

Abstract
The functions of platelets and fibrinogen in protecting tumor cells from natural killer cytotoxicity have been discussed for more than 20 years. However, their exact roles and relationships in the process are still not clear. In this study, we show that tumor cells prefer to adhere to fibrinogen than to platelets, and fibrinogen can enhance the adhesion of tumor cells to platelets. β3 integrin plays an important role in the adhesion of B16F10 to platelets enhanced by fibrinogen. In the presence of thrombin, fibrinogen forms dense fibrin(ogen) layers around tumor cells. Tumor cells can induce platelets to aggregate and form thrombin. Platelets, as well as thrombin, can help fibrinogen protect tumor cells from lethal contact with natural killer cells and natural killer cytotoxicity. Hirudin, a specific inhibitor of thrombin, can reverse the effect of platelets on fibrinogen in blocking natural killer cytotoxicity. Our results suggest that fibrinogen helps platelets to adhere to tumor cells, and platelets in turn promote more fibrinogen to aggregate around tumor cells by forming thrombin. They facilitate each other in protecting tumor cells from natural killer cytotoxicity. (Cancer Sci 2009; 100: 859–865)
Hematogenous metastasis is a highly regulated and dynamic process, in which cancerous cells separate from the primary tumor and migrate into the bloodstream, and the surviving tumor cells eventually produce colonies at remote sites after circulation.( 1 ) Most tumor cells are killed during circulation in blood vessels( 2 , 3 ) and natural killer (NK) cells are largely responsible for the killing. The antimetastasis function of NK cells has been shown in different models of experimental as well as spontaneous metastasis.( 4 , 5 ) Although the exact mechanisms by which NK cells recognize and destroy tumor cells are still in unclear, it is apparent that direct contact between NK cells and tumor cells is required.( 6 ) Consequently, it is important to understand how tumor cells avoid lethal contact with NK cells.
Several lines of experiments have shown that some coagulation factors can facilitate hematogenous dissemination of tumor cells.( 7 , 8 ) These factors help the stroma formation, angiogenesis, growth, or metastasis of tumors.( 9 , 10 , 11 ) Among all the effects of coagulation factors on tumor progress, the role of platelets and fibrinogen in protecting tumor cells has been investigated for more than 20 years. Some reports claimed that platelets could bind to tumor cells tightly and aggregate around tumor cells to form physical barriers, which could block the effector–target interaction and inhibit NK cytotoxicity.( 12 , 13 ) Others asserted that fibrinogen aggregated around tumor cells to form dense fibrin layers that could block the effector–target interaction and impede NK cytotoxicity.( 14 , 15 , 16 ) Recently, Palumbo et al. showed that fibrinogen and platelets are both necessary in protecting tumor cells.( 17 ) However, the exact roles and relationships of platelets and fibrinogen in the process remain elusive. Here we show that tumor cells prefer to adhere to fibrinogen than to platelets, and fibrinogen enhances the interaction between tumor cells and platelets. Tumor cells can induce platelets to form thrombin. In the presence of thrombin, fibrinogen form dense fibrin(ogen) layers around tumor cells, which can help tumor cells escape NK cytotoxicity.
Materials and Methods
Cells and mice. Mice melanoma cell line B16F10 and lymphoma cell line YAC‐1 were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Iscove's modified Dulbecco's medium IMDM (Invitrogen Grand Island, NY, USA) supplemented with 10% heat‐inactivated fetal bovine serum at 37°C in the presence of 5% CO2. B16F10 cells were harvested with 0.2% ethylenediaminetetraacetic acid (EDTA). Subsequently, the tumor cells were washed twice with phosphate‐buffered saline (PBS), then resuspended in serum‐free IMDM culture medium. Specific pathogen‐free C57BL/6 J mice (male, 6–8 weeks old) were obtained from the animal center of Jilin University (Changchun, China). The study protocols were approved by the Animal Care and Use Committee of the Institute of Genetics and Cytology, School of Life Science, Northeast Normal University, Changchun, P.R. China.
Preparation of platelets. Blood of the mice injected with prostaglandin E1 (PGE‐1) was collected retroorbitally, then immediately mixed with sodium citrate anticoagulant (0.38% w/v). Platelet‐rich plasma was prepared by centrifugation of whole blood at 300 g for 10 min, and platelet‐poor plasma (PPP) and platelets were separated by centrifugation of platelet‐rich plasma at 1300 g for 10 min. Erythrocytes were lysed for 2 min with lysis buffer (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM EDTA) if a trace of red color was found in the pellet. The platelets were adjusted to a density of 2 × 108/mL by adding HEPES–Tyrode's buffer (134 mM NaCl, 12 mM NaHCO3, 2.9 mM KCl, 0.34 mM NaH2PO4, 5 mM HEPES, 5 mM glucose, 1% bovine serum albumin [BSA], pH 7.4) for further use. To avoid any unwanted activation of platelets, all of the buffers used contained 2 µM PGE‐1.( 18 ) The platelets can be kept in a stable unactivated state for more than 5 h. PGE‐1 was removed by washing twice with Tyrode's buffer before use. Compared with the fresh platelets extracted by the method of traditional two‐step centrifugation without PGE‐1, the aggregation and activation of platelets prepared in our experiments were not change significantly after thrombin stimulation.
