

Abstract
The mutation of Apc is an important early genetic event in colon carcinogenesis. However, it remains to be clarified what kinds of cooperative genes are required for complete carcinogenesis. To identify cooperative genes for the ApcMin mutation the authors carried out retroviral insertional mutagenesis (RIM) using Min mouse‐derived IMCE colon epithelial cells. Anchorage‐independent transformed colonies were induced by retroviral infection only in IMCE cells, while no transformation was found in young adult mouse colon (YAMC) cells that are normal for Apc. One hundred and fifty‐seven retroviral integration sites (RIS) were identified in 101 independent transformants, and four common integration sites (CIS), Dnah3, Ahnak, Stk17b and Rbm9, were observed. Upregulation of Dnah3 and Ahnak, and truncation of Dnah3 due to the viral integration, was revealed. In addition, Dnah3‐overexpressing IMCE cells showed impairment of microtubule function. These data suggest the importance of cytoskeletal function in Apc‐related tumor development and the usefulness of RIM in non‐hematopoietic tissues, providing new insight into the early stage of colon carcinogenesis. (Cancer Sci 2008; 99: 979–985)
Carcinogenesis is a multistep molecular process that requires combinations of oncogenic mutations, loss‐of‐function mutations of tumor suppressors, epigenetic alterations of gene expression and structural abnormalities of chromosomes.( 1 ) It is therefore important to understand how individual genetic alterations cooperate with each others. Moreover, the mutated genes and their cofactors in cancers are cell‐type‐specific. Thus, it is desirable to clarify such cell‐type‐specific oncogenic processes and the functions of the disease genes in the proper cell types.
Retroviral insertional mutagenesis (RIM) is a powerful tool by which to identify causative oncogenes and tumor suppressor genes, and many cancer disease genes have been identified using the method.( 2 , 3 ) More importantly, the method has recently been used to identify cooperative genes for certain oncogenic stimuli.( 4 , 5 , 6 ) However, the method has mainly been applied for hematologic neoplasms, with a few exceptions,( 7 , 8 ) because most murine retroviruses are strong mutagens specific for hematopoietic cells. To exclude such disadvantages due to hematopoietic tropism of retroviruses in vivo, the authors have devised a novel system of RIM that facilitates the analysis of oncogenic transformation of the colon epithelium in vitro.
Loss‐of‐function mutations of the Apc gene play a critical role in both human and murine colon carcinogenesis.( 9 ) It has been noted that the Apc mutation is an early oncogenic event in human colon carcinogenesis, and that mutational accumulation of disease genes including K‐ras, SMAD2, SMAD4 and p53 occurs.( 10 , 11 , 12 , 13 ) However, it remains unclear what genes specifically cooperate with the Apc mutation in certain steps of colon carcinogenesis. To identify cooperative genes for the Apc mutation that are important for colon cancer development, IMCE and young adult mouse colon (YAMC) cells were infected with the murine stem cell virus (MSCV) retrovirus.( 14 , 15 ) YAMC is an immortalized colon epithelial cell line derived from the H‐2kb‐tsA58 transgenic mouse (Immorto mouse) that bears the interferone‐inducible and temperature‐sensitive simian virus 40 (tsSV40) large T antigen as a transgene. IMCE cells are obtained from the F1 mouse cross between the Immorto mouse and the Min mouse that has a germline mutation of Apc.( 16 , 17 , 18 , 19 ) After MSCV infection, the IMCE cells showed colony formation in soft agar; however, the retroviral infection did not transform YAMC cells. This result suggests that retroviral integrations might cooperate with the Apc mutation for transformation of colon epithelial cells in vitro. The authors have identified 157 retroviral integration sites (RIS) and four common integration sites (CIS) in 101 transformed YAMC colonies. The candidate cooperative genes for the Apc mutation have been isolated in these sites. The present study presents a novel application of RIS for cancer research and new molecular mechanisms of colon carcinogenesis.
Materials and Methods
Cell lines. The epithelial cell lines derived from the colonic mucosa of the Immorto mouse (YAMC cell) and from an F 1 Immorto–Min mouse hybrid (IMCE cell), carrying Apc min mutation, were gifts from Dr Robert H. Whitehead and have been described elsewhere.( 14 , 15 ) These cells are untransformed colonic epithelial cell lines that are conditionally immortalized by introducing a tsSV40 large T antigen. The YAMC and IMCE cells were maintained in RPMI‐1640 medium (Sigma‐Aldrich, St Louis, MO, USA) supplemented with 5% fetal bovine serum (FBS; Cell Culture Technologies, Canada), 100 mM 4‐(2‐hydroxyethyl)‐1‐piperazine‐ethanesulphonic acid (HEPES; Nakarai Tesque, Kyoto, Japan), 1 IU of insulin (Sigma‐Aldrich), 5 IU of γ‐interferon (Sigma‐Aldrich) and penicillin/streptomycin (Sigma‐Aldrich), in a CO incubator at 33°C.
