‐dependent manner, the inhibitory effects of RAI against p53 action were observed only with bax‐promoter and NF‐κB‐dependent gene expression, not with the p21‐promoter. These observations were consistent with previous reports.( 1 , 5 ) However, it is confirmed that both long (95 kDa) and short (39 kDa) RAI proteins have a similar inhibiting activity. Similar results were observed with another human cell line, Saos2, containing no wild‐type p53 protein (data not shown).
Figure 2.
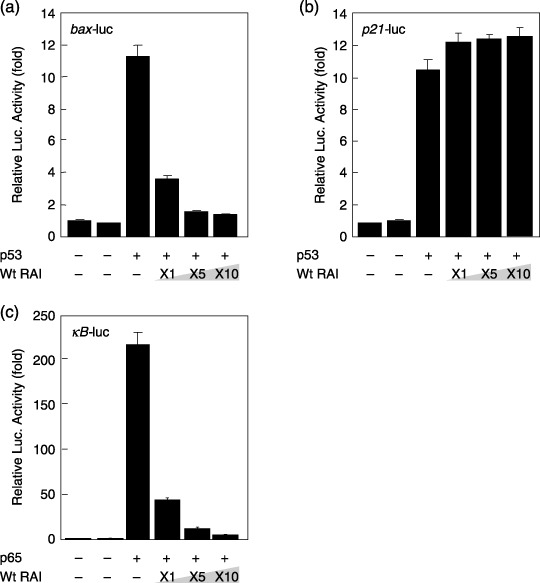
Inhibitory effects of RelA‐associated inhibitor (RAI) on p53‐ and p65‐dependent gene expression. (a) A total of 293 cells were transfected with bax‐luc together with pC53‐SN3 and various amounts of pFLAG‐CMV2‐RAI. (b) Saos2 cells were transfected with p21‐luc together with pC53‐SN3 and various amounts of pFLAG‐CMV2‐RAI. (c) A total of 293 cells were transfected with κB‐luc together with pGL3‐p65 and various amounts of pFLAG‐CMV2‐RAI. The luciferase activity was normalized by Renilla luciferase activity, which was cotransfected as an internal control. The data are represented as the fold increase in luciferase activities (mean ± SD) relative to control transfection of three independent experiments.
Prediction of the 3D structure of the p53–RAI protein complex by molecular docking. The amino acid sequences of RAI and 53BP2 are similar (52% identical amino acid residues) and the 3D structure of the 53BP2–p53 protein complex (1tsr) has been solved by X‐ray crystallography.( 22 , 23 ) Thus, we predicted the 3D structure of RAI in a protein–protein complex with p53 by superimposing it on that of 53BP2. In order to fine tune the 3D structure of the p53–RAI complex, we further searched for its 3D structure using GreenPepper molecular docking simulation, thus culminating in the prediction of the p53–RAI complex as illustrated in Fig. 3, in which two different views of a representative structure of the p53–RAI complex are shown. Table 1 indicates the detailed interactions between the amino acids located on p53 and RAI. For example, this model indicates that mutation‐prone amino acids of p53, such as R248 and R273, make direct contact with RAI. Another mutation‐prone amino acid of p53, R175, does not appear to be involved in the protein complex with RAI. These findings were consistent with previous report of the in vivo protein–protein interaction between p53 and RAI (iASPP).( 24 ) These findings suggest that p53 mutations appear to occur in order to prevent the interaction with RAI, thus highlighting the crucial importance of RAI in carcinogenesis.
Figure 3.
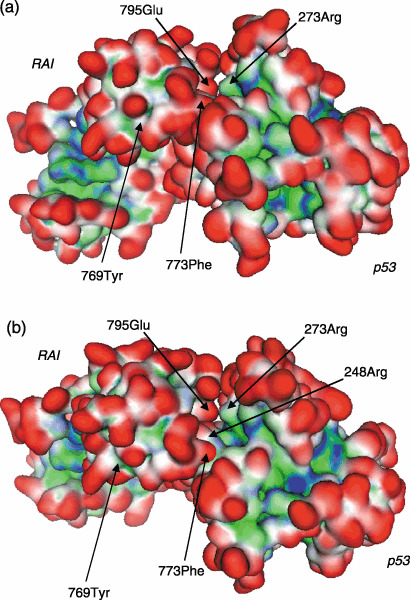
Computer‐assisted molecular docking and interaction between p53 and RelA‐associated inhibitor (RAI) in silico. Based on the atomic coordinates of p53–DNA (1tsr) and p53–p53‐binding protein (53BP2) (1ycs) determined by X‐ray crystallography and the predicted structure of RAI, the 3‐dimensional structure of the p53–RAI complex was predicted using the superimpose method and molecular docking software with the integrated Molecular Operating Environment software. Two different views of the p53–RAI protein complex are shown from different angles: (a) 0° and (b) –45°. Representative interacting amino acids are shown by arrows (for details, see Fig. 4). The molecular surface is indicated in red for exposed, blue for hydrophilic, and green for hydrophobic regions.