Platelet aggregation assay. Tumor cell‐induced platelet aggregation was monitored by a four‐channel light aggregometer (LBY‐NJ; Precil Group, Beijing, China). Platelet samples (2.5 × 108/mL in Tyrode's buffer containing 1 mM CaCl2, 300 µL) with a trace of PPP (1/400) were prewarmed for 5 min at 37°C before the addition of aggregating factors. Thrombin (at a final concentration of 1 U/mL) was used as the control agonist for all of the assays. Tumor cell‐induced platelet aggregation was initiated by the addition of tumor cells at a volume ratio of 1/100 (at final concentrations of 10–106/mL), and monitored for 10 min.
Thrombin chromogenic assay. Thrombin in the culture medium was evaluated by S‐2160 (Bz‐Phe‐Val‐Arg‐pNA, Testozyme; Chromogenix, Umea, Sweden) using a chromogenic thrombin assay. Fifty microliters of tumor cells at different concentrations in IMDM was cultured with 50 µL platelets (in PBS added with 1 mM Ca2+) or PBS (added with 1 mM Ca2+) and PPP (1/400) in 96‐well culture plates. The color reaction was allowed to develop for 10 min after addition of 50 µL of 0.5 mM S‐2160 (at 1 mM Tris, pH 8.1), then stopped by adding 20 µL glacial acetic acid. Absorbance of the released color product was measured with an enzyme‐linked immunosorbent assay reader at a wavelength of 405 nm. The results were obtained in triplicate, and the amount of thrombin generated was reflected by the absorbance.
Laminar flow adhesion assay. Platelet monolayers were prepared as described previously.( 19 ) For fibrinogen layers, 100 µL fibrinogen (1 mg/mL) was dropped onto the surfaces of circular glass slides, and the coated slides were incubated at 4°C overnight. Nonspecific binding was blocked with 0.1% BSA at 37°C for 10 min.
Tumor cell adhesion to platelets or fibrinogen under flow conditions was measured by a flow chamber assay as reported previously.( 20 ) Tumor cells were washed, resuspended and adjusted to 1 × 106 cells/mL in serum‐free medium containing 0.1% BSA. Platelet (or fibrinogen)‐coated circular glass slides were assembled to a flow chamber (GlycoTech, Rockville, MD, USA) and mounted on the stage of an inverted microscope (Olympus Optical, Tokyo, Japan) equipped with a camera (Panasonic, Yokohama, Japan) connected to a personal computer by a television monitor card. Surface‐adhered platelets were incubated with 100 µL thrombin (1 U/mL in PBS/0.1% BSA) at 37°C for 1 min. After washing the platelet layer with PBS/0.1% BSA for approximately 2 min, tumor cells were perfused through the chamber for 3 min at appropriate flow rates to obtain wall shear stresses of 0.3–1.2 dyn/cm2 at 22°C using a syringe pump (Cole‐Parmer Instrument Co., Vernon Hills, IL, USA), thereby mimicking the flow mechanical environment of microcirculation in postcapillary venules. Interactions between tumor cells and surface‐adherent platelets (or fibrinogen) were visualized in real time by phase‐contrast video microscopy. The numbers of bound cells were quantified from digital recordings of 10 randomly selected fields of the views obtained.
Flow cytometric assay. Tumor cells were washed twice and resuspended in PBS (containing 1 mM Ca2+). For the platelets or fibrinogen binding assay, platelets stained with PKH26 (red; Sigma, St. Louis, MO, USA; #PKH26PCL), and fibrinogen conjugated with Alexa Fluor488 (green; Molecular Probes, Invitrogen, OR, USA; #F13191) were used. Each aliquot (0.1 mL, 5 × 106 cells/mL) of cells was incubated with the same volume of platelets or fibrinogen at 37°C for 20 min. As the fluorescent molecules used were different, we set controls (containing 2 mM EDTA) for each case. After washing three times with PBS, the aliquots were resuspended in 0.5 mL PBS for immediate flow cytometric analysis (Coulter Epics XL; Beckman‐Coulter, FL, USA).
NK cytotoxicity assay. Cytotoxic activity of NK cells was tested by a 4‐h calcein acetyoxymethyl (AM)‐release assay.( 21 , 22 ) Briefly, NK cells (DX5 positive cells are more than 90%) were separated from splenic cells with a specific mice NK cell separation buffer (1.076 g/mL; Hao Yang Biological Manufacture Co, Tianjin, China; #LTS1068‐2). The NK cells cultured with target tumor cells were prelabeled with calcein AM (Molecular Probes; #32805 W), at an effector : target ratio of 50:1, in triplicate at 37°C for 4 h. Fluorescence released into the IMDM was used to calculate the cytotoxicity in percentage = (A–B)/(C – B) × 100 formula, in which A is calcein released in the test well, B is spontaneous release from target cells without addition of effector cells, and C is total release. For platelet or fibrinogen blocking experiments, traces of PPP (at a final concentration of 1/200) and fibrinogen (2 mg/mL) or platelets (2 × 109/mL) were added into the cytotoxicity assay system.
Statistics. Data were expressed as the mean ± standard deviation. Statistical significance of differences between means was determined by the analysis of variance. If means were shown to be significantly different, multiple comparisons by pairs were carried out using Tukey's test. Probability values of P < 0.05 (*), P < 0.01 (**) or P < 0.001 (***) were selected to be statistically significant.