Retrovirus infection and soft agar assay. The MSCV–EGFP retrovirus plasmid is composed of MSCV long‐terminal repeats (LTR), the enhanced green fluorescent protein (EGFP) gene and a neomycin‐resistance gene driven by the cytomegalovirus (CMV) promoter (Fig. 1b). The plasmid was transfected to Plat‐E packaging cells (a gift from Dr Toshio Kitamura) using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacture's protocol. The culture supernatant was used to infect the YAMC and IMCE cells. Two milliliters of the supernatant were added to the YAMC and IMCE cells (1 × 106 cells in a 10‐cm diameter dish) together with 4 µg/mL of polybrene (Sigma‐Aldrich) and incubated for 2 h at 33°C. The cultures were incubated for a further 48 h at 33°C. The infected cells were then harvested and seeded in soft agar including G418 for selection at a density of 1 × 104 cells per well in a 6‐well plate. After 2 weeks of incubation the colonies were counted and isolated.
Figure 1.
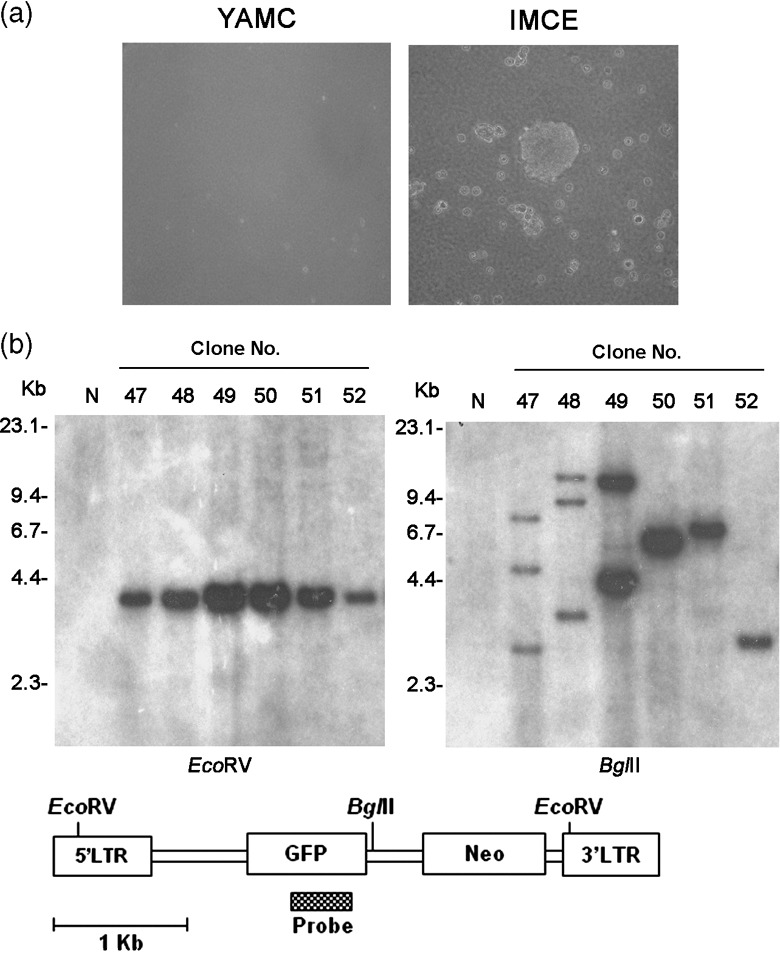
Transformation of IMCE cells by retrovirus. (a) Morphology of transformed colonies in soft agar. Retrovirus‐infected IMCE cells show anchorage‐independent growth. (b) Southern blot analysis of transformants using a green fluorescent protein (GFP) probe. Genomic DNA was digested with BglII or EcoRV, which cut the retrovirus once or at both long terminal repeat (LTR), respectively. Retroviruses of the expected size were confirmed (left), and monoclonal or oligoclonal integrations were detected (right). Genomic DNA of the C57BI/6 J mouse was used as control (N). YAMC, young adult mouse colon.
Isolation of retroviral integration sites. Retroviral integration sites were identified using the inverse polymerase chain reaction (IPCR) or splinkerette PCR (SPCR) approach as described previously,( 5 ) with slight modifications.
IPCR. Genomic DNA samples were digested with EcoRI, BamHI, BglII, NcoI, HindIII or SacI, self‐ligated, and subjected to nested IPCR. The PCR primers for each restriction digestion are available on request. The PCR products were analyzed using agarose gel electrophoresis, subcloned into plasmids, and subjected to sequence analysis.
SPCR. Genomic DNA samples were digested with BstYI. After inactivation of the enzyme, digested the DNA was ligated to a splinkerette oligonucleotide. The ligated mixtures were digested with EcoRV. The virus/genomic junction fragments were isolated after nested PCR amplification. The PCR products were analyzed as for IPCR.