Table 1.
Interacting amino acids involved in the binding between p53 and RelA‐associated inhibitor (RAI) proteins
| p53 | RAI | Force |
|---|---|---|
| 178His | 723Thr | HB |
| 183Ser | 725Ser | HB |
| 243Met | 798Trp | HB |
| 773Phe | HYD | |
| 779Phe | HYD | |
| 798Trp | HYD | |
| 810Val | HYD | |
| 815Phe | HYD | |
| 244Gly | 772Glu | HB |
| 246Met | 773Phe | HYD |
| 248Arg | 775Asp | ION |
| 769Tyr | HB | |
| 249Arg | 772Glu | ION |
| 251Ile | 773Phe | HYD |
| 273Arg | 795Glu | ION |
HB, hydrogen bonding; HYD, hydrophobic interaction; ION, ionic interaction.
Fig. 4 illustrates the representative interacting surfaces between p53 and RAI. Among the mutation‐prone amino acids of p53, R273 makes direct contact with E795 of RAI by ionic interaction (Fig. 4A), and R248 of p53 with D775 and Y769 of RAI by ionic interaction and hydrogen bonding, respectively (Fig. 4B). Similarly, a hydrophobic interaction between M246 of p53 and F773 of RAI is shown (Fig. 4C). There are five distinct hydrogen bonds and three ionic interactions between p53 and RAI that are involved in recognizing partner molecules and stabilizing the p53–RAI complex. In addition, there are seven predicted hydrophobic interactions between these two proteins, which are considered primarily responsible for stabilizing the p53–RAI complex. Interestingly, these RAI amino acids did not make direct contact with another interacting partner of RAI, such as p65. Table 2 indicates the interactions among the amino acids on p65 and RAI. It is noted that all of the amino acid residues on RAI predicted to make direct contact with p53 do not appear to interact with p65, thus the protein–protein interactions between RAI and p53 or p65 are distinct.
Figure 4.
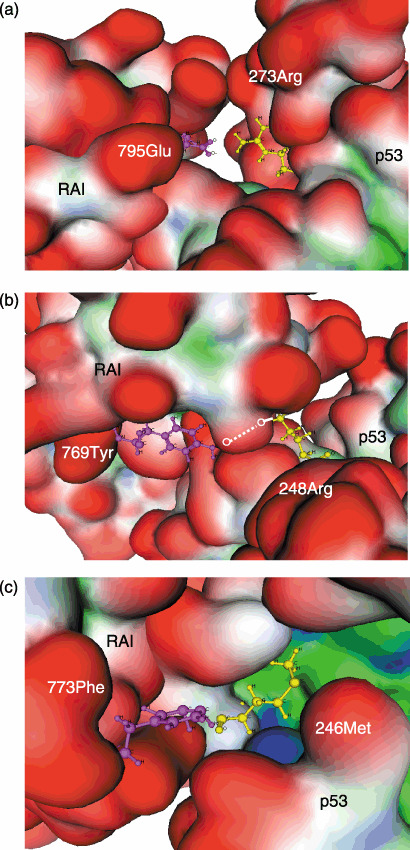
Interacting amino acid residues on the p53 and RelA‐associated inhibitor (RAI) proteins. Representative amino acid pairs are shown, including those representing (a) ionic interactions between p53 (R273) and RAI (E795), (b) hydrogen bonding between p53 (R248) and RAI (Y769), and (c) hydrophobic interactions between p53 (M246) and RAI (F773). These interacting amino acid residues are shown by the ball and stick model in magenta (RAI: E795, Y769, F773) and yellow (p53: R273, R248, M246). Other regions are shown by surface representation. The hydrogen bonding between p53 (R248) and RAI (Y769) is shown by a dotted line. The 3‐dimensional structure of the p53–RAI complex was predicted by molecular docking using GreenPepper software. The definition of colors is as described in Figure 3.
