

Abstract
Sorafenib is an orally active multikinase inhibitor that targets serine and threonine, and tyrosine kinases that are involved in tumor‐cell signal transduction and tumor angiogenesis. This phase I trial was conducted to evaluate the pharmacokinetics (PK), safety, and preliminary efficacy of sorafenib in Japanese patients with hepatocellular carcinoma (HCC) with underlying liver dysfunction. Patients with unresectable HCC, Child–Pugh status A or B, and adequate organ functions were treated. A single dose of sorafenib was administered, followed by a 7‐day wash‐out period, after which patients received either sorafenib 200 mg (cohort 1) or 400 mg (cohort 2) twice daily. The PK were investigated after a single dose and during steady state. The efficacy was evaluated using the Response Evaluation Criteria in Solid Tumors. A total of 27 patients were evaluated for PK, safety, and efficacy. Although both area under the concentration–time curve for 0–12 h and maximal concentration at steady state were slightly lower in Child–Pugh B patients than in Child–Pugh A patients, the difference was not considered to be clinically relevant. Common adverse drug events included elevated lipase, amylase, rash or desquamation, diarrhea, and hand–foot skin reaction. A dose‐limiting toxicity of hand–foot skin reaction was observed in one patient (cohort 2). Among the 24 patients evaluable for tumor response, one patient (4%) achieved a partial response, 20 (83%) had stable disease, and three (13%) had progressive disease. Sorafenib demonstrated a favorable tolerability and safety profile in Japanese HCC patients. Moreover, promising preliminary antitumor activity has been observed. Finally, there were no clinically relevant differences in PK between Child–Pugh A and B patients. (Cancer Sci 2008; 99: 159–165)
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide. Surgery and local ablation therapy, including radiofrequency, are considered curative treatment for HCC.( 1 , 2 , 3 ) Transcatheter arterial chemoembolization (TACE) has been applied to patients with advanced incurable HCC.( 3 , 4 , 5 ) The majority of patients, however, have recurrence or metastasis after these treatments. Although systemic therapy, including chemotherapeutic agents, is available for metastatic or TACE‐refractory advanced HCC, the prognosis remains poor. No standard systemic therapy that prolongs survival has been identified.
Sorafenib (BAY 43‐9006; Bayer HealthCare Pharmaceuticals, West Haven, CT, USA) was discovered based on its potent activity against Raf kinase in a battery of biochemical, cellular, and in vivo assays.( 6 , 7 ) Extensive mechanism of action studies have shown that sorafenib may inhibit tumor growth through multiple mechanisms: by inhibiting tumor‐cell proliferation that is dependent on activation of the mitogen‐activated protein kinase (MAPK) pathway, and by inhibiting tumor angiogenesis through inhibition of vascular endothelial growth factor receptor (VEGFR)‐2 and platelet‐derived growth factor receptor (PDGFR)‐β. Some evidence points to the MAPK signal‐transduction pathway as playing an important role in tumor growth and progression in HCC.( 8 ) Published data suggest that vascular endothelial growth factor (VEGF) also plays a critical role in angiogenesis of HCC, which is important for the growth and progression of HCC.( 9 ) Sorafenib has been investigated in various solid tumors in clinical studies( 10 , 11 , 12 , 13 , 14 , 15 ) and has been approved in many countries for the treatment of renal cell carcinoma. Promising results with sorafenib were recently observed in a phase II study in HCC patients.( 15 )
Various factors, such as liver function or disease extension, influence treatment selection and prognosis for HCC.( 2 , 3 , 16 ) Etiology, underlying condition, and treatment for HCC vary across countries or regions.( 2 , 3 , 17 ) Most HCC patients in Japan have hepatitis or cirrhosis due to hepatitis B or C virus( 2 ) and suffer from complications of liver dysfunction, with potential changes in the activity of metabolic enzymes, a reduction in blood flow in the liver, or protein‐binding ability due to low serum albumin. However, the degree of influence of these factors on the pharmacokinetics (PK) and tolerability of sorafenib in Japanese patients with HCC is unknown. A phase I study in Japanese patients with advanced solid tumors was conducted before the present study,( 18 ) and found that sorafenib at 400 mg b.i.d. was well tolerable and recommended for phase II studies based on safety and efficacy data. To investigate the effect of liver dysfunction and its complications on the PK, safety, and tolerability of sorafenib in Japanese patients with HCC, a phase I study was conducted. The primary objective of the present study was to evaluate the PK of sorafenib, and the secondary objectives were to evaluate the safety and tolerability of sorafenib, tumor response, time to progression (TTP), and overall survival in Japanese patients with HCC.
Materials and Methods
Patient eligibility. The eligibility criteria for enrolment in the study were: (1) histologically confirmed HCC; (2) unresectable and incurable with ablation therapy or TACE; (3) age ≥ 20 years; (4) Eastern Cooperative Oncology Group performance status of 0 or 1; (5) adequate bone marrow (absolute neutrophil count ≥ 1500 cells/mm3, platelet count ≥ 75 000 cells/mm3, and hemoglobin ≥ 8.0 g/dL), coagulation (prothrombin time ≤ 1.5 × upper limit of normal [ULN] and activated partial thromboplastin time ≤ 1.5 × ULN), renal function (serum creatinine concentration ≤ 1.5 × ULN), and hepatic function (serum total bilirubin level ≤ 3.0 mg/dL, serum aspartate and alanine transaminase levels ≤ 5.0 × ULN); (6) cirrhotic status of Child–Pugh A or B; (7) life expectancy of at least 12 weeks; and (8) written informed consent from the patient.
