

Abstract
Hyperleptinemia is a common feature of obese women who have a higher risk of endometrial cancer than women with normal weights, and epidemiologic studies have suggested a correlation between obesity and endometrial carcinoma. Therefore, understanding of the molecular mechanism involved in leptin signaling transduction is important in endometrial cancer prevention and treatment. In this study, both isoforms of the leptin receptor (Ob‐R), the long form (Ob‐Rb) and short form (Ob‐Ra), were detected as being expressed in six endometrial cancer cell lines with various differentiation status by western blotting, and Ob‐Ra was found to be more abundant than Ob‐Rb in these cells. Moreover, the expressions of both isoforms were inversely correlated with histoprognostic grading. We also showed that leptin stimulated cell proliferation and induced activations of signal transducers and activators of transcription 3 (STAT3), extracellular signal‐regulated kinase (ERK1/2), AKT, and cyclooxygenase (COX)‐2 in endometrial cancer cells dose‐dependently by [3H] thymidine incorporation assay and western blotting. Leptin‐stimulation resulted in increased expression of COX‐2 mRNA and prostaglandin E2 (PGE2) production of endometrial cancer cells by reverse transcription–polymerase chain reaction and enzyme immunoassay, respectively, which was effectively blocked by pharmacological inhibitors of Janus tyrosine kinase 2 (JAK2), AG490; of mitogen‐activated protein kinase (MAPK) kinase, U0126; of phosphatidylinositol 3‐kinase (PI3K), LY294002; and of COX‐2, NS398. These results suggest that leptin promotes cell proliferation of endometrial cancer cells via the aforementioned multiple signal‐transduction pathways. Leptin‐induced functional activation of COX‐2 is JAK2/STAT3‐, MAPK/ERK‐, and PI3K/AKT‐dependent, indicating that COX‐2 may be a critical factor of endometrial carcinogenesis in obesity. (Cancer Sci 2009; 100: 389–395)
Endometrial cancer is the most common gynecologic malignancy in the United States, and the occurrence has been increasing in Asian countries. It comprises about 4% of all cancer in women globally and occurs predominantly after the menopause. The role of obesity as one of the main risk factors in both premenopausal and postmenopausal women has been firmly established.( 1 , 2 ) Leptin, the 16 kDa secreted protein of the obese (Ob) gene by white adipose tissue, has been recently known to stimulate the proliferation of cancer cells in various organs, such as in the breast, ovary, prostate, and colon,( 3 , 4 , 5 , 6 ) leading to consideration of this protein as a carcinogenetic factor.
Leptin exerts its biological action through the leptin receptor, termed Ob‐R, which belongs to the cytokine receptor superfamily.( 7 ) So far, six different isoforms of the leptin receptor have been discovered. Ob‐Rb, the long form, and Ob‐Ra, the short form, are the two major isoforms present in mammalian cells.( 8 , 9 ) It is now well established that the long isoform Ob‐Rb, mainly expressed in hypothalamus, is responsible for signal transduction through the activation of Janus‐activated kinase 2 (JAK2)/signal transducers and activators of transcription 3 (STAT3) and the mitogen‐activated protein kinase (MAPK)/extracellular signal‐regulated kinase (ERK) signaling pathways.( 10 , 11 ) On the other hand, the short isoform Ob‐Ra, which is more abundant in peripheral tissues than Ob‐Rb, mainly activates MAPK and seems to be responsible for mitogenic activity.( 10 , 11 ) Ample evidence has also demonstrated that Ob‐Rb can be found in many peripheral tissues and a variety of cancer cells, including endometrial and ovarian cancers.( 3 , 6 , 12 , 13 , 14 , 15 , 16 ) Thus far, the functions of both Ob‐Rb and Ob‐Ra in the human peripheral system are not completely understood. Moreover, it has been recently reported that leptin stimulates proliferation of various types of human cancer cells via multiple signaling pathways, such as phosphatidylinositol 3‐kinase (PI3K)/AKT, steroid receptor coactivator (SRC)‐1 and cyclooxygenase (COX)‐2, in addition to JAK2/STAT3 and MAPK/ERK.( 3 , 6 , 16 , 17 , 18 , 19 , 20 ) Therefore, leptin may be an important factor in the increased incidence of human cancer in obesity acting through both isoforms of Ob‐Rb and Ob‐Ra, and the interactions among these signals activated by leptin stimulation need to be further identified.
COX, a key enzyme in the conversion of arachidonic acid to prostaglandins (PGs) and other eicosanoids, exists as two isoforms, COX‐1 and COX‐2. Accumulating evidence indicates that overexpression of the human COX‐2 gene induces tumorigenesis, increases metastasis potential, and promotes angiogenesis.( 21 , 22 , 23 ) NS398, a selective COX‐2 inhibitor, inhibits growth and induces apoptosis in cultured endometrial, colon, and other types of cancer cells.( 24 , 25 , 26 , 27 ) Thus, COX‐2 has recently been thought to be a target for the prevention and/or treatment of human cancer.( 28 , 29 , 30 , 31 ) Previous studies have found that the induced expressions of COX‐2 mRNA and protein can be directly mediated by the MAPK and PI3K pathways.( 32 , 33 , 34 ) Furthermore, limited data have demonstrated that initial stimulation with leptin in OE33 esophageal adenocarcinoma cells induces JAK2 activation and subsequent activation of MAPK and AKT signals followed by increased COX‐2 mRNA and PGE2 production, suggesting that JAK2/STAT, MAPK, and PI3K/AKT signals may be required to increase COX‐2 mRNA levels and consequently increase PGE2 production.( 20 )
A few studies show that leptin may promote a proliferative response and invasiveness in human endometrial cancer cells by activating the aforementioned multiple signal‐transduction pathways,( 35 ) and the aberrantly expressed leptin receptor in human endometrial cancer tissues is possibly involved in the pathogenesis of endometrial cancer.( 13 ) However insufficient data regarding the effect of leptin on human endometrial cancer cells are available, and the mechanisms involved remain unclear. In the present study, we show that COX‐2 mRNA and protein expressions, as well as consequent PGE2 production, were increased by leptin, and that the JAK2/STAT3‐, MAPK/ERK‐, and PI3K/AKT‐dependent COX‐2 pathway are involved in the cell proliferative effect of leptin on human endometrial cancer cells.