Table 2.
Interacting amino acids involved in the binding between p65 and RelA‐associated inhibitor (RAI) proteins
| p65 | RAI | Force |
|---|---|---|
| 19Pro | 796Thr | HB |
| 24Ile | 731Phe | HYD |
| 50Arg | 726Asp | ION |
| 110Ilet | 731Phe | HYD |
| 253Arg | 659Glu | ION |
| 297Arg | 639Glu | ION |
HB, hydrogen bonding; HYD, hydrophobic interaction; ION, ionic interaction.
Effects of RAI mutants. In order to confirm the biological effects of the RAI amino acids that were predicted to interact with p53, we created a series of RAI mutants in which these interface amino acids were substituted with Ala or other amino acids with opposite electrostatic characteristics (Table 3). The target amino acid residues for site‐directed mutagenesis were chosen based on the cancer‐related incidence of mutations of the interacting amino acid residues on p53. Thus, we created seven RAI mutants: RAI‐m1 (F773A), RAI‐m2 (D775A), RAI‐m3 (D775R), RAI‐m4 (Y769A), RAI‐m5 (E795A), RAI‐m6 (E795D), and RAI‐m7 (D775A; Y769A). The biological effects of wild‐type RAI and these RAI mutants were assessed for their effects on the p53‐mediated transactivation of the bax gene promoter as there was no significant effect on gene expression from the p21 promoter (Fig. 2b).
Table 3.
List of RelA‐associated inhibitor (RAI) mutants used in this study
| Mutant | AA position | AA (wild type) | AA (mutant) | AA (p53) | Force |
|---|---|---|---|---|---|
| RAI‐m1 | 773 | Phe | Ala | Met or Ile † | HYD |
| RAI‐m2 | 775 | Asp | Ala | Arg | ION |
| RAI‐m3 | 775 | Asp | Arg | Arg | ION |
| RAI‐m4 | 769 | Tyr | Ala | Arg | HB |
| RAI‐m5 | 795 | Glu | Ala | Arg | ION |
| RAI‐m6 | 795 | Glu | Arg | Arg | ION |
| RAI‐m7 | 769 | Tyr | Ala | Arg | HB |
| 775 | Asp | Ala | Arg | ION |
The 773F amino acid of RAI is predicted to interact with three amino acid residues, 243Met, 246Met, and 251Ile, of p53 via hydrogen bonding. AA, amino acid; HB, hydrogen bonding; HYD, hydrophobic interaction; ION, ionic interaction.
Fig. 5 demonstrates the effects of RAI mutants in blocking the transcriptional activity of p53 with the bax promoter. Whereas RAI‐m1 (F773A) exhibited a similar inhibitory effect on the p53‐mediated transactivation of bax expression (Fig. 5a), both RAI‐m2 (D775A) and RAI‐m3 (D775R) lost the inhibitory action on p53‐mediated bax expression (Fig. 5b,c). D775 of RAI interacts with R248 of p53 (Table 1) and, in fact, a greater extent of mutation effect was obtained with RAI‐m3 (D775R) over RAI‐m2 (D775A), thus confirming the prediction that these amino acids interact with each other possibly through an ionic interaction. It was also suggested that a single amino acid substitution at the molecular contact surface of the hydrophobic interaction could not significantly block the interaction. In addition, substitution of Y769 of RAI, interacting with R248 of p53 by hydrogen bonding, to the non‐hydrogen‐bonding amino acid Ala lost the inhibitory effect of RAI substantially (Fig. 5d). Because substitution of RAI E795 (which interacts with p53 R273 by ionic interaction) did not dramatically reduce the inhibitory action of RAI (Fig. 5e,f), it is assumed that there may be other surface interactions with this mutation‐prone p53 region, as predicted by molecular docking (Fig. 4a). As shown in Fig. 6, RAI‐m7 (D775A; Y769A), containing a double substitution at D775 and Y769 (both to A), exhibited a complete lack of inhibitory activity of RAI, which was more significant than that of RAI‐m2 (D775A), thus confirming the above observations. These mutants were tested on p53‐mediated gene expression from the p21 promoter. However, no significant inhibition of the p53‐mediated induction of p21 gene expression was observed (data not shown).