Results
Tumor cells prefer to adhere to fibrinogen than to platelets. A great deal of work has reported that both platelets and fibrinogen can bind to tumor cells and form physical barriers between tumor cells and NK cells, which block the effector–target interaction and inhibit NK cytotoxicity.( 12 , 14 ) So it is necessary to compare the adhesion of fibrinogen and platelets to tumor cells before determining whether either or both of them are responsible for protecting tumor cells from being killed by NK cells. To this end, B16F10 and YAC‐1 cells were incubated with PKH26‐labeled platelets (activated by 1 U/mL thrombin for 1 min before mixing with tumor cells) in physiological density, or Alexa Fluor488‐conjugated fibrinogen, and the percentages of tumor cells adhered to platelets or fibrinogen were determined by flow cytometry. As shown in Figure 1(A), the percentage of B16F10 cells adhered to platelets was 95.2% before washing, but it decreased to 3.2% after washing. The percentages of B16F10 cells adhered to fibrinogen were over 99% both before and after washing, but the fluorescent density was decreased dramatically after washing. Statistical results of control and washed samples are shown in Figure 1(B). Compared with the positive cells (approximately 1%) in each control, only 1.1% of YAC‐1 cells were adhered to platelets, whereas 99.1% of YAC‐1 cells were adhered to fibrinogen. Similarly, only 2.26% of B16F10 cells were adhered to platelets, and 98.6% of B16F10 cells were adhered to fibrinogen.
Figure 1.
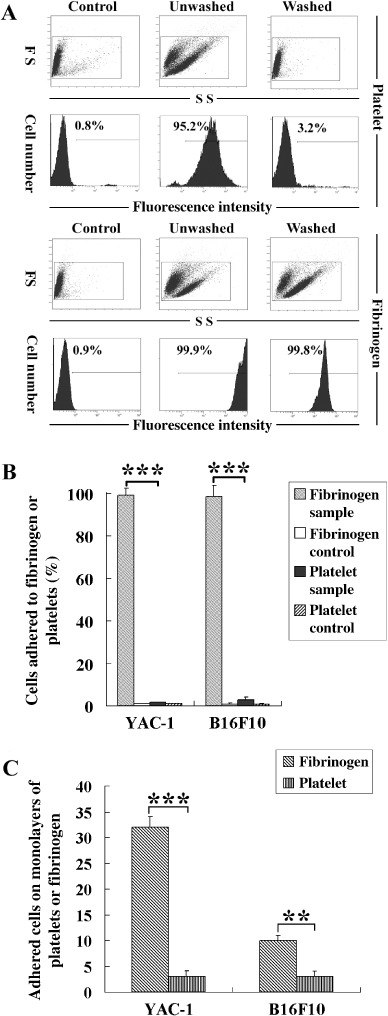
Tumor cells prefer to adhere to fibrinogen than to platelets. (A) One hundred microliters of 2 mg/mL fibrinogen (Alexa Fluor488 conjugated, green) or 2 × 108/mL platelets (PKH26 dyed, red) were incubated with 100 µL of 2 × 105 YAC‐1 or B16F10 cells in phosphate‐buffered saline (PBS) containing Ca2+ (sample) or ethylenediaminetetraacetic acid (control) for 30 min. Tumor cells adhered to fibrinogen or platelets were counted by fluorescence‐activated cell sorting before and after washing three times with PBS. FS, Forward Scatter; SS, Side Scatter. (B) Statistical results (control and washed samples) from flow cytometry. (C) Fibrinogen or platelet monolayers on glass slides were washed with PBS/0.1% bovine serum albumin for 2 min, tumor cells (1 × 106/mL) were perfused over the layers at the shear stress of 1.2 dyn/cm2, and the number of tumor cells adhered to fibrinogen or platelets were counted in 10 randomly selected fields after 3 min. All of the experiments were repeated more than three times. **P < 0.01; ***P < 0.001.
To mimic flow conditions in the vessels, a flow chamber assay was carried out. Fibrinogen or platelets were coated on the surface of glass slides, and tumor cells were allowed to flow over the slides at the shear stress of 1.2 dyn/cm2. The adhesive ability was reflected by the number of tumor cells adhered to the surface of the slides. As shown in Figure 1(C), many more B16F10 and YAC‐1 cells adhered to fibrinogen than to platelets. These results indicate that tumor cells prefer to adhere to fibrinogen than to platelets and the adhesion between tumor cells and fibrinogen is stronger than that between tumor cells and platelets.
Fibrinogen enhances interaction between tumor cells and platelets. Previous studies showed that fibrinogen could enhance platelet adhesion to platelets and leukocyte adhesion to endothelial cells by acting as a molecular bridge.( 23 ) To test if fibrinogen could enhance the adhesion of platelets to tumor cells, tumor cells were incubated with 2.0 mg/mL fibrinogen for 30 min before being perfused over the platelet layers. As shown in Figure 2(A), the adhesion of YAC‐1 cells preincubated with fibrinogen on the platelet monolayers was dramatically increased, compared with the cells preincubated with PBS under different shear stresses. Similar results were obtained from the assay of B16F10 (Fig. 2B). These data indicate that fibrinogen enhances the adhesion between tumor cells and platelets.
Figure 2.