Materials and Methods
Reagents. The human recombinant leptin was purchased from R&D Systems (Minneapolis, MN, USA) and stored as stock solution in phosphate‐buffered saline (PBS) at –20°C. AG490, a JAK2 inhibitor, and U0126, an inhibitor of mitogen‐activated protein kinase kinase (MEK1/2) were purchased from CalBiochem‐NovaBiochem (La Jolla, CA, USA). LY294002, a PI3K inhibitor, was purchased from Cell Signaling Technology (Danvers, MA, USA). NS398, a selective COX‐2 inhibitor, was purchased from Sigma Chemical Corporation (St. Louis, MO, USA). Eagle's minimum essential medium (Eagle's MEM) and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY, USA). Anti‐Ob‐R (H‐300) for the detection of the leptin receptor, both Ob‐Rb and Ob‐Ra isoforms, anti‐COX‐2, anti‐STAT3, anti‐ERK1/2, and goat antimouse IgG horseradish peroxidase (HRP)‐conjugated second antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti‐phosphorylated STAT3 Tyrosine705 and Serine 727 (P‐STAT3 Tyr705 and Ser727), and antiphosphorylated ERK1/2 Threonine 202/Tyrosine 204 (P‐ERK1/2) were purchased from BD Biosciences Pharmingen (San Diego, CA, USA). Anti‐AKT and antiphosphorylated AKT Serine 473 were purchased from Cell Signaling Technology. Anti‐β‐actin and donkey antirabbit IgG HRP‐conjugated second antibody and the enhanced chemiluminescence (ECL) system were purchased from Sigma Chemical Corporation and Amersham Biosciences (Buckinghamshire, UK), respectively. The enzyme immunoassay (EIA) kit for the measurement of PGE2 levels was purchased from Assay Designs (Ann Arbor, MI, USA).
Cell culture and treatment. The human endometrial cancer cell lines used were Ishikawa( 36 ) and ECC‐1,( 37 ) both derived from well‐differentiated endometrial adenocarcinoma; HEC‐1A and a substrain HEC‐1B derived from moderately differentiated endometrial adenocarcinoma;( 38 , 39 ) RL95‐2 derived from moderately differentiated adenosquamous carcinoma of the endometrium;( 40 ) and AN3CA derived from undifferentiated endometrial adenocarcinoma.( 41 ) Of six cell lines, Ishikawa and AN3CA were gifts from Dr Teruhiko Tamaya (Gifu University School of Medicine, Japan); others were purchased from the America Type Culture Collection (Manassas, VA, USA). The cell lines were maintained in Eagle's MEM supplemented with 10% heat‐inactivated (56°C, 30 min) FBS. Monolayer cultures were incubated at 37°C in a 95% humidified atmosphere containing 5% CO2. The pharmaceutical inhibitors were dissolved in 100% dimethylsulfoxide (DMSO) and then diluted in medium to a final concentration of DMSO at 0.1%. For treatment, cells were seeded at a density of 8 × 10 000 cells/mL of medium in tissue culture dishes. After 18–24 h of serum starvation, the medium was replenished with fresh serum‐free medium containing treatments as indicated. Cultures were treated with human recombinant leptin alone at 100 ng/mL or with dose escalation. For combined treatment, cells were pretreated with AG490 at 50 µM, or U0126 at 10 µM, or LY294002 at 10 µM, or NS398 at 10 µM for 1 h followed by combination with a constant 100 ng/mL dose of leptin. Throughout the indicated duration of each treatment, cells treated were harvested and saved for the following experiments.
[3H] Thymidine incorporation assay. [3H] Thymidine incorporation assay was performed to analyze the proliferation index in endometrial cells. Six hours prior to termination of the indicated experiment, cells in each 24‐well plate were pulsed with 1 Ci of [3H] thymidine. After incubation, the cells were washed three times with ice‐cold PBS, followed by precipitation with 0.5 mL 10% trichloroacetic acid for 30 min at 4°C. The precipitate was washed in methanol twice and solubilized in 250 µL of 0.5 N sodium hydroxide, and the incorporated radioactivity was measured in the tri‐Carb Liquid Scintillation. Each experiment was performed in quadruplicate.
Reverse transcription–polymerase chain reaction (RT‐PCR). Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and chloroform extraction according to the manufacturer's instructions. Two‐microgram total RNA served as template for first‐strand cDNA synthesis in a reaction using oligo (dT) primers and M‐MLV reverse transcriptase. The cDNAs obtained were further amplified by PCR using the following primers: 5′‐ATTTGATTGACAGTCCACC‐3′ (sense) and 5′‐TACATCATCAGACCAGGCA‐3′ (antisense) for human COX‐2 and 5′‐TGAAGGTCGGAGTCAACGGATTTGGT‐3′ (sense) and 5′‐CATGTGGGCCATGAGGTCCACCAC‐3′ (antisense) for the human housekeeping gene glyceraldehydes‐3‐phosphate dehydrogenase (GAPDH). The PCR was performed for 40 cycles (94°C for 1 min, 62°C for 1 min, and 72°C for 2 min) for COX‐2 (amplifies a fragment of 550 bp) and 25 cycles (94°C for 1 min, 58°C for 1 min, and 72°C for 2 min) to amplify the human GAPDH (amplifies a fragment of 983 bp). A 0.5 µg portion of cDNA was used in a standard 25 µL PCR mixture with 1 µM each of the primers mentioned above. 0.5 mM dNTP; 0.4 µL Taq DNA polymerase (5 units/µL), and 2.2 mM magnesium chloride. The PCR products were analyzed by electrophoresis in 2% agarose gel in the presence of ethidium bromide.