Figure 5.
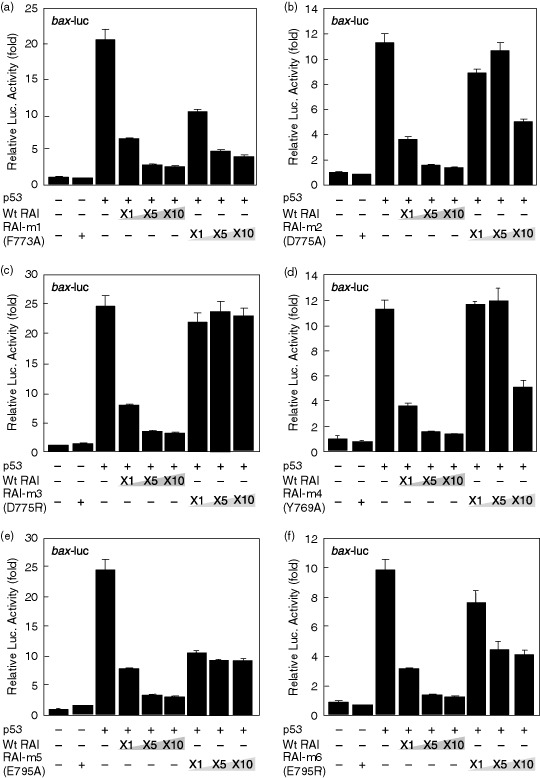
Effects of p53‐dependent gene expression by RelA‐associated inhibitor (RAI) and its mutants. A total of 293 cells were transfected with the bax‐luc reporter plasmid together with the pC53‐SN3 plasmid and various amounts of plasmids that express wild‐type RAI (pFLAG‐CMV2‐RAI) or RAI mutants, including (a) RAI‐m1 (F773A), (b) RAI‐m2 (D775A), (c) RAI‐m3 (D775R), (d) RAI‐m4 (Y769A), (e) RAI‐m5 (E795A), or (f) RAI‐m6 (E795R). The luciferase activity was normalized to Renilla luciferase activity, which was cotransfected as an internal control. The data are presented as the fold increase in luciferase activities (mean ± SD) relative to control transfection of three independent experiments.
Figure 6.
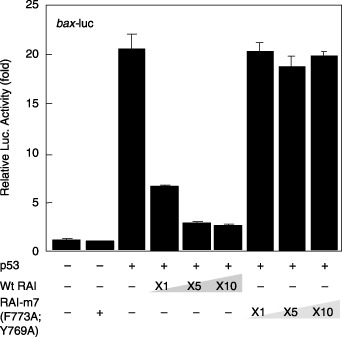
Effects of p53‐dependent gene expression by the RelA‐associated inhibitor (RAI) double mutants. The experiments were carried out as for Figure 5. Note that there was no significant inhibition by RAI‐m7 (D775A; Y769A).
Effects of RAI mutants on NF‐κB. Finally, the effects of these RAI mutants were examined for p65‐mediated gene expression from the NF‐κB‐responsive luciferase reporter gene. As shown in Fig. 7, either RAI‐m3 (D775R) or RAI‐m7 (D775A; Y769A) did not significantly lose the inhibitory action of RAI, which was expected from the molecular docking simulation where no common amino acid residue was involved in the interaction between RAI and p53 or p65 (1, 2).
Figure 7.