Southern blotting. Southern blot analysis was carried out to assess the clonal insertion of the retrovirus. The genomic DNA was digested with appropriate restriction enzymes, subjected to agarose gel electrophoresis and transferred to a Hybond‐N nylon filter (GE Healthcare, UK). The filters were hybridized with 32P‐labeled GFP cDNA or mouse genomic DNA fragments flanking to integration sites as probes.
Northern blotting. Poly (A) + RNA was extracted from the transformants using a FastTrack 2.0 kit (Invitrogen), size‐fractionated using a formaldehyde–agarose gel, transferred to a Hybond‐N nylon filter and probed with the Dnah3 or Ahnak cDNA fragments. The Gapdh probe was used as a control.
Quantitative real‐time reverse transcription (RT)‐PCR and standard RT‐PCR. Expression of Dnah3 and Ahnak was quantitated using a 7500 Fast Real‐time PCR system (Applied Biosystems, Foster City, CA, USA) and SYBR‐Green reagents according to the manufacture's protocol. Poly (A) + RNA samples were reverse transcribed to cDNA using an ImProm‐II Reverse Transcription System (Promega, Madison, WI, USA). The specific forward and reverse primers were designed as follows: Dnah3 forward, 5′‐GCTTATGTCATCCCCCCCTATG‐3′; Dnah3 reverse, 5′‐GGTTTCCTGGTTGTCCTTGGTG‐3′; Ahnak forward, 5′‐GGGATGATGGAGTCTTTGTTCAGG‐3′; Ahnak reverse, 5′‐TTCAGCAACTGGGTCACCTCAC‐3′; Gapdh forward, 5′‐TGTCGTGGAGTCTACTGGTGTCTTC‐3′, Gapdh reverse, 5′‐GGAGATGATGACCCTTTTGGCTC‐3′.
Standard RT‐PCR was used for detection of a Dnah3/retrovirus transcript. PCR was performed using cDNA samples as described above, and a retrovirus‐forward (virus‐F; 5′‐CATCCGAATCGTGGACTC‐3′), Dnah3 exon 50 forward (50‐F; 5′‐CCAAGAAACTTCTGGTGCAGGCA‐3′, Dnah3 exon52 reverse (52‐R; 5′‐ACACTCCCCAGTAATCTTCTATC‐3′, Dnah3 reverse (Dnah3‐R; 5′‐CTATGATGCTACACAGCTGC‐3′). Amplified bands were separated on 1% Tris‐phosphate‐EDTA (TPE) agarose gels.
5′‐rapid amplification of cDNA end (5′‐RACE). 5′‐RACE was performed using the SMART RACE cDNA Amplification kit (Clontech, Palo Alto, CA, USA) with Dnah3 gene‐specific primers 5Rex1 (5′‐GACAGGGGTGCCAAACTGCAGGGC‐3′) and 5Rex8 (5′‐GGTCAGCCACGTAGCCCCCCAGGGGC‐3′) and adaptor primers UPM Long (5′‐CTAATACGACTCACTATAGGGCAAGCAGT‐GGTATCAACGCAGAGT‐3′), UPM Short (5′‐CTAATACGACTCACTATAGGGC‐3′) and NUP (5′‐AAGCAGTGGTATCAACGCAGAGT‐3′). The 5′‐RACE products were subcloned into plasmids and sequenced.
Microtubule polymerization. The IMCE and transformed cells (E39) were incubated on ice for 1 h to depolymerize the microtubules. Then the cold medium was removed and fresh medium at 39°C was added. The cells were incubated for 0, 1, 2 min and fixed with methanol. Immunofluorescence staining of the cells was carried out using anti‐α‐tubulin (T5168; Sigma‐Aldrich). The proportion of cells with microtubules asters in the preparations was counted.
Results
Retroviral infection transforms the IMCE cell. The colonic epithelial cell lines, YAMC and IMCE, were derived from the Immorto mouse, YAMC, and an Immorto‐Min mouse hybrid, respectively. While the YAMC cell has normal Apc alleles (Apc +/+) the IMCE cell has a mutant Apc allele (Apc min/+). Both cell lines are non‐transformed and cannot form anchorage independent colonies in soft agar.( 15 ) The YAMC and IMCE cells were transduced with the MSCV–GFP retrovirus and after 48 h seeded into soft agar containing G418 for 2 weeks at 33°C. The number of colonies was counted, and an average of 35 colonies per 1 × 104 transduced cells IMCE was obtained; in contrast no colony appeared in transduced YAMC cells (Table 1 and , Fig. 1a).
Table 1.
Transformation of IMCE cells
| Cell | Retrovirus | No. of colonies/well† |
|---|---|---|
| YAMC | – | 0 |
| + | 0 | |
| IMCE | – | 0 |
| + | 35 ± 10.8 |
Number of colonies per 1 × 104 cells (average ± standard error) was determined after 2 weeks of growth in soft agar. YAMC, young adult mouse colon.