Western blot analysis. Western blot analysis was performed as previously described.( 25 ) The cells were washed twice with ice‐cold PBS and lyzed with buffer A (10 mM Tris‐HCl [pH 7.4], 1% Triton X‐100, 0.1% sodium deoxycholate, 0.1% sodium dodecylsufate [SDS], 150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol [DTT], 0.5 mM phenylmethylsulfonyl fluoride [PMSF], 10 µg/mL leupeptin, 5 µg/mL aprotinin). After incubation for 30 min on ice, the suspensions were centrifuged (15 000 g, 30 min). The supernatants were removed and stored at –80°C until analysis using gel electrophoresis. The protein concentration was determined by Bio‐Rad DC protein estimation assay according to the manufacturer's instructions. Sixty to 100 µg of whole cell proteins were separated using 10% polyacrylamide gels and transferred to nitrocellulose membranes. After blocking of the membranes with TBS‐T (10 mM Tris‐HCl [pH 7.4], 150 mM NaCl, and 0.1% Tween 20) containing 5% bovine serum albumin at 4°C overnight, the membranes were incubated with the indicated antibodies at room temperature for 90 min, and then with the second antirabbit or antimouse antibodies at room temperature for 60 min. Each protein was detected using the ECL system. β‐Actin was used as an internal control.
PGE2 assay. Endometrial cancer cells were cultured on 12‐well tissue culture plates in serum‐free medium for 18–24 h and treated with leptin and/or the inhibitor for 24 h in duplicate. Aliquots (0.5 mL) of culture medium were withdrawn and stored at –20°C until analysis using an EIA kit as reported previously.( 25 ) Immediately before assay, the samples were centrifuged (7500 g, 5 min) to remove any cellular debris. The supernatant was transferred to fresh tubes and kept on ice. The PGE2 assay was performed according to the manufacturer's instructions. PGE2 concentration was corrected by protein content on each culture plate. The protein content was determined as described above. The results obtained as pg PGE2/µg protein were transformed to relative values of the control taking the samples from cells not treated as 1.
Statistical analysis. Each experiment was repeated three times. Data are presented as mean ± SD. Statview 5.0 software was used for statistical analyses. Statistical comparison among the groups was performed using anova, followed by Fisher's least significant difference test. Differences were considered to be statistically significant when P was less than 0.05.
Results
Expression of leptin receptor in endometrial cancer cells. Firstly, we investigated the expression of leptin receptor in six lines of endometrial cancer cells with various differentiation status by western blotting. Both Ob‐Rb and Ob‐Ra were expressed in all of the six lines, but the Ob‐Rb expressed in each line was relatively weaker than the Ob‐Ra (Fig. 1a). In these endometrial carcinoma cells, both isoforms were strongly expressed in well‐differentiated Ishikawa and ECC‐1 cells; and weakly expressed in moderately differentiated RL95‐2, HEC‐1B, HEC‐1 A cells and undifferentiated AN3CA cells. There was a reverse correlation between expression of leptin receptor and the carcinoma differentiation status among the six lines as reported by Yuan et al.( 12 ) The expression of leptin receptor in endometrial cancer tissues was highly correlated with the differentiation status of the cancer tissues, with the more‐differentiated status expressing higher levels of leptin receptor than the poorly differentiated ones.
Figure 1.
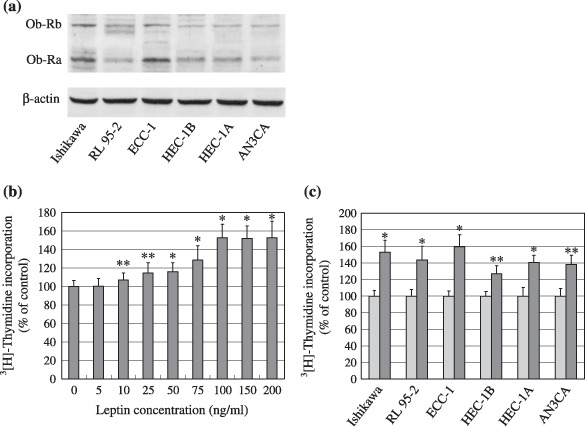
Expression of the leptin receptor and effect of leptin on the proliferation of endometrial cancer cells. (a) Western blot analysis of leptin receptor expression, long form (Ob‐Rb, 120 kDa) and short form (Ob‐Ra, 90 kDa), in endometrial cancer cells. Equal loading and transfer were shown by repeat probing with β‐actin. (b) Serum‐starved Ishikawa cells were treated for 24 h with increasing concentrations of leptin and (c) six lines of serum‐starved endometrial cancer cells were treated with a 100 ng/mL dose of leptin for 24 h. DNA syntheses were determined by [3H] thymidine incorporation assay. *P < 0.01 and **P < 0.05 compared with untreated control cells, respectively.
Leptin stimulates proliferation of endometrial cancer cells. Leptin added in serum‐free culture medium significantly stimulated DNA synthesis dose‐dependently in six lines for 24 h of treatment, and the maximum effect was at 100 ng/mL which was used for further characterization (Fig. 1b and data not shown). Higher concentrations of leptin (150 and 200 ng/mL) did not exert an additional effect on the six lines at 24 h. As low as a 10 ng/mL dose of leptin for 24 h resulted in increased proliferation in these cells (Fig. 1b and data not shown). In addition, the effect of leptin (100 ng/mL) on the proliferation of six cell lines, as shown in Fig. 1(c), demonstrated a positive correlation with the expression of leptin receptor, with the higher level of leptin receptor being more sensitive to leptin‐stimulation than the lower ones.