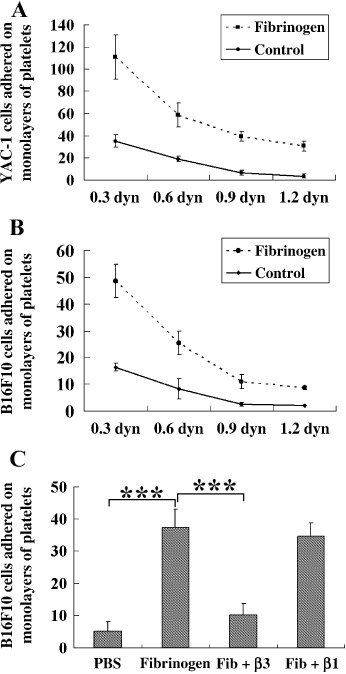
Fibrinogen enhances the adhesion between tumor cells and platelets. (A,B) Platelet monolayers on glass slides were washed with phosphate‐buffered saline (PBS)/0.1% bovine serum albumin for 2 min. YAC‐1 (A) and B16F10 (B) cells were preincubated with or without fibrinogen (2 mg/mL) for 30 min before being used, then the treated cells were perfused over the layers under a series of shear stress from 0.3 to 1.2 dyn/cm2. The adhered tumor cells on platelet monolayers were counted in 10 randomly selected fields after 3 min. (C) B16F10 cells were preincubated with the function blocking antibodies (10 µg/mL) against mice β3 or β1 integrin before being incubated with fibrinogen (Fib). After incubation, B16F10 cells were allowed to flow over the platelet monolayers under 0.3 dyn/cm2 shear stress, and the adhered cells were counted as above. All of the experiments were repeated more than three times. ***P < 0.001.
It is well known that fibrinogen is the physiological ligand of some integrins on B16F10 cells and platelets, and αIIbβ3 integrin plays a major role in the adhesion of platelets to fibrinogen.( 24 ) To determine which kind of integrin is involved in the adhesion of B16F10 cells to fibrinogen, the function blocking antimice integrin β3 (2C9.G2; Santa Cruz Biotechnology, Santa Cruz, CA, USA; #10806) or β1 (HMb1‐1; Biolegend, San Diego, CA, USA; #111634) antibody was added before incubation of B16F10 cells with fibrinogen. As shown in Figure 2(C), B16F10 cells pretreated with antiβ3 antibody greatly decreased their ability to adhere to platelet monolayers even in the presence of fibrinogen, whereas antiβ1 antibody did not affect the role of fibrinogen on adhesion. These results suggest that β3 integrin is mainly involved in the interactions between tumor cells and platelets, enhanced fibrinogen.
Fibrinogen forms dense layers around tumor cells when thrombin present. It has been reported that fibrin plays an important role in the protection of murine tumor cells from NK cytotoxicity.( 25 ) Fibrinogen is the natural substrate of thrombin, and can be converted to fibrin. So thrombin should be essential for the protective shields of fibrin(ogen). To test this hypothesis, B16F10 cells were incubated with fibrinogen (conjugated with Alexa Fluor488) at a concentration of 1 mg/mL in the wells containing thrombin (Fig. 3A(a,b)), or not (Fig. 3A(c,d)), and the interactions between B16F10 cells and fibrinogen were observed under fluorescence microscope. As shown in Figure 3(A), dense fibrin(ogen) layers (a thickness of more than 2 µm) around B16F10 cells were formed in the wells containing 1 U/mL thrombin, but only thin layers (a thickness of less than 0.5 µm) were formed on the cells without thrombin. We then counted the B16F10 cells coated with dense fibrin(ogen) layers. As shown in Figure 3(B), the dense fibrin(ogen) layers were formed on the surfaces of approximately 10% of B16F10 cells in the wells containing thrombin, but very few cells were coated with dense fibrin(ogen) layers in the wells without thrombin.
Figure 3.
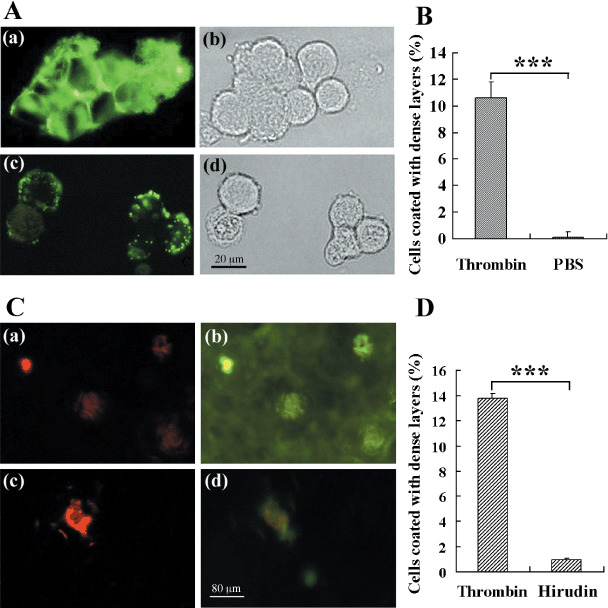
Thrombin aggregates fibrinogen around tumor cells. One hundred microliters of fibrinogen (2 mg/mL, conjugated with Alexa Fluor488, green) was mixed with 100 µL B16F10 cells (1 × 106/mL). After the addition of 1 U/mL thrombin (A(a,b)), or phosphate‐buffered saline (PBS) (A(c,d)), the mixture was incubated at 37°C for 30 min. After washing three times with PBS, the tumor cells were checked under a fluorescence microscope. (B) Thirty randomly selected photographs were counted for the tumor cells coated with dense fibrin(ogen) layers. (C) PKH26‐dyed B16F10 cells (red; 2 × 105/mouse) were injected into the mice after the injection of fibrinogen conjugated with Alexa Fluor488 (green; 1 mg/mouse), and the fibrin layers formed around tumor cells in the capillary were observed. PBS (a,b) or hirudin (c,d) (10 mg/kg) were injected with tumor cells. (D) B16F10 cells coated with fibrin layers were numbered from 30 randomly selected digital microscope photographs of the frozen sections of mouse lung. ***P < 0.001.