The Southern blot analysis showed that most of the transformed IMCE colonies were monoclonal and that each colony had an average of 1.5 copies of the retroviruses (Fig. 1b). The wild type allele of Apc was preserved in all the colonies (data not shown). These results suggest that the IMCE cells were transformed by retroviral integration and that Apc heterozygous mutation is important for transformation.
Identification of RIS and CIS. To analyze the RIS of the transformed IMCE cells, host–virus junction sequences were amplified using inverse PCR or splinkerette PCR. A total of 157 RIS in 101 colonies were isolated (Table S1). A BLAT search of the flanking DNA sequences of each RIS using a public database (http://www.genome.ucsc.edu/) revealed the genomic location and the candidate target genes of the RIS. Four CIS (defined as integration sites found at least two independent colonies no more than 100 kb apart) were identified (Table 2). These were Dnah3, Ahnak, Stk17b and Rbm9, for the Apc mutation was isolated near these CIS.
Table 2.
Common integration sites and candidate cooperative genes
| Candidate genes | Gene products | Chromosome locus | No. of integrations (n = 157) |
|---|---|---|---|
| Dnah3 | Dynein axonemal heavy chain 3 | 7qF2 | 2 |
| Ahnak | Ahnak/desmoyokin nucleoprotein | 19qA | 2 |
| Stk17b | Serine/threonine kinase 17b | 1qC1.1 | 2 |
| Rbm9 | RNA‐binding motif protein 9 | 15qE1 | 2 |
Upregulation of the Dnah3 and Ahnak genes by retrovirus integrations. The position and orientation of retroviral integrations in the E39 and A8 clones at the Dnah3 locus in mouse chromosome 7qF2 is shown in Fig. 2a. DNA rearrangements were detected in both clones using the appropriate probes (Fig. 2b), indicating that the integrations at the Dnah3 locus occur in the majority of both clones and that retroviral integrations of E39 and A8 are located at the 49 and 43 introns, respectively. The expression levels of Dnah3 were analyzed using quantitative RT‐PCR and northern blot (Fig. 2c,d). Quantitative RT‐PCR of the E39 and A8 clones showed 4287‐fold and 35‐fold enhanced expression, respectively, compared with the F5 clone that does not have integrations at the Dnah3 locus. The Dnah3 upregulation of both clones was further confirmed by the northern blot analysis. In addition, abnormal >10 kb, 7.5 kb and 6 kb transcripts were detected in E39 (Fig. 2d); in contrast the wild‐type 8 kb transcript was detected in parental IMCE, F5 and A8 clones. These results indicate that retroviral integrations at the Dnah3 loci enhanced Dnah3 expression and suggest that Dnah3 might be important for retrovirus‐induced transformation.
Figure 2.

Retroviral integrations at the Dnah3 locus result in Dnah3 upregulation. (a) A physical map of retroviral integrations (arrowheads) within the Dnah3 gene. K; KpnI. E; EcoRV. White boxes indicate Dnah3 exon 41 through 59. (b) Southern blot analyses using the probes indicated in (a). DNA rearrangements are shown in E39 and A8 clones. (c‐d) Overexpression of Dnah3 by retroviral integrations. Retroviral integrations are indicated as + or –. (c) Quantitative real‐time reverse transcription‐polymerase chain reaction analysis. The Y‐axis indicates the expression level of Dnah3 relative to Gapdh. (d) Northern blot analysis using a Dnah3 cDNA fragment as a probe. The quality and quantity of RNA was confirmed by reprobing the same blot with Gapdh.
The physical map of the Ahnak locus in 19qA is shown in Fig. 3a. The A22 and C8 clones had retroviral integrations within intron 4 and 2.5 kb downstream of Ahnak, respectively. Retroviral integrations at the Ahnak loci were again confirmed using Southern blot analysis (Fig. 3b, left), although the lesser intensity of the rearranged bands suggested that these cell lines might contain minor populations that do not have the integrations. As shown in Fig. 3b, right, A22 and C8 clones increased enhancement of Ahnak expression 2.5‐ and 3.5‐fold, respectively, compared with the E24 clone, without an integration at this locus. Collectively, these results suggest that Dnah3 and Ahnak are targets for retroviral insertions upon IMCE transformation.
Figure 3.