Leptin activates STAT3, ERK1/2, AKT, and COX‐2 in endometrial cancer cells. To investigate the intracellular signaling mechanisms responsible for the proliferative effect of leptin on endometrial cancer cells, we further examined the changes in signal phosphorylation of STAT3, ERK1/2, AKT, and COX‐2 expression in the Ishikawa cells after leptin stimulation in time‐/dose–course. As shown in Fig. 2, the phosphorylation of STAT3 on both Tyr705 and ser727 residues was increased after 5 min of a 100 ng/mL dose of leptin treatment, was maximal at 30 min, and then declined progressively. Similarly, leptin significantly increased the immediate phosphorylation of ERK1/2. The maximal activation of ERK1/2 appeared at 5–15 min after exposure to a 100 ng/mL dose of leptin, and declined thereafter (Fig. 2a). The pattern of leptin‐stimulated activation of AKT demonstrated in Fig. 2a was same as that of activated STAT3. Additionally, dose–course studies have shown that as low as 25 ng/mL to a maximal 100 ng/mL dose of leptin significantly increased STAT3 (on Tyr705 and Ser727 residues), ERK1/2, and AKT phosphorylation after 10 min of treatment (Fig. 2b), and higher concentrations (150 and 200 ng/mL) of leptin did not result in additional effect on activations of these signals (data not shown). While the total protein levels of STAT3, ERK1/2, and AKT were not changed in Ishikawa cells treated with leptin (Fig. 2a,b), our observation that stimulation of Ishikawa cells with leptin led to immediate increase in STAT3, ERK1/2, and AKT phosphorylation was consistent with a previous report.( 35 ) Importantly, the increased COX‐2 protein was evident within 4 h after leptin treatment, reached a maximum at 24 h, and the increase remained detectable for up to 48 h (Fig. 2a and data not shown). COX‐2 protein was also measured in cells treated with leptin with a dose‐gradient for 24 h. A concentration‐dependent increase in COX‐2 expression by leptin stimulation was also observed (Fig. 2b), whereas COX‐1 protein expression was constitutively expressed and did not change after leptin treatment (data not shown).
Figure 2.
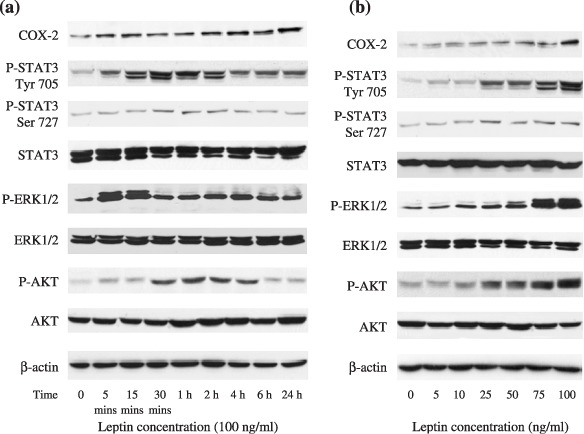
Multiple intracellular pathways involved in the growth‐stimulatory effect of leptin on endometrial cancer cells. Serum‐starved Ishikawa cells were treated with leptin (100 ng/mL) for various intervals of time (a) and treated for 10 min with increasing concentrations of leptin (b). Protein expressions of cyclooxygenase (COX)‐2, total and phosphorylated forms of signal transducers and activators of transcription 3 (STAT3), extracellular signal‐regulated kinase (ERK1/2), as well as AKT were detected by western blot analysis. Equal loading and transfer were shown by repeat probing with β‐actin.
Leptin induces functional activation of COX‐2 in endometrial cancer cells. The main aim of this study was to determine whether leptin induced functional activation of COX‐2 in endometrial cancer cells. For this purpose, six cell lines of endometrial cancer cells were treated with 100‐ng/mL leptin for 4 or 24 h and COX‐2 mRNA or protein expression was detected by RT‐PCR or western blotting, respectively. Stimulation of these cells with leptin induced a marked increase in COX‐2 mRNA and protein (Fig. 3a,b). The bioactivity of COX‐2 is to catalyze PGE2 production. We also found that the stimulation of leptin significantly induced PGE2 production compared with untreated cells (Fig. 3c). Moreover, the leptin‐induced increase in PGE2 production was similar to that for COX‐2 expression, indicating that the increase in COX‐2 expression has biological a function which might be involved in the leptin‐stimulated proliferation of endometrial cancer cells.
Figure 3.
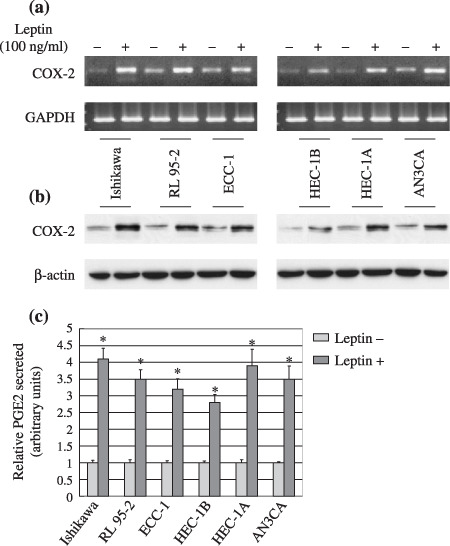
Leptin induced cycooxygenase (COX)‐2 mRNA/protein expression and prostaglandin E2 (PGE2) secretion in endometrial cancer cells. Six lines of serum‐starved endometrial cancer cells were stimulated for 4 or 24 h. (a) COX‐2 mRNA expression was detected by reverse transcription–polymerase chain reaction and glyceraldehydes‐3‐phosphate dehydrogenase (GAPDH) was used as an internal control. (b) COX‐2 protein expression and (c) PGE2 production were measured by western blot analysis and enzyme immunoassay, respectively. Results expressed as PGE2 production relative to untreated control cells. *P < 0.01 compared with untreated cells.