To further confirm the important role of thrombin in the process, we compared the formation of dense fibrin(ogen) layers around tumor cells in the capillary of mice injected with PBS or hirudin (10 mg/kg), a specific inhibitor of thrombin. PKH26‐dyed B16F10 cells (red; 2 × 105/mouse) were injected into the mice after the injection of fibrinogen conjugated with Alexa Fluor488 (green; 1 mg/mouse). The frozen sections of lung were observed under a fluorescence microscope 10 min after the injections. As shown in Figure 3(C), fibrinogen aggregated to form dense layers around tumor cells 10 min after they enter blood (Fig. 3C(a,b)), and hirudin injection made the layers obscure (Fig. 3C(c,d)). We then counted the B16F10 cells coated with dense fibrin(ogen) layers in vivo from 30 randomly selected photographs. As shown in Figure 3(D), the dense fibrin(ogen) layers were formed on the surfaces of approximately 14% of B16F10 cells in control mice, but only 1.8% were formed in the mice injected with hirudin. These results indicate that fibrinogen forms dense layers around tumor cells in a thrombin‐dependent manner.
Tumor cells can induce platelets to aggregate and form thrombin. As presented above, thrombin is essential for the formation of dense fibrin(ogen) layers around tumor cells. It is well known that platelets play a critical role in localizing and controlling the burst of thrombin generation that leads to fibrin clot formation during the coagulation process.( 26 ) It has been reported that platelets can be activated and aggregated by tumor cells.( 27 , 28 ) To test if B16F10 cells can induce platelets to aggregate and form thrombin, platelet aggregative and thrombin chromogenic assays were carried. In aggregative assays, tumor cells at different densities were added into the platelet samples. Results from the light aggregometer showed that B16F10 cells could induce platelets to aggregate in a concentration‐dependent manner (Fig. 4A). In thrombin chromogenic assays, platelets or PPP, or both of them, were added into the wells containing S‐2160 and different concentrations of B16F10 cells. Then the mixtures were incubated and checked by an enzyme‐linked immunosorbent assay reader. As shown in Figure 4(B), in the wells containing B16F10 cells, PPP and platelets, the density of thrombin formed was correlated with the quantity of tumor cells, whereas the density of thrombin changed neither in the wells containing B16F10 cells and platelets, nor in the wells containing B16F10 cells and PPP. The results indicate that tumor cells can induce platelets to form thrombin which might be responsible for aggregating more fibrin(ogen) around tumor cells.
Figure 4.
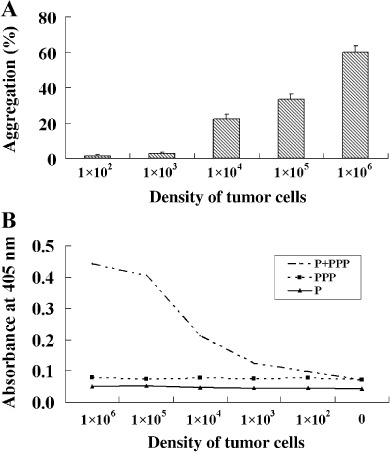
Tumor cells can induce platelets to aggregate and form thrombin. (A) Platelet (2.5 × 108/mL) aggregation induced by B16F10 cells at different densities was monitored by a light aggregometer. (B) B16F10 cells were checked by a chromogenic assay for their ability to induce platelets to form thrombin. The absorbance at 405 nm reflected the level of thrombin formed in the wells. P, wells containing platelets and tumor cells but without platelet‐poor plasma; PPP, wells containing tumor cells and PPP but without platelets; P+PPP, wells containing tumor cells, platelets and platelet‐poor plasma.
Fibrinogen blocks the effector–target cell interactions in a thrombin or platelet‐dependent manner. As NK cytotoxicity depends on effector–target cell adhesion and conjugate formation, interaction blocking can help tumor cells survive.( 6 ) To determine the inhibitory effects of fibrinogen or platelets on the interactions between tumor cells and NK cells, a static effector–target adhesion assay was carried out. B16F10 cells growing on the bottom surface of 96‐well culture plates were incubated with NK cells (calcein AM dyed) at 37°C for 1 h. Then the plates were checked for effector–target cell adhesion by a plate reader. As shown in Figure 5(A), in the absence of thrombin, the blocking effect of platelets was less than 5%, and the blocking effect of fibrinogen was approximately 20%. Platelets and fibrinogen could work together to block approximately 80% of adhesion, and hirudin could reverse the blocking effect. In the presence of thrombin (1 U/mL), platelets still could not block the adhesion, whereas fibrinogen could block the adhesion to the level of 25% (Fig. 5B). Similarly, when both fibrinogen and platelets were added, the adhesion could be blocked by up to 25%, and hirudin could still reverse the blocking effect. The results show that fibrinogen blocks the effector–target cell adhesion in a thrombin‐dependent manner. However, platelets can not block the adhesion, whether thrombin is present or not, implying that the major role of platelets in this process is to provide thrombin for fibrinogen.
Figure 5.