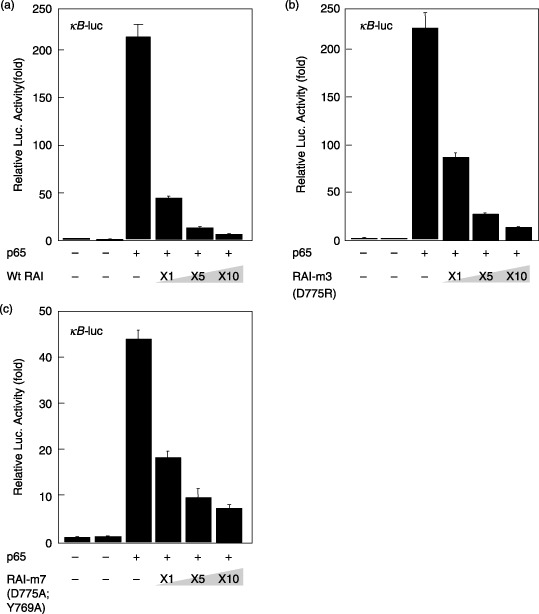
Effects of p65‐dependent gene expression by wild‐type RelA‐associated inhibitor (RAI) and its mutants. A total of 293 cells were transfected with the κB‐luc plasmid together with the pGL3‐p65 plasmid and various amounts of plasmids that express wild‐type RAI (pFLAG‐CMV2‐RAI) or RAI mutants, including (a) RAI‐m1 (F773A), (b) RAI‐m2 (D775A), (c) RAI‐m3 (D775R), (d) RAI‐m4 (Y769A), (e) RAI‐m5 (E795A), and (f) RAI‐m6 (E795R). The luciferase activity was normalized to Renilla luciferase activity, which was cotransfected as an internal control. The data are presented as the fold increase in luciferase activities (mean ± SD) relative to control transfection of three independent experiments.
Discussion
Because of the dual functions of RAI as an interacting inhibitor protein of p53 and the p65 subunit of NF‐κB, the biological roles of RAI are mysteriously ambivalent.( 6 , 7 ) The p53 protein acts as a tumor suppressor and is primarily involved in cell cycle arrest and apoptosis induction through transactivating the p21 and bax genes, respectively.( 25 , 26 ) In contrast, the p65 subunit of NF‐κB, a cellular homolog of the c‐Rel oncogene, acts as a pro‐carcinogenic protein and is involved in immunoinflammatory responses and progression of carcinogenesis, as evidenced by constitutive activation of NF‐κB in some tumors and leukemia as well as chronic inflammatory processes.( 7 ) Although there has been no report of mutations in the rai gene in cancer or leukemia, RAI is reported to be overexpressed in leukemia cells that are refractory to chemotherapy.( 8 ) Because RAI inhibits p53‐mediates transactivation of bax but not that of p21,( 5 , 6 ) it appears that RAI makes the cells containing DNA damage (presumably caused by irradiation or other DNA‐damaging reagents) arrest the cell cycle rather than inducing irreversible apoptosis, thus protecting cancerous or precancerous cells. However, RAI inhibits the transcriptional activity of NF‐κB by directly blocking its DNA binding activity,( 1 , 4 ) thus preventing tumor promotion and its progression.
In the present study, we have analyzed the 3D structures of RAI in complex with p53 and p65 in order to understand the structural background of the RAI–p53 interaction by adopting a computer‐assisted molecular docking simulation. We found that RAI interacts with p53 and p65 with distinct amino acid residues, and that substitutions of amino acid residues on RAI that ruined the interaction with p53 did not substantially damage the interaction with p65. These findings also confirmed the predicted 3D structure of the p53–RAI complex. Although we attempted to demonstrate the physical binding between p53 and various RAI mutants in accordance with the method of Bergamaschi et al.( 5 ) we were not able to demonstrate the binding reproducibly either in vitro or in vivo (data not shown). This is probably due to the possibility that the interaction between p53 and RAI requires additional proteins.
Since the establishment of the protein data bank, there has been an accumulation of structural data obtained by X‐ray crystallography and nuclear magnetic resonance. Also, a number of bioinformatic procedures have been developed to predict the 3D structures of biomolecules such as proteins and nucleic acids. In the present study, we utilized the general software Molecular Operating Environment for analysis of the 3D structures of biomolecules, and the molecular docking‐simulation software GreenPepper for prediction of 3D structures of protein–protein complexes. We used this software to predict the amino acids that are involved in the molecular interaction between RAI and p53 or p65. We then confirmed the computer‐assisted prediction of the 3D structure by carrying out site‐directed mutagenesis of interacting amino acids by substituting them with neutrally charged amino acid, such as Ala, or oppositely charged amino acids by considering the mode of protein–protein interaction.