Leptin‐stimulated proliferation of endometrial cancer cells is associated with JAK2/STAT3‐, MAPK/ERK‐, and PI3K/AKT‐dependent COX‐2 bioactivity. To confirm whether activation of COX‐2 is involved in leptin‐induced proliferation of endometrial cancer cells and mediated by the JAK2/STAT3, MEK/ERK1/2, and PI3K/AKT pathways, six cell lines were pretreated with 50 µM of AG490, 10 µM of U0126, 10 µM of LY294002, or 10 µM of NS398 for 1 h followed by combination with a constant 100 ng/mL dose of leptin for 24 h as described in ‘Materials and Methods’. As shown in Fig. 4(a), leptin‐induced proliferation was abolished by the selective COX‐2 inhibitor NS398. Pharmacological inhibition of JAK2 with AG490, MEK with U0126, and PI3K with LY 294002 also abolished leptin‐induced proliferation. Furthermore, the effect of leptin‐increased COX‐2 protein expression and PGE2 secretion were blocked by COX‐2 inhibitor NS398, similarly abrogated by inhibition of the JAK2, ERK, and PI3K pathways (Fig. 4b,c). Of note, these pharmacological inhibitors used at the indicated concentrations in this study for 24‐h treatment alone, with the exception of NS 398, could markedly inhibit the respective targets (Fig. 4b). Interestingly, AG490 treatment blocked leptin‐induced activation of both ERK1/2 and AKT; while blockage of the MEK/ERK1/2 pathway by U0126 not only down‐regulated the constitutive P‐STAT3 Ser727 level, but also abolished leptin‐induced P‐STAT3 Ser727 overexpression (Fig. 4b), indicating that leptin‐induced phosphorylation of ERK1/2 and AKT was dependent on JAK2/STAT3 activation as well as a potential interaction between the MAPK/ERK and JAK2/STAT3 pathways. Together, these data suggest that the increased biological function of COX‐2 is downstream from the activated JAK2/STAT3, MAPK/ERK, and PI3K/AKT pathways responsible for leptin‐induced proliferation of endometrial cancer cells.
Figure 4.
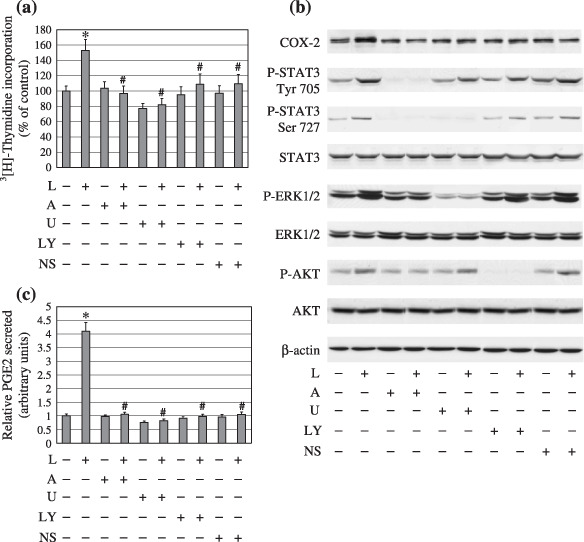
Leptin‐induced proliferation and functionally active cyclooxygenase (COX)‐2 of endometrial cancer cells were abolished in the presence of signal transducers and activators of transcription 3 (STAT3), extracellular signal‐related kinase (ERK1/2), AKT, and COX‐2 inhibitors. Six lines of serum‐starved endometrial cancer cells were pretreated with various inhibitors for 1 h followed by combining with/without stimulation of a 100 ng/mL dose of leptin for 24 h. (a) DNA syntheses. (b) Protein expressions of COX‐2, total and phosphorylated forms of STAT3 and ERK1/2, as well as AKT, and (c) prostaglandin E2 (PGE2) production were determined by [3H] thymidine incorporation assay, western blot analysis and enzyme immunoassay, respectively. Equal loading and transfer were shown by repeat probing with β‐actin. *P < 0.01 compared with untreated cells and #P < 0.01 compared with leptin treatment.
Discussion
In the present study, stimulatory effect of leptin was shown on various lines of human endometrial cancer cells. Leptin treatment activated the JAK2/STAT3, MAPK/ERK, and PI3K/AKT pathways, and importantly induced downstream COX‐2 expression as well as increased PGE2 production. Our findings therefore provide a molecular mechanism–based insight on numerous epidemiological studies that have indicated that elevated serum leptin levels are associated with obesity which may promote endometrial cancer development. Namely, our results might explain why weight gain, which is usually accompanied by higher serum leptin concentrations, has been found to be associated with increased endometrial cancer risk.( 42 , 43 , 44 , 45 )
Aberrant plasma leptin expression levels are noticed in patients with endometrial cancer.( 12 , 42 , 46 ) The biological actions of leptin on target tissues are carried out through interaction with its specific receptors, Ob‐R. However, the roles of the leptin receptor in human cancers are still unclear, although it has been known that leptin acts mostly through the long isoform Ob‐Rb. As for Ob‐Ra, Ob‐Rb is also reported to be present in various peripheral organs, such as in the lung, kidney, liver, intestine, and insulin‐secreting pancreatic B cells, and is expressed in the hypothalamus.( 47 ) Previous reports showed that both Ob‐Rb and Ob‐Ra mRNAs and proteins but not leptin itself were expressed in endometrium.( 48 ) In this study, Ob‐Ra protein has been detected to be more abundant than Ob‐Rb in six endometrial cancer cell lines which agreed well with the observation( 12 ) that an increase of the relative concentrations of Ob‐Ra to Ob‐Rb has been found in human endometrial cancer tissues. In addition, our data demonstrated a positive correlation between the growth‐stimulatory effect of leptin and Ob‐R expression in endometrial cancer cells. With RT‐PCR and immunoblotting, Yuan et al.( 12 ) showed that Ob‐Ra, not Ob‐Rb, is the major leptin receptor isoform in normal endometrial tissue, and its expression decreases significantly in endometrial cancer tissues, indicating that the action of leptin in the endometrial carcinogenesis may be mediated by Ob‐Ra, while Ob‐Rb may be involved in the promoting effect of leptin on endometrial cancer cells. In another study( 49 ) on 45 primary human endometrial‐type endometrial carcinomas, Ob‐R immunostaining was detected as present in almost 60% of well‐ and moderately differentiated tumors compared to only 17% of poorly differentiated neoplasms, indicating that Ob‐R expression is associated with histological grading. In agreement with these studies on human endometrial cancer tissues,( 12 , 49 ) we showed that the expressions of two main isoforms of the leptin receptor were inversely correlated with histoprognostic grading, suggesting that endometrial cancer responds to leptin stimulation via an autocrine pathway; high leptin receptors are biological markers of a more differentiated phenotype.