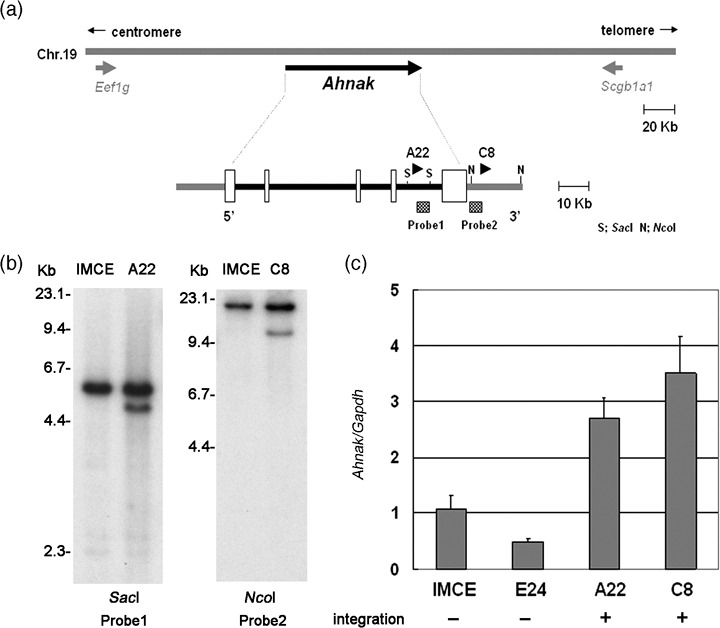
Retroviral integrations at the Ahnak locus. (a) A physical map of retroviral integrations at the Ahnak locus. Arrowheads indicate the location of retroviral integration. S; SacI, N; NcoI. White boxes indicate Ahnak exons. (b) Southern blot using the probes indicated in (a). Rearranged bands were detected in both A22 and C8 clones. (c) Quantitative real‐time reverse transcription‐polymerase chain reaction. Expression levels of Ahnak relative to Gapdh are indicated.
Fusion transcript between Dnah3 and the retroviral sequence. Truncated and/or fusion transcripts are frequently observed when a retrovirus is inserted within a gene.( 2 ) A 5′‐RACE experiment using an E39 cDNA with Dnah3‐specific and adaptor primers identified a retroviral packaging sequence fused to Dnah3 exon 50 in the same transcriptional orientation (Fig. 4). RT‐PCR using retrovirus (Virus‐F) and Dnah3 exon 52 (52‐R) primers confirmed the presence of the same fusion transcript (Fig. 4a, top left). In addition, the wild‐type Dnah3 transcripts were detected both in IMCE and the E39 clone using Dnah3‐specific 50‐F and 52‐R primers (Fig. 4a, top right).
Figure 4.
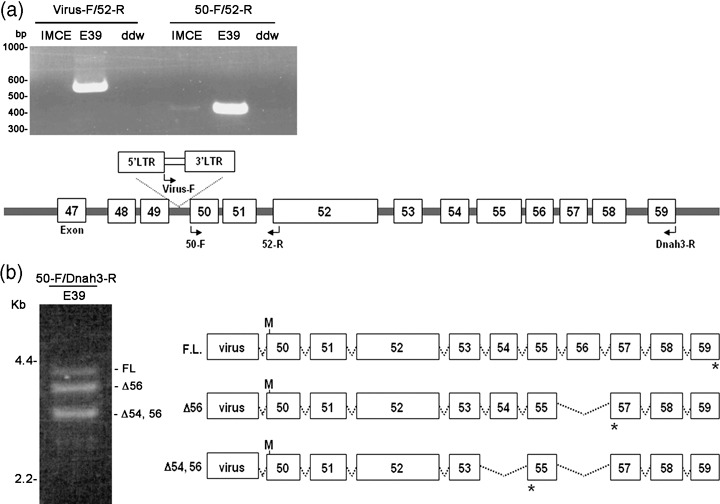
Fusion transcript between Dnah3 and retrovirus. (a) A fusion transcript identified using reverse transcription‐polymerase chain reaction (RT‐PCR). Left, RT‐PCR using retrovirus (virus‐F) and Dnah3 exon 52 (52‐R) primers. A fusion transcript is found in the E39 clones. In contrast, the wild‐type Dnah3 transcripts are detected both in IMCE and the E39 clone using Dnah3‐specific 50‐F and 52‐R primers. (b) Alternative transcripts observed in the E39 clone. RT‐PCR shows three different products. Structures of the retrovirus–Dnah3 chimeric transcripts are shown in the right panel. M, putative translation initiation; *, termination codons.
As a result of the fusion between Dnah3 and MSCV it is predicted that a truncated Dnah3 protein is synthesized using an alternative translation initiation site within exon 50 (Fig. 4b). Moreover, three types of Dnah3 transcripts downstream of exon 50 in the E39 clone were observed (Fig. 4b, left). The structure of three transcripts, the full‐length transcript, the transcript lacking exon 56 (Δ56), and that lacking exon 54 and 56 (Δ54; 56) due to alternative splicing is shown in Fig. 4b, right. Dnah3 encodes dynein heavy chain protein and the deduced amino acids of these Dnah3/retrovirus chimera are 1298, 1247 and 1166, containing a microtubules‐binding domain, and functions as a motor protein.( 20 , 21 ) None of these three transcripts was detected in the parental IMCE, probably due to the low expression of Dnah3 (data not shown). The Dnah3 truncated products by the retroviral integration might execute some functions in IMCE transformation.