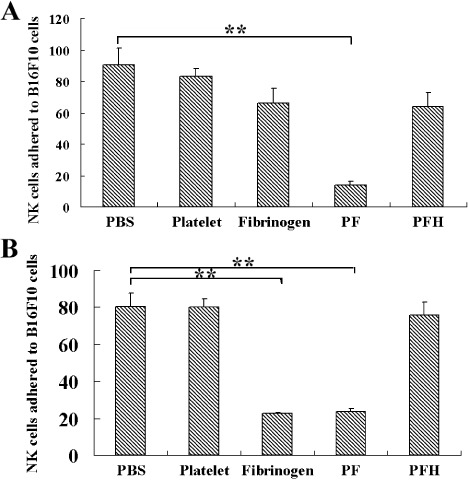
Fibrinogen blocks the effector–target cell interaction in a thrombin‐ or platelet‐dependent manner. 100 µL of B16F10 cells (2 × 105/mL) was seeded in each well of the 96‐well culture plate and cultured overnight. After that, 50 µL of platelets (2 × 108/mL), fibrinogen (4 mg/mL), and calcein acetyoxymethyl‐dyed natural killer (NK) cells (2 × 107/mL) were added to the wells, the culture plate was incubated at 37˚C for 1 h. After washing three times with phosphate‐buffered saline (PBS), the fluorescence at 515 nm was checked by a plate reader, which could reflect the quantity of NK cells adhered to B16F10 cells. Thrombin (1 U/mL) was present in (B) but not in (A). All of the wells contained a trace of platelet‐poor plasma (1/100). PBS, wells added with PBS for control; Platelets, wells containing platelets; Fibrinogen, wells containing fibrinogen; PF, wells containing platelets and fibrinogen; PFH, wells containing platelets and fibrinogen after hirudin treatment (0.1 mg/mL). **P < 0.01.
Thrombin or platelets are required for role of fibrinogen in blocking NK cytotoxicity. To examine the ability of platelets and fibrinogen in the protection of tumor cells, tumor cells preincubated with fibrinogen or platelets, or both, were added into the cytotoxic system. As shown in Figure 6, platelets could not inhibit NK cytotoxicity efficiently even in the presence of thrombin (Fig. 6A, YAC‐1). Fibrinogen could inhibit NK cytotoxicity in a concentration‐dependent manner when thrombin was present (Fig. 6B, YAC‐1). The lysis of tumor cells was efficiently impeded in the wells containing platelets and fibrinogen, whereas the addition of hirudin in these wells could reverse the blockage to NK cytotoxicity (Fig. 6C). Furthermore, similar to the reports of others,( 29 ) hirudin could block the pulmonary metastasis of B16F10 cells in mice significantly (Fig. 6D). These results indicate that fibrinogen can block NK cytotoxicity dramatically in a thrombin‐dependent manner, and platelets can facilitate fibrinogen blockage of NK cytotoxicity by providing thrombin.
Figure 6.
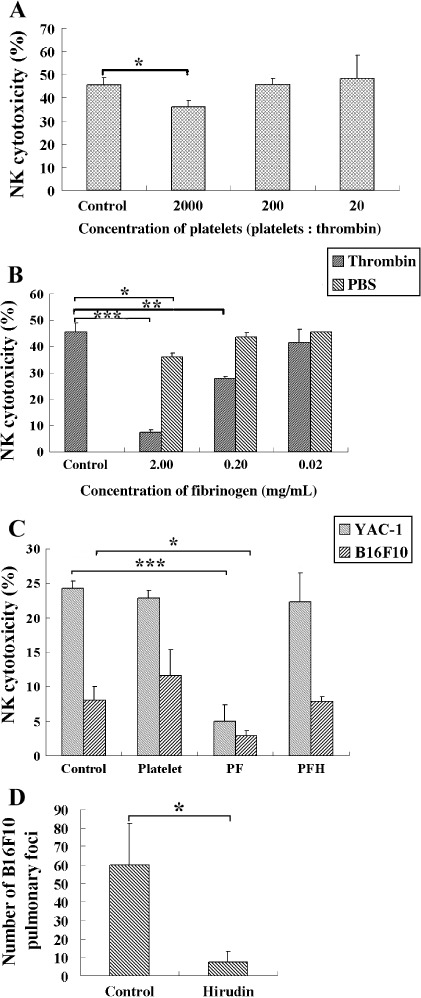
Thrombin or platelets are required for the role of fibrinogen in blocking natural killer (NK) cytotoxicity. Fifty microliters of calcein acetyoxymethyl‐dyed YAC‐1 cells (1 × 105/mL) was mixed with 50 µL NK cells (1 × 107/mL) for NK cytotoxic assay. To check the effect of platelets and fibrinogen on NK cytotoxicity, 50 µL of different concentrations of platelets (A) or fibrinogen (B) was added into the systems. (C) To determine the role of platelets on the blocking effect of fibrinogen on NK cytotoxicity, 50 µL platelets (2 × 108/µL) or fibrinogen (2 µg/µL) was added to wells containing 1 U/µL thrombin, or without thrombin (PF). All of the wells contained a trace of platelet‐poor plasma (1/100). PFH, wells containing fibrinogen and platelets after hirudin treatment (10 mg/kg). (D) Hirudin (10 mg/kg) or phosphate‐buffered saline (PBS) was injected with B16F10 cells (2 × 105/mouse) into the lateral tail vein. After 14 days the animals were killed and the lungs removed. The metastatic nodes on the surface of lung were numbered under an anatomical lens. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
A number of published reports have indicated that platelets and fibrinogen could protect tumor cells from NK cytotoxicity, and the basis of the protective function is their strong adhesion to tumor cells.( 12 , 13 , 14 , 15 , 16 ) Here we compare the adhesion of platelets and fibrinogen to tumor cells. Our flow cytometric results show that tumor cells prefer to adhere to fibrinogen rather than platelets (Fig. 1B). These results are further confirmed by a flow chamber assay that can mimic the flow conditions of tumor cells in the vessels (Fig. 1C). Compared with the reports of others, we have provided evidence that fibrinogen has more potential as the inhibitor of NK cytotoxicity to tumor cells.