Leptin is a cytokine mainly produced by adipose tissue. It has been reported to stimulate the proliferation of various cancer cells.( 50 ) Hyperleptinemia is a common feature of obese women who have a higher risk of endometrial cancer than women with normal weights, and epidemiologic studies have suggested a correlation between obesity and endometrial carcinoma. Therefore, understanding of the molecular mechanism involved in leptin signaling transduction is important in endometrial cancer prevention and treatment. We showed in this study that the JAK2/STAT3, MAPK/ERK, PI3K/AKT, and COX‐2 signaling pathways are implicated in the cell proliferative effect of leptin. We also emphasized a new role for leptin, linking COX‐2, PGE2, and obesity. This finding has potential clinical implications for endometrial carcinogenesis and management of obesity. Most recently, Ogunwobi et al.( 20 ) found that leptin stimulates proliferation and inhibits apoptosis in Barrett's esophageal adenocarcinoma cells by COX‐2‐dependent, PGE2‐mediated transaction of the epidermal growth factor receptor (EGFR) and c‐Jun NH2‐terminal kinase activation. However, there has been relatively few data to date that elucidate the effect of leptin and its receptor on human endometrial cancer, and no further studies concerning leptin and COX‐2 expression and function. Leptin has been reported to increase PGF2α but inhibit PGE2 production( 51 ) in the rabbit oviduct, while leptin has been shown to increase COX‐2 expression and PGE2 release in murine macrophage J774A.1 cells.( 52 ) In this study, we showed that leptin stimulates COX‐2 activation dose‐ and time‐dependently which derived from leptin‐mediated COX‐2 mRNA. Although COX‐2 is a downstream signal of multiple cell survival pathways, to our knowledge this is the first study of the involvement of COX‐2 on the effect of leptin on endometrial caner cells. In addition, our results are consistent with the data of other authors( 19 ) who have reported that leptin‐induced activation of ERK and AKT was JAK2‐dependent in Barrett's esophageal adenocarcinoma cells. Importantly, we found the potential interaction between STAT3 and ERK, which therefore provides evidence that ERK‐mediated phosphorylation of Ser727 is required for full stimulation of STAT3 by leptin.( 53 )
In this study, the selective COX‐2 inhibitor NS398 alone did not down‐regulate COX‐2 expression and PGE2 production of human endometrial cancer cells which is consistent with our previous study,( 25 ) whereas NS398 could block leptin‐induced COX‐2 expression and PGE2, indicating that NS398, similar to other pharmacological inhibitors, can abrogate leptin‐stimulated COX‐2 activity. Previous studies( 16 , 20 , 54 ) on various human cancer cells have demonstrated that activated EGFR results in a marked induction of COX‐2 protein and PGE2 release; and leptin induces delayed PGE2‐mediated activation of EGFR which downstream of COX‐2 is essential to the leptin effects. Moreover, transactivation of EGFR is involved in leptin‐induced activation of JAK2 and ERK1/2. Thus, we hypothesize that NS398 abolished leptin‐induced COX‐2 overexpression and PGE2 production through PGE2‐mediated downstream EGFR activation in the ‘feedback loop’, on which further study is needed in the future.
Taken together, our data have demonstrated that the leptin receptor, both forms of Ob‐Rb and Ob‐Ra, are inversely associated with histological grading in various differentiated endometrial cancer cells. The growth‐stimulatory effect of leptin on endometrial cancer cells was strongly correlated with Ob‐R expression and mediated via complex multiple‐cell survival pathways. We have, for the first time, shown that the increased functional COX‐2 by leptin in endometrial cancer cells is an important downstream target and therefore blocking its action could be a rational therapeutic strategy.
Acknowledgments
We thank Dr Yu‐Yan Zhu for technical advice and her help in the preparation of this manuscript. We are grateful to Drs Li‐Yuan Chu and Sheng‐Fu Hong for the helpful discussion and proofreading the manuscript.
References
- 1. Bray F, Dos Santos Silva I, Moller H, Weiderpass E. Endometrial cancer incidence trends in Europe: underlying determinants and prospects for prevention. Cancer Epidemiol Biomarkers Prev 2005; 14: 1132–42. [DOI] [PubMed] [Google Scholar]
- 2. Modesitt SC, Van Nagell JR Jr. The impact of obesity on the incidence and treatment of gynecologic cancers: a review. Obstet Gynecol Surv 2005; 60: 683–92. [DOI] [PubMed] [Google Scholar]
- 3. Choi JH, Park SH, Leung PC, Choi KC. Expression of leptin receptors and potential effects of leptin on the cell growth and activation of mitogen‐activated protein kinases in ovarian cancer cells. J Clin Endocrinol Metab 2005; 90: 207–10. [DOI] [PubMed] [Google Scholar]
- 4. Onuma M, Bub JD, Rummel TL, Iwamoto Y. Prostate cancer cell–adipocyte interaction: leptin mediates androgen‐independent prostate cancer cell proliferation through c‐Jun NH2‐terminal kinase. J Biol Chem 2003; 278: 42660–7. [DOI] [PubMed] [Google Scholar]
- 5. Rouet‐Benzineb P, Aparicio T, Guilmeau S et al . Leptin counteracts sodium butyrate‐induced apoptosis in human colon cancer HT‐29 cells via NF‐kappaB signaling. J Biol Chem 2004; 279: 16495–502. [DOI] [PubMed] [Google Scholar]