Impaired microtubule polymerization. The microtubule polymerization rate was visualized by the size and intensity of the asters in IMCE and E39 cells that over‐express Dnah3. Appearance of the asters is delayed in E39, and the difference between E39 and IMCE was remarkable at 1 min after temperature change (Fig. 5 and Table 3). In the IMCE cell, the aster formation was observed in 63% of population, whereas it was shown in only 25% of the E39 cell. These findings suggest that overexpression of Dnah3 might promote cytoskeletal dysfunction induced by Apc heterozygous mutation.
Figure 5.

Microtubule polymerization in IMCE and E39 cells. Immunofluorescence of microtubules in IMCE and E39 cells after depolymerization. Polymerization rate of microtubules after ice treatment for 1 h was studied by adding medium at 39°C for 1 and 2 min followed by staining with anti‐tubulin antibody. Maximally depolymerized microtubules are shown after 1 h ice treatment (0 min). The new microtubules start to grow from the centrosome in the cell to form a small star‐like structure, called aster (original magnification, ×40).
Table 3.
The frequency of cells with visible microtubule asters
| Cell | Time (min) | ||
|---|---|---|---|
| 0 | 1 | 2 | |
| IMCE (%) | <1 | 63.4 | >80 |
| E39 (%) | <1 | 25.0 | 75.5 |
Other RIS. Additionally, retroviral integrations in two independent clones each at the Stk17b and Rbm9 loci were confirmed by the Southern blot analysis (data not shown). Retroviral integrations were observed within intron 2 and 26.7 kb upstream of the Stk17b gene, and 28.7 and 78.0 kb upstream of the Rbm9 gene. The expression levels of Stk17b and Rbm9 were examined using northern blot and quantification of RT‐PCR. In both Stk17b and Rbm9 loci only one of each of two clones was upregulated (3.6‐fold in Stk17b and 2.5‐fold in Rbm9) while the expression of the other clones remained unchanged (data not shown).
Several interesting genes were identified in the IMCE transformants although they were isolated only once in the present study. A selection of these loci is listed in Table 4 and includes Max, Stat6, Ephb3, Map3k1, Fgfr2, Upa, Fbln5, Pdcd8, Cd226, and Cdc25a, which are implicated in the development of colon cancer and/or growth modification of colon cancer cells.
Table 4.
Other retroviral integration sites
| Candidate genes | |
|---|---|
| Transcription | Max, Stat6 |
| Signaling | Ephb3, Map3k1, Fgfr2, Upa |
| Cytoskeleton | Fbln5, Pdcd8, Cd226 |
| Cell cycle | Cdc25a |
Discussion
Apc mutations are early genetic events in human colon carcinogenesis.( 9 ) In familial adenomatous polyposis patients, one allele of the Apc gene is mutated, and most colon adenomas in these patients show deletions or mutations of the wild‐type allele.( 22 , 23 ) Subsequently, multiple genetic alterations including K‐ras and p53 are observed during transformation of adenomas toward more malignant tumors.( 10 ) However, many important genes that are involved in colon carcinogenesis remain to be identified, and prospective studies to identify cooperative oncogenic events with Apc heterozygous mutation have not been sufficiently achieved.
RIM is a powerful tool by which to identify oncogenes and tumor suppressors that are important in carcinogenesis, particularly in leukemogenesis.( 2 , 3 , 4 ) However, the method has limited application for non‐hematopoietic tissues in vivo. In the present study an in vitro RIM assay was devised to identify the important genes responsible for the transformation of colon epithelial cells. MSCV infection transformed Min mouse‐derived IMCE cells but not YAMC cells with normal Apc status, indicating that transformation requires both retroviral integrations in some specific loci and the Apc heterozygous mutation. This suggests that retroviral integrations may affect the genes cooperative with the Apc mutation. Transduction efficiency was more than 90% and the transformation rate was 3 × 10−3. It is rather surprising that only four CIS were isolated and most of the RIS were identified only once in our cohort. In addition, 44 of 153 present RIS are included in the Retroviral Tagged Cancer Gene Database (RTCGD; http://rtcgd.abcc.ncifcrf.gov/) and only seven are CIS in the RTCGD. The difference of RIS distribution might be attributable to the non‐hematopoietic nature of the IMCE cell. However, a recent study showed there is no significant difference of RIS distribution between human CD34+ cells and HeLa cells,( 24 ) suggesting many retroviral integrations identified in the present study have some biological effects on the growth of IMCE cells.
In the present study none of the transformant clones showed loss of the Apc wild‐type allele (data not shown), suggesting that the anchorage‐independent colony formation of IMCE cells in soft agar might not be an equivalent step to adenoma development. Interestingly, it has been reported that gap junctional intercellular communication (GJIC) is decreased in heterozygous mutations of Apc.( 25 ) As a result of Apc heterozygous mutation of connexin‐43 is decreased, which may be related to microtubule malfunction. In addition, the interaction between the COOH‐terminus of APC and EB1 plays an essential role in the regulation of microtubule polymerization.( 26 ) In this context it is intriguing that the abnormal Dnah3 transcript is identified in the IMCE transformant, because Dnah3 is a component of dynein motor complex, the function of which is microtubule‐dependent.( 27 , 28 ) Indeed, the formation of cytoplasmic asters of E39 cells over‐expressing Dnah3 was sluggish compared with IMCE cells. The genetic changes identified here might be those in the very early stages of colon cancer development before adenoma development, nevertheless, they could induce in vitro transformation of the IMCE cell that retains the wild‐type Apc allele.