As expected, whereas most of the RAI mutants created lost the inhibitory, some showed only subtle changes (Table 3; Fig. 5). For example, RAI‐m1 (F773A) did not lose much inhibitory action. Because F773 is involved in the hydrophobic patch formed between p53 and RAI at multiple surfaces such as those involving M243, M246, and I251 of p53, it is considered that a sole mutation of F773 did not change the interaction even though this particular amino acid residue is involved in hydrophobic patches. In contrast, RAI‐m3 (D775R) lost the interaction with p53 almost completely, suggesting that the presumable ionic interaction between RAI D775 and p53 R248 might be crucial and possibly involved in the primary molecular interaction between these two molecules. Moreover, p53 R248 was predicted to interact with both D775 and Y769 of RAI through ionic interactions and hydrogen bonding, respectively. Interestingly, p53 R248 is the most frequently mutated amino acid associated with cancer.( 27 , 28 ) Similarly, RAI‐m5 (E795A) lost the interaction to a significant extent, although not completely. Its counterpart amino acid on p53, R273, is the second most‐frequent site of p53 mutation in cancer.( 27 , 28 ) Thus, our findings shown here demonstrate the feasibility of the computational methods adopted in this study in predicting the protein–protein interaction.
In summary, we have carried out the computer‐assisted prediction of the 3D structure of the protein complex between p53 and RAI. We were able to confirm the predicted structure by creating a series of RAI mutants in which the interacting amino acids involved in protein–protein binding to p53 were mutated. These mutants lost the ability to inhibit the biological function of p53 in transactivating bax gene expression. The findings demonstrated here clearly indicate feasibility and accuracy of such computational methods in interpreting the biological significance of such interacting amino acids and predicting mutants that lose the biological activity. Moreover, further elucidation of the structural characteristics of the protein complex should predict effective small chemical compounds that could interfere with the RAI–p53 interactions.
Acknowledgments
We thank Drs Hirofumi Arakawa, Toshiyuki Miyashita, and Toshiyuki Sakai for the generous gifts of plasmids.
References
- 1. Yang JP, Hori M, Sanda T, Okamoto T. Identification of a novel inhibitor of nuclear factor‐κB, Rel‐A‐associated inhibitor. J Biol Chem 1999; 274: 15 662–70. [DOI] [PubMed] [Google Scholar]
- 2. Takahashi N, Kobayashi S, Jiang X et al . Expression of 53BP2 and ASPP2 proteins from TP53BP2 gene by alternative splicing. Biochem Biophys Res Commun 2004; 315: 434–8. [DOI] [PubMed] [Google Scholar]
- 3. Yang JP, Hori M, Takahashi N, Kawabe T, Kato H, Okamoto T. NF‐κB subunit p65 binds 53BP2 and inhibits cell death induced by 53BP2. Oncogene 1999; 18: 5177–86. [DOI] [PubMed] [Google Scholar]
- 4. Takada N, Sanda T, Okamoto H et al . RelA‐associated inhibitor blocks transcription of human immunodeficiency virus type 1 by inhibiting NF‐κB and Sp1 actions. J Virol 2002; 76: 8019–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bergamaschi D, Samuels Y, O’Neil N et al . iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet 2003; 33: 162–7. [DOI] [PubMed] [Google Scholar]
- 6. Samuels Y, O’Connor DJ, Bergamaschi D et al . ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 2001; 8: 781–94. [DOI] [PubMed] [Google Scholar]
- 7. Okamoto T, Sanda T, Asamitsu K. NF‐κB signaling and carcinogenesis. Curr Pharm Des 2007; 13: 447–62. [DOI] [PubMed] [Google Scholar]
- 8. Zang X, Wang M, Zhou C, Chen S, Wang J. The expression of iASPP in acute leukemias. Leuk Res 2005; 29: 179–83. [DOI] [PubMed] [Google Scholar]
- 9. Herron BJ, Rao C, Liu S et al . A mutation in NF‐κB interacting protein 1 results in cardiomyopathy and abnormal skin development in wa3 mice. Hum Mol Genet 2005; 14: 667–77. [DOI] [PubMed] [Google Scholar]