- 6. Yin N, Wang D, Zhang H et al . Molecular mechanisms involved in the growth stimulation of breast cancer cells by leptin. Cancer Res 2004; 64: 5870–5. [DOI] [PubMed] [Google Scholar]
- 7. White DW, Tartaglia LA. Leptin and OB‐R: body weight regulation by a cytokine receptor. Cytokine Growth Factor Rev 1996; 7: 303–9. [DOI] [PubMed] [Google Scholar]
- 8. Tartaglia LA, Dembski M, Weng X et al . Identification and expression cloning of a leptin receptor, OB‐R. Cell 1995; 83: 1263–71. [DOI] [PubMed] [Google Scholar]
- 9. Fei H, Okano HJ, Li C et al . Anatomic localization of alternatively spliced leptin receptors (Ob‐R) in mouse brain and other tissues. Proc Natl Acad Sci USA 1997; 94: 7001–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bjorbaek C, Uotani S, Da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem 1997; 272: 32686–95. [DOI] [PubMed] [Google Scholar]
- 11. Yamashita T, Murakami T, Otani S, Kuwajima M, Shima K. Leptin receptor signal transduction. OBRa and OBRb of fa type. Biochem Biophys Res Commun 1998; 246: 752–9. [DOI] [PubMed] [Google Scholar]
- 12. Yuan SS, Tsai KB, Chung YF et al . Aberrant expression and possible involvement of the leptin receptor in endometrial cancer. Gynecol Oncol 2004; 92: 769–75. [DOI] [PubMed] [Google Scholar]
- 13. Yuan SS, Chung YF, Chen HW et al . Aberrant expression and possible involvement of the leptin receptor in bladder cancer. Urology 2004; 63: 408–13. [DOI] [PubMed] [Google Scholar]
- 14. Bado A, Levasseur S, Attoub S et al . The stomach is a source of leptin. Nature 1998; 394: 790–3. [DOI] [PubMed] [Google Scholar]
- 15. Caprio M, Fabbrini E, Isidori AM, Aversa A, Fabbri A. Leptin in reproduction. Trends Endocrinol Metab 2001; 12: 65–72. [DOI] [PubMed] [Google Scholar]
- 16. Shida D, Kitayama J, Mori K, Watanabe T, Nagawa H. Transactivation of epidermal growth factor receptor is involved in leptin‐induced activation of janus‐activated kinase 2 and extracellular signal‐regulated kinase 1/2 in human gastric cancer cells. Cancer Res 2005; 65: 9159–63. [DOI] [PubMed] [Google Scholar]
- 17. Hu X, Juneja SC, Maihle NJ, Cleary MP. Leptin – a growth factor in normal and malignant breast cells and for normal mammary gland development. J Natl Cancer Inst 2002; 94: 1704–11. [DOI] [PubMed] [Google Scholar]
- 18. Pai R, Lin C, Tran T, Tarnawski A. Leptin activates STAT and ERK2 pathways and induces gastric cancer cell proliferation. Biochem Biophys Res Commun 2005; 331: 984–92. [DOI] [PubMed] [Google Scholar]
- 19. Ogunwobi OO, Beales IL. The anti‐apoptotic and growth stimulatory actions of leptin in human colon cancer cells involves activation of JNK mitogen activated protein kinase, JAK2 and PI3 kinase/Akt. Int J Colorectal Dis 2007; 22: 401–9. [DOI] [PubMed] [Google Scholar]
- 20. Ogunwobi O, Mutungi G, Beales IL. Leptin stimulates proliferation and inhibits apoptosis in Barrett's esophageal adenocarcinoma cells by cyclooxygenase‐2‐dependent, prostaglandin‐E2‐mediated transactivation of the epidermal growth factor receptor and c‐Jun NH2‐terminal kinase activation. Endocrinology 2006; 147: 4505–16. [DOI] [PubMed] [Google Scholar]
- 21. Liu CH, Chang SH, Narko K et al . Overexpression of cyclooxygenase‐2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem 2001; 276: 18563–9. [DOI] [PubMed] [Google Scholar]
- 22. Tsujii M, Kawano S, DuBois RN. Cyclooxygenase‐2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci U S A 1997; 94: 3336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998; 93: 705–16. [DOI] [PubMed] [Google Scholar]
- 24. Gao J, Niwa K, Takemura M et al . Significant anti‐proliferation of human endometrial cancer cells by combined treatment with a selective COX‐2 inhibitor NS398 and specific MEK inhibitor U0126. Int J Oncol 2005; 26: 737–44. [PubMed] [Google Scholar]
- 25. Gao J, Niwa K, Sun W et al . Non‐steroidal anti‐inflammatory drugs inhibit cellular proliferation and upregulate cyclooxygenase‐2 protein expression in endometrial cancer cells. Cancer Sci 2004; 95: 901–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li M, Wu X, Xu XC. Induction of apoptosis in colon cancer cells by cyclooxygenase‐2 inhibitor NS398 through a cytochrome c‐dependent pathway. Clin Cancer Res 2001; 7: 1010–16. [PubMed] [Google Scholar]
- 27. Minter HA, Eveson JW, Huntley S, Elder DJ, Hague A. The cyclooxygenase 2‐selective inhibitor NS398 inhibits proliferation of oral carcinoma cell lines by mechanisms dependent and independent of reduced prostaglandin E2 synthesis. Clin Cancer Res 2003; 9: 1885–97. [PubMed] [Google Scholar]
- 28. Milella M, Metro G, Gelibter A, Pino SM, Cognetti F, Fabi A. COX‐2 targeting in cancer: a new beginning? Ann Oncol 2008; 19: 1209–10. [DOI] [PubMed] [Google Scholar]
- 29. Sarkar FH, Adsule S, Li Y, Padhye S. Back to the future: COX‐2 inhibitors for chemoprevention and cancer therapy. Mini Rev Med Chem 2007; 7: 599–608. [DOI] [PubMed] [Google Scholar]
- 30. Sinicrope FA. Targeting cyclooxygenase‐2 for prevention and therapy of colorectal cancer. Mol Carcinog 2006; 45: 447–54. [DOI] [PubMed] [Google Scholar]