Dnah3 encodes a dynein heavy chain protein consisting of 4116 amino acids that is a component of the dynein motor complex (DMC), which is important for intracellular trafficking on microtubules.( 26 , 29 , 30 , 31 ) It contains two ATP binding sites within a central P‐loop motif and a C‐terminal coiled‐coil domain as a putative protein‐binding domain.( 27 , 28 , 29 , 30 , 31 ) As a result of protein truncation due to retroviral integration, the aberrant Dnah3 lacks the N‐terminal two‐thirds but retains a C‐terminal domain that is putatively important for association with microtubules.( 20 , 21 ) Thus, the mutant Dnah3 may affect DMC functions such as cell motility, trafficking the molecules/organelles and/or mitosis, retaining its ability to bind microtubules.
Ahnak encodes a 600‐kDa giant propeller‐like protein that associates with calcium channel proteins.( 32 ) Its carboxyl‐terminal region interacts with actin filaments on muscle contracture.( 33 ) Although the significance of upregulated expression of Ahnak in IMCE colonies is not clear, it may affect the cytoskeletal network of colon epithelial cells. Interaction between the cytoskeletal components and APC is important for tumor initiation and progression,( 34 ) and its role in mitotic spindle on chromosomal alignment have been emphasized.( 35 , 36 , 37 ) The identification of Dnah3 and Ahnak as CIS in the present study suggests that the Apc heterozygous mutation may cooperate with these genes through abnormal cytoskeletal function in the early stage of colon tumor development.
Several genes were identified only once in this study, however, they are nonetheless intriguing because of their function in cellular proliferation and differentiation. Moreover, the genes listed in Table 3 are in fact implicated in colon carcinogenesis and/or affected in growth of colon epithelial cells. Many RIS identified in IMCE cells are not reported in the RTCGD and are unique to the present study. This may be due to the difference between hematopoietic cells and epithelial cells. Further studies are needed to clarify the different effects of retroviral integration on various cell types, and modification of retroviral sequences may enable the application of RIM to non‐hematopoietic tumors in vivo.
Supporting information
Table S1. Retroviral integration sites in transformed IMCE cells.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This work was supported by KAKENHI (Grant‐in‐Aid for Scientific Research) on Young Scientists (B) from the Japan Society for the Promotion of Science and on Priority Areas ‘Integrative Research Toward the Conquest of Cancer’ the Ministry of Education, Culture, Sports, Science and Technology of Japan. The authors would like to thank Dr Robert H. Whitehead for YAMC and IMCE, Dr Toshio Kitamura for Plat‐E cell, and Drs Takeshi Kuwata and Toru Hirota for helpful discussions.
References
- 1. Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 2001; 1: 222–31. [DOI] [PubMed] [Google Scholar]
- 2. Jonkers J, Berns A. Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochem Biophys Acta 1996; 1287: 29–57. [DOI] [PubMed] [Google Scholar]
- 3. Mikkers H, Berns A. Retroviral insertional mutagenesis: tagging cancer pathways. Adv Cancer Res 2003; 88: 53–99. [DOI] [PubMed] [Google Scholar]
- 4. Nakamura T. Retroviral insertional mutagenesis identifies oncogene cooperation. Cancer Sci 2005; 96: 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jin G, Yamazaki Y, Takuwa M et al . Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood 2007; 109: 3998–4005. [DOI] [PubMed] [Google Scholar]
- 6. Iwasaki M, Kuwata T, Yamazaki Y et al . Identification of cooperative genes for NUP98‐HOXA9 in myeloid leukemogenesis using a mouse model. Blood 2005; 105: 784–93. [DOI] [PubMed] [Google Scholar]
- 7. Peters G, Lee AE, Dickson C. Activation of cellular gene by mouse mammary tumour virus may occur early in mammary tumor development. Nature 1984; 309: 273–5. [DOI] [PubMed] [Google Scholar]
- 8. Johansson FK, Brodd J, Eklof C et al . Identification of candidate cancer‐causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci USA 2004; 101: 11334–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Powell SM, Zilz N, Beazer‐Barclay Y et al . APC mutations occur early during colorectal tumorigenesis. Nature 1992; 359: 235–7. [DOI] [PubMed] [Google Scholar]
- 10. Fodde R, Smits R, Clevers H. Apc, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 2001; 1: 55–67. [DOI] [PubMed] [Google Scholar]