A previous work reported that tumor cells arrested in pulmonary vasculature were associated with platelets and fibrin(ogen),( 30 ) however, our results from flow chamber assays show that the direct adhesion of tumor cells and platelets hardly occurs under high shear force (Fig. 1C). As fibrinogen could adhere strongly to both tumor cells and platelets,( 24 , 31 ) we wonder if fibrinogen functions as a molecular bridge to link tumor cells and platelets, similar to its roles in platelet–platelet or platelet–leukocyte adhesion.( 23 ) Our results show that the stronger adhesion between tumor cells and platelets occurs after tumor cells are incubated with fibrinogen (Fig. 2A,B), implying that fibrinogen can enhance the adhesion between tumor cells and platelets in vessels by connecting them. We also show that among the integrin ligands of fibrinogen expressed on the surface of B16F10 cells and platelets,( 32 , 33 )β3 integrin is more important than β1 integrin in mediating the interaction between B16F10 cells and platelets enhanced by fibrinogen (Fig. 2C).
In the presence of thrombin, fibrinogen can aggregate to form fibrin, reported to play an important role in protecting tumor cells from NK cytotoxicity.( 25 ) In this study, we found that fibrinogen forms a dense layer around tumor cells when thrombin is present (Fig. 3). The layers can dramatically block the formation of effector–target conjugate (Fig. 5) and NK cytotoxicity (Fig. 6) by approximately 80%. Although the protection of platelets to tumor cells has been reported previously( 12 ) we show here that platelets can block cytotoxicity by no more than 25% even in the presence of thrombin (Fig. 6B). Our results suggest that the dense fibrin(ogen) layers, but not platelet layers, play a main role in protecting tumor cells from NK cytotoxicity. However, fibrinogen can not work alone. Without thrombin, fibrinogen can not be converted to fibrin, and the layers of fibrinogen formed on tumor cells is very thin and incomplete (Fig. 3). Under these conditions, the inhibitory effects of the thin layers on the formation of effector–target conjugate and NK cytotoxicity are no more than 30% (5, 6). So the formation of thrombin around tumor cells is essential for the fate of tumor cells during the hematogenous phase.
It is well known that platelets play a major role in localizing and controlling the burst of thrombin generation that leads to fibrin clot formation( 26 ) and platelets can be activated by tumor cells.( 27 , 28 ) Here we show that tumor cells can induce platelets to aggregate and form thrombin in a density‐dependent manner (Fig. 4). We further show that platelets can help fibrinogen to block the effector–target interaction (Fig. 5) and NK cytotoxicity (Fig. 6) although they can not block the effector–target interaction (Fig. 5), nor protect tumor cells directly (Fig. 6). These data indicate that the exact role of platelets in protecting tumor cells is to form thrombin for fibrinogen.
In summary, this study explores the roles and relationships of platelets and fibrinogen in the protection of tumor cells. We have shown that platelets and fibrinogen facilitate each other in the formation of protective coats around tumor cells. Fibrinogen enhances the adhesion of platelets to tumor cells, whereas the aggregation of dense fibrin(ogen) around tumor cells depends on the thrombin formed by platelets. The exact role of platelets is to provide thrombin for fibrinogen. Our data have provided some novel information for expanding our knowledge of platelets and fibrinogen in protecting tumor cells from NK cytotoxicity.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (30570927, 30670471), the Specialized Research Fund for the Doctoral Program of Higher Education (2006200003), and the Natural Science Foundation of Jilin Province (200705488‐1).
References
- 1. Liotta LA. Cancer cell invasion and metastasis. Sci Am 1992; 266: 54–9, 62–3. [DOI] [PubMed] [Google Scholar]
- 2. Fidler I. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125I‐5 iodo‐2‐deoxyuridine. J Natl Cancer Inst 1970; 45: 773–9. [PubMed] [Google Scholar]
- 3. Mehdi AB, Tozawa K, Fisher AB, Shientag L, Lee A, Muschel RJ. Intravascular origin of metastasis from the proliferation of endothelium‐attached tumor cells: a new model for metastasis. Nat Med 2000; 6: 100–2. [DOI] [PubMed] [Google Scholar]
- 4. Gorelik E, Wiltrout RH, Okumura K, Habu S, Herberman RB. Role of NK cells in the control of metastatic spread and growth of tumor cells in mice. Int J Cancer 1982; 30: 107–12. [DOI] [PubMed] [Google Scholar]
- 5. Hanna N. The role of natural killer cells in the control of tumor growth and metastasis. Biochim Biophys Acta 1985; 780: 213–26. [DOI] [PubMed] [Google Scholar]
- 6. Arnon TI, Achdout H, Lieberman N et al . The mechanisms controlling the recognition of tumor‐ and virus‐infected cells by NKp46. Blood 2004; 103: 664–72. [DOI] [PubMed] [Google Scholar]
- 7. Rickles FR, Falanga A. Molecular basis for the relationship between thrombosis and cancer. Thromb Res 2001; 102: V215–24. [DOI] [PubMed] [Google Scholar]