- 31. Zha S, Yegnasubramanian V, Nelson WG, Isaacs WB, De Marzo AM. Cyclooxygenases in cancer: progress and perspective. Cancer Lett 2004; 215: 1–20. [DOI] [PubMed] [Google Scholar]
- 32. Adderley SR, Fitzgerald DJ. Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2‐mediated induction of cyclooxygenase‐2. J Biol Chem 1999; 274: 5038–46. [DOI] [PubMed] [Google Scholar]
- 33. Matsuura H, Sakaue M, Subbaramaiah K et al . Regulation of cyclooxygenase‐2 by interferon gamma and transforming growth factor alpha in normal human epidermal keratinocytes and squamous carcinoma cells. Role of mitogen‐activated protein kinases. J Biol Chem 1999; 274: 29138–48. [DOI] [PubMed] [Google Scholar]
- 34. Rodriguez‐Barbero A, Dorado F, Velasco S, Pandiella A, Banas B, Lopez‐Novoa JM. TGF‐beta1 induces COX‐2 expression and PGE2 synthesis through MAPK and PI3K pathways in human mesangial cells. Kidney Int 2006; 70: 901–9. [DOI] [PubMed] [Google Scholar]
- 35. Sharma D, Saxena NK, Vertino PM, Anania FA. Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal‐transduction pathways. Endocr Relat Cancer 2006; 13: 629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nishida M, Kasahara K, Kaneko M, Iwasaki H. Establishment of a new endometrial adenocarcinoma cell line, Ishikawa cells, containing estrogen and progesterone receptors. Acta Obstet Gynaecol Jpn 1985; 37: 1103–11. [PubMed] [Google Scholar]
- 37. Clarke CL, Feil PD, Satyaswaroop PG. Progesterone receptor regulation by 17 beta‐estradiol in human endometrial carcinoma grown in nude mice. Endocrinology 1987; 121: 1642–8. [DOI] [PubMed] [Google Scholar]
- 38. Kuramoto H. Studies of the growth and cytogenetic properties of human endometrial adenocarcinoma in culture and its development into an established line. Acta Obstet Gynaecol Jpn 1972; 19: 47–58. [PubMed] [Google Scholar]
- 39. Kuramoto H, Tamura S, Notake Y. Establishment of a cell line of human endometrial adenocarcinoma in vitro. Am J Obstet Gynecol 1972; 114: 1012–19. [DOI] [PubMed] [Google Scholar]
- 40. Way DL, Grosso DS, Davis JR, Surwit EA, Christian CD. Characterization of a new human endometrial carcinoma (RL95‐2) established in tissue culture. In Vitro 1983; 19: 147–58. [DOI] [PubMed] [Google Scholar]
- 41. Dawe CJ, Banfield WG, Morgan WD, Slatick MS, Curth HO. Growth in continuous culture, and in hamsters, of cells from a neoplasm associated with acanthosis nigricans. J Natl Cancer Inst 1964; 33: 441–56. [PubMed] [Google Scholar]
- 42. Petridou E, Belechri M, Dessypris N et al . Leptin and body mass index in relation to endometrial cancer risk. Ann Nutr Metab 2002; 46: 147–51. [DOI] [PubMed] [Google Scholar]
- 43. Rose PG. Endometrial carcinoma. N Engl J Med 1996; 335: 640–9. [DOI] [PubMed] [Google Scholar]
- 44. Trentham‐Dietz A, Nichols HB, Hampton JM, Newcomb PA. Weight change and risk of endometrial cancer. Int J Epidemiol 2006; 35: 151–8. [DOI] [PubMed] [Google Scholar]
- 45. Xu WH, Xiang YB, Zheng W et al . Weight history and risk of endometrial cancer among Chinese women. Int J Epidemiol 2006; 35: 159–66. [DOI] [PubMed] [Google Scholar]
- 46. Tessitore L, Vizio B, Pesola D et al . Adipocyte expression and circulating levels of leptin increase in both gynaecological and breast cancer patients. Int J Oncol 2004; 24: 1529–35. [PubMed] [Google Scholar]
- 47. Attoub S, Noe V, Pirola L et al . Leptin promotes invasiveness of kidney and colonic epithelial cells via phosphoinositide 3‐kinase‐, rho‐, and rac‐dependent signaling pathways. Faseb J 2000; 14: 2329–38. [DOI] [PubMed] [Google Scholar]
- 48. Kitawaki J, Koshiba H, Ishihara H, Kusuki I, Tsukamoto K, Honjo H. Expression of leptin receptor in human endometrium and fluctuation during the menstrual cycle. J Clin Endocrinol Metab 2000; 85: 1946–50. [DOI] [PubMed] [Google Scholar]
- 49. Bogusiewicz M, Semczuk A, Gogacz M, Skomra D, Jakowicki JA, Rechberger T. Lack of correlation between leptin receptor expression and PI3‐K/Akt signaling pathway proteins immunostaining in endometrioid‐type endometrial carcinomas. Cancer Lett 2006; 238: 61–8. [DOI] [PubMed] [Google Scholar]
- 50. Housa D, Housova J, Vernerova Z, Haluzik M. Adipocytokines and cancer. Physiol Res 2006; 55: 233–44. [DOI] [PubMed] [Google Scholar]
- 51. Zerani M, Boiti C, Dall’Aglio C et al . Leptin receptor expression and in vitro leptin actions on prostaglandin release and nitric oxide synthase activity in the rabbit oviduct. J Endocrinol 2005; 185: 319–25. [DOI] [PubMed] [Google Scholar]
- 52. Raso GM, Pacilio M, Esposito E, Coppola A, Di Carlo R, Meli R. Leptin potentiates IFN‐gamma‐induced expression of nitric oxide synthase and cyclo‐oxygenase‐2 in murine macrophage J774A.1. Br J Pharmacol 2002; 137: 799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. O’Rourke L, Shepherd PR. Biphasic regulation of extracellular‐signal‐regulated protein kinase by leptin in macrophages: role in regulating STAT3 Ser727 phosphorylation and DNA binding. Biochem J 2002; 364: 875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Coffey RJ, Hawkey CJ, Damstrup L et al . Epidermal growth factor receptor activation induces nuclear targeting of cyclooxygenase‐2, basolateral release of prostaglandins, and mitogenesis in polarizing colon cancer cells. Proc Natl Acad Sci USA 1997; 94: 657–62. [DOI] [PMC free article] [PubMed] [Google Scholar]