- 10. Zang X, Diao S, Rao Q et al . Identification of a novel isoform of iASPP and its interaction with p53. J Mol Biol 2007; 368: 1162–71. [DOI] [PubMed] [Google Scholar]
- 11. Baker SJ, Markowitz S, Fearon ER, Willson JV, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild‐type p53. Science 1990; 249: 912–15. [DOI] [PubMed] [Google Scholar]
- 12. Sato T, Asamitsu K, Yang JP et al . Inhibition of human immunodeficiency virus type 1 replication by a bioavailable serine/threonine kinase inhibitor, fasudil hydrochloride. AIDS Res Hum Retroviruses 1998; 14: 293–8. [DOI] [PubMed] [Google Scholar]
- 13. Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995; 80: 293–9. [DOI] [PubMed] [Google Scholar]
- 14. Nakano K, Mizuno T, Sowa Y et al . Butyrate activates the WAF1/Cip1 gene promoter through Sp1 site in a p53‐negative human colon cancer cell line. J Biol Chem 1997; 272: 22 199–206. [DOI] [PubMed] [Google Scholar]
- 15. Asamitsu K, Tetsuka T, Kanazawa S, Okamoto T. RING finger protein AO7 supports NF‐κB‐mediated transcription by interacting with the transactivation domain of the p65 subunit. J Biol Chem 2003; 278: 26 879–87. [DOI] [PubMed] [Google Scholar]
- 16. Suganuma M, Kawabe T, Hori H, Funabiki T, Okamoto T. Sensitization of cancer cells to DNA damage‐induced cell death by specific cell cycle G2 checkpoint abrogation. Cancer Res 1999; 59: 5887–91. [PubMed] [Google Scholar]
- 17. Tetsuka T, Uranishi H, Imai H et al . Inhibition of nuclear factor‐κB‐mediated transcription by association with the amino‐terminal enhancer of split, a Groucho‐related protein lacking WD40 repeats. J Biol Chem 2000; 275: 4383–90. [DOI] [PubMed] [Google Scholar]
- 18. Uranishi H, Tetsuka T, Yamashita M et al . Involvement of the prooncoprotein TLS (translocated in liposarcoma) in nuclear factor‐κB p65‐mediated transcription as a coactivator. J Biol Chem 2001; 276: 13 395–401. [DOI] [PubMed] [Google Scholar]
- 19. Kondo T, Ishida H, Yamauchi R, Watanabe E, Hirose C, Go M. GreenPepper: The docking prediction software of protein complex. In: The 15th International Conference on Genome Informatics , Posters and Software Demonstrations, p. 128, 2004. Available from URL: http://www.jsbi.org/molecules/journal/index.php/GIW04/GIW04Poster.html
- 20. Palm PN, Krippahl L, Wampler JE, Moura JJG. BIGGER: A new (soft) docking algorithm for predicting protein interactions. Proteins 2000; 39: 372–84. [PubMed] [Google Scholar]
- 21. Glaser F, Steinberg DM, Vakser IA, Ben‐Tal N. Residue frequencies and pairing preferences at protein–protein interfaces. Proteins 2001; 43: 89–102. [PubMed] [Google Scholar]
- 22. Gorina S, Pavletich NP. Structure of the p53 tumor suppressor bound to the Ankyrin and SH3 domains of 53BP2. Science 1996; 274: 1001–5. [DOI] [PubMed] [Google Scholar]
- 23. Naumovski L, Cleary ML. The p53‐binding protein 53BP2 also interacts with Bcl2 and impedes cell cycle progression at G2/M. Mol Cell Biol 1996; 16: 3884–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Slee EA, Gillotin S, Bergamaschi D et al . The N‐terminus of a novel isoform of human iASPP is required for its cytoplasmic localization. Oncogene 2004; 23: 9007–16. [DOI] [PubMed] [Google Scholar]
- 25. Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl‐2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993; 74: 609–19. [DOI] [PubMed] [Google Scholar]
- 26. Harper JW, Adams GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk‐interacting Cip1 is a potent inhibitor of G1 cyclin‐dependent kinases. Cell 1993; 75: 805–16. [DOI] [PubMed] [Google Scholar]
- 27. Soussi T, Lozano G. p53 mutation heterogeneity in cancer. Biochem Biophys Res Commun 2005; 331: 834–42. [DOI] [PubMed] [Google Scholar]
- 28. Soussi T. p53 alterations in human cancer: more questions than answers. Oncogene 2007; 26: 2145–56. [DOI] [PubMed] [Google Scholar]