- 8. Terraube V, Pendu R, Baruch D et al . Increased metastatic potential of tumor cells in von Willebrand factor‐deficient mice. J Thromb Haemost 2006; 4: 519–26. [DOI] [PubMed] [Google Scholar]
- 9. Dvorak HF, Nagy JA, Berse B et al . Vascular permeability factor, fibrin, and the pathogenesis of tumor stroma formation. Ann NY Acad Sci 1992; 667: 101–11. [DOI] [PubMed] [Google Scholar]
- 10. Nierodzik ML, Karpatkin S. Thrombin induces tumor growth, metastasis, and angiogenesis: evidence for a thrombin‐regulated dormant tumor phenotype. Cancer Cell 2006; 10: 355–62. [DOI] [PubMed] [Google Scholar]
- 11. Langer F, Amirkhosravi A, Ingersoll SB et al . Experimental metastasis and primary tumor growth in mice with hemophilia A. J Thromb Haemost 2006; 4: 1056–62. [DOI] [PubMed] [Google Scholar]
- 12. Nieswandt B, Hafner M, Echtenacher B et al . Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 1999; 59: 1295–300. [PubMed] [Google Scholar]
- 13. Weiler H. A platelets cloak for tumor cells. Blood 2005; 105: 5–6. [Google Scholar]
- 14. Gunji Y, Lewis J, Gorelik E. Fibrin formation inhibits the in vitro cytotoxic activity of human natural and lymphokine‐activated killer cells. Blood Coagul Fibrinolysis 1990; 1: 663–72. [PubMed] [Google Scholar]
- 15. Atagi S, Sone S, Fukuta K et al . Inhibition by fibrin coagulation of lung cancer cell destruction by human interleukin‐2‐activated killer cells. Jpn J Cancer Res 1992; 83: 1088–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palumbo JS, Potter JM, Kaplan LS, Talmage K, Jackson DG, Degen JL. Spontaneous hematogenous and lymphatic metastasis, but not primary tumor growth or angiogenesis, is diminished in fibrinogen‐deficient mice. Cancer Res 2002; 62: 6966–72. [PubMed] [Google Scholar]
- 17. Palumbo JS, Talmage KE, Massari JV et al . Platelets and fibrin (ogen) increase metastatic potential by impeding natural killer cell‐mediated elimination of tumor cells. Blood 2005; 105: 178–85. [DOI] [PubMed] [Google Scholar]
- 18. Menitove JE, Frenzke M, Aster RH. Use of PGE1 for preparation of platelet concentrates. Transfusion 1986; 26: 346–50. [DOI] [PubMed] [Google Scholar]
- 19. Gao YG, Wei M, Zheng S, Ba XQ, Hao S, Zeng XL. Chemically modified heparin inhibits the in vitro adhesion of nonsmall cell lung cancer cells to P‐selectin. J Cancer Res Clin Oncol 2006; 132: 257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei M, Tai GH, Gao YG et al . Modified heparin inhibits P‐selectin‐mediated cell adhesion of human colon carcinoma cells to immobilized platelets under dynamic flow conditions. J Biol Chem 2004; 279: 29202–10. [DOI] [PubMed] [Google Scholar]
- 21. Roden MM, Lee KH, Panelli MC, Marincola FM. A novel cytolysis assay using fluorescent labeling and quantitative fluorescent scanning technology. J Immunol Meth 1999; 226: 29–41. [DOI] [PubMed] [Google Scholar]
- 22. Neri S, Mariani E, Meneghetti A, Cattini L, Facchini A. Calcein‐acetyoxymethyl cytotoxicity assay: standardization of a method allowing additional analyses on recovered effector cells and supernatants. Clin Diagn Lab Immunol 2001; 8: 1131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leung L, Nachman R. Molecular mechanisms of platelet aggregation. Annu Rev Med 1986; 37: 179–86. [DOI] [PubMed] [Google Scholar]
- 24. Bennett JS. Platelet–fibrinogen interactions. Ann NY Acad Sci 2001; 936: 340–54. [DOI] [PubMed] [Google Scholar]
- 25. Gunji Y, Gorelik E. Role of fibrin coagulation in protection of murine tumor cells from destruction by cytotoxic cells. Cancer Res 1988; 48: 5216–21. [PubMed] [Google Scholar]
- 26. Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol 2002; 22: 1381–9. [DOI] [PubMed] [Google Scholar]
- 27. Heinmöller E, Weinel RJ, Heidtmann HH et al . Studies on tumor‐cell‐induced platelet aggregation in human lung cancer cell lines. J Cancer Res Clin Oncol 1996; 122: 735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jurasz P, Alonso‐Escolano D, Radomski MW. Platelet–cancer interactions: mechanisms and pharmacology of tumour cell‐induced platelet aggregation. Br J Pharmacol 2004; 143: 819–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palumbo JS, Kombrinck KW, Drew AF et al . Fibrinogen is an important determinant of the metastatic potential of circulating tumor cells. Blood 2000; 96: 3302–9. [PubMed] [Google Scholar]
- 30. Im JH, Fu W, Wang H, Bhatia SK et al . Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res 2004; 64: 8613–9. [DOI] [PubMed] [Google Scholar]
- 31. Zaidi TN, McIntire LV, Farrell DH, Thiagarajan P. Adhesion of platelets to surface‐bound fibrinogen under flow. Blood 1996; 88: 2967–72. [PubMed] [Google Scholar]
- 32. Felding‐Habermann B, Ruggeri ZM, Cheresh DA. Distinct biological consequences of integrin alpha v beta 3‐mediated melanoma cell adhesion to fibrinogen and its plasmic fragments. J Biol Chem 1992; 267: 5070–7. [PubMed] [Google Scholar]
- 33. Leavesley DI, Ferguson GD, Wayner EA, Cheresh DA. Requirement of the integrin beta 3 subunit for carcinoma cell spreading or migration on vitronectin and fibrinogen. J Cell Biol 1992; 117: 1101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]