- 11. Yamada Y, Mori H. Multistep carcinogenesis of the colon in Apc Min/+ mouse. Cancer Sci 2007; 98: 6–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–67. [DOI] [PubMed] [Google Scholar]
- 13. Morin PJ, Sparks AB, Korinek V et al . Activation of β‐catenin‐Tcf signaling in colon cancer by mutagen in β‐catenin or APC. Science 1997; 275: 1787–90. [DOI] [PubMed] [Google Scholar]
- 14. Whitehead RH, Joseph JL. Derivation of conditionally immortalized cell lines containing the Min mutation from the normal colonic mucosa and other tissues of an ‘immortomouse’/Min hybrid. Epith Cell Biol 1994; 3: 119–25. [PubMed] [Google Scholar]
- 15. D’abaco GM, Whitehead RH, Burgess AW. Synergy between Apc min and an activated ras mutation is sufficient to induce colon carcinomas. Mol Cell Biol 1996; 16: 884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jat PS, Noble MD, Ataliotis P et al . Direct derivation of conditionally immortal cell lines from an H‐2Kb‐tsA58 transgenic mouse. Proc Natl Acad Sci USA 1991; 88: 5096–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whitehead RH, Vaneeden PE, Noble MD, Ataliotis P, Jat PS. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H‐2Kb‐tsA58 transgenic mice. Proc Natl Acad Sci USA 1993; 90: 587–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990; 247: 322–4. [DOI] [PubMed] [Google Scholar]
- 19. Su LK, Kinzler KW, Vogelstein B et al . Multiple intestinal neoplasia caused by a mutation in the murine homolog of the Apc gene. Science 1992; 256: 668–70. [DOI] [PubMed] [Google Scholar]
- 20. Gee MA, Heuser JE, Vallee RB. An extended microtubule‐binding structure within the dynein motor domain. Nature 1997; 390: 636–9. [DOI] [PubMed] [Google Scholar]
- 21. Koonce MP. Identification of a microtubule‐binding domain in a cytoplasmic dynein heavy chain. J Biol Chem 1997; 272: 19714–18. [DOI] [PubMed] [Google Scholar]
- 22. Ichii S, Takeda S, Horii A et al . Detailed analysis of genetic alterations in colorectal tumors from patients with and without familial adenomatous polyposis (FAP). Oncogene 1993; 8: 2399–405. [PubMed] [Google Scholar]
- 23. Levy DB, Smith KJ, Beazer‐Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer Res 1994; 54: 5953–8. [PubMed] [Google Scholar]
- 24. Cattoglio C, Facchini G, Sartori D et al . Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007; 110: 1770–8. [DOI] [PubMed] [Google Scholar]
- 25. Husoy T, Cruciani V, Knutsen HK, Mikalsen SO, Olstorn HB, Alexander J. Cells heterozygous for the ApcMin mutation have decreased gap junctional intercellular communication and connexin43 level, and reduced microtubule polymerization. Carcinogenesis 2003; 24: 643–50. [DOI] [PubMed] [Google Scholar]
- 26. Nakamura M, Zhou ZX, Lu KP. Critical role for the EB1 and APC interaction in the regulation of microtubule polymerization. Curr Biol 2001; 11: 1062–7. [DOI] [PubMed] [Google Scholar]
- 27. Milisav I. Dynein and dynein‐related genes. Cell Motil Cytoskeleton 1998; 39: 261–72. [DOI] [PubMed] [Google Scholar]
- 28. Ross JL, Wallace K, Shuman H, Goldman YE, Holzbaur E. Processive bidirectional motion of dynein‐dynactin complexes in vitro. Nat Cell Biol 2006; 8: 562–70. [DOI] [PubMed] [Google Scholar]
- 29. Vallee RB, Sheetz MP. Targeting of motor proteins. Science 1996; 271: 1539–44. [DOI] [PubMed] [Google Scholar]
- 30. Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 1998; 279: 519–26. [DOI] [PubMed] [Google Scholar]
- 31. Vale RD. The molecular motor toolbox for intracellular transport. Cell 2003; 112: 467–80. [DOI] [PubMed] [Google Scholar]
- 32. Komuro A, Masuda Y, Kobayashi K et al . The AHNAKs are a class of giant propeller‐like proteins that associate with calcium channel proteins of cardiomyocytes and other cells. Proc Natl Acad Sci USA 2004; 101: 4053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haase H, Pagel I, Khalina Y et al . The carboxyl‐terminal ahnak domain induces actin bundling and stabilizes muscle contraction. FASEB J 2004; 18: 839–41. [DOI] [PubMed] [Google Scholar]
- 34. Näthke I. Cytoskeleton out of the cupboard: colon cancer and cytoskeletal changes induced by loss of APC. Nat Rev Cancer 2006; 6: 967–74. [DOI] [PubMed] [Google Scholar]
- 35. Kiyosue MY, Tsukita S. Where is APC going? J Cell Biol 2001; 154: 1105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Human Mol Genet 2001; 10: 721–33. [DOI] [PubMed] [Google Scholar]
- 37. Green RA, Wollman R, Kaplan KB. APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol Biol Cell 2005; 16: 4609–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Retroviral integration sites in transformed IMCE cells.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item