Exclusion criteria included clinically evident congestive heart failure, serious cardiac arrhythmias, active or symptomatic coronary artery disease or ischemia, active clinically serious infections, seizure disorder requiring medication, history of organ allograft, prior malignancy (any cancer treated curatively >3 years prior to entry was not excluded), metastatic brain or meningeal tumors, anticancer therapy within 3 months of study entry, and pregnancy or lactation for women. This protocol was approved by the National Cancer Center's institutional review board for clinical investigation with the provisions of the Declaration of Helsinki, Good Clinical Practice guidelines, and local laws and regulations.
Treatment methods. The dose for the first cohort was 200 mg bid sorafenib, and the dose for the second cohort was escalated to 400 mg bid. To investigate the PK profile of sorafenib, including its elimination phase, a single dose was given as one‐time administration followed by a 7‐day wash‐out period. Subsequently, the drug was given twice daily for 28 days without a resting period (cycle 1). Either 200 mg or 400 mg sorafenib was given to all patients orally twice daily, in the morning and in the evening (every 12 h as far as possible). Patients were allowed to continue on sorafenib after cycle 1 if they consented to continue, and no intolerable adverse event was experienced, as assessed by investigators. Treatment was continued until disease progression, intolerable adverse event, or consent of withdrawal.
Examination and observation for safety was conducted every 2 weeks, and administration of the drug was to be terminated immediately when the patient met the criteria for removal from the study, described in this protocol with due consideration for the patient's safety.
Study design. The present study was a non‐randomized, uncontrolled, non‐blinded, single‐center phase I study to investigate the PK, safety, and tolerability of sorafenib in Japanese patients with HCC. The dose level investigated in this study was 200 mg bid for the first cohort and 400 mg bid for the second cohort. Twelve patients, including six with Child–Pugh A and six with Child–Pugh B, were to be enrolled in each cohort. Tolerability was evaluated at the end of cycle 1 by Child–Pugh classification. If less than two out of six patients experienced dose‐limiting toxicity (DLT) in the 200‐mg bid cohort, the study would proceed to the 400‐mg bid cohort. DLT that needed dose modification was defined as: (1) grade 3 and grade 4 non‐hematological toxicity, except for pancreatic enzyme abnormality and hand–foot skin reaction; (2) grade 4 pancreatic enzyme elevation with values that persisted on two consecutive determinations with a 3‐day interval, or clinical and/or imaging findings of pancreatitis, or pancreatic adverse event considered to be life threatening, or having a high risk of serious or chronic disorders; (3) severe hand–foot skin reaction, moist desquamation, ulceration, blistering, or severe pain of the hands or feet, or severe discomfort that caused the patient to be unable to work or carry out the activities of daily living; (4) grade 4 neutropenia (absolute neutrophil count less than 500/µL) for 7 days duration; (5) grade 4 neutropenia of any duration with fever of 38.5°C and above; and (6) platelet count < 25 000 cells/mm3. Toxicity was graded according to the National Cancer Institute common toxicity criteria version 2.0. The independent safety committee for this study gave advice on the evaluation of tolerability of the dose level and the cohort transition.
Pharmacokinetics. All patients who received at least one dose of study medication were included in the PK analysis. Blood samples for the determination of plasma concentrations of sorafenib (and its metabolites) were collected prior to drug administration, as well as 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 36, 48, 72, 96, and 120 h after single‐dose administration. For the first cycle, blood was sampled prior to the first dosing on days 1, 4, 7, 10, 14, 21, and 28, along with 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, and 12 h after the first dose on days 14 and 28. Urine voided up to 48 h after single administration was collected.
Concentrations of sorafenib and its metabolites in plasma and urine were determined using validated liquid chromatography and tandem mass spectrometry methods. Plasma PK parameters were calculated by non‐compartment analysis by the KINCALC program (Bayer HealthCare Pharmaceuticals).( 10 ) Primary plasma PK parameters were area under the concentration–time curve (AUC), AUC for 0–12 h (AUC0–12), and maximal concentration (Cmax). Plasma concentrations and PK parameters were analyzed by dose and Child–Pugh classification.
Clinical assessments. Physical examination, complete blood cell counts, serum chemistries, and urinalysis were carried out at baseline and at least twice monthly after initiating treatment with sorafenib. Patients underwent dynamic computed tomography (CT) to evaluate tumor response at baseline, the end of cycle 1, and every two cycles thereafter. CT was carried out by obtaining contiguous transverse sections with the helical scanning method at a section thickness of 5 mm. Tumor evaluation was assessed using the Response Evaluation Criteria in Solid Tumours (RECIST).( 19 )
Statistical analysis. The data were analyzed using SAS (SAS Institute, Cary, NC, USA). The safety and efficacy were evaluated on an intention‐to‐treat basis. Progression‐free survival was calculated from the first day of treatment until evidence of tumor progression, clinical progression, or death due to any cause. Overall survival was calculated from the first day of treatment until death due to any cause. Survival data were analyzed using the Kaplan–Meier method.
Results
Patient characteristics and treatments. From April 2004 through January 2005, a total of 27 patients were enrolled in the present study. Thirteen patients were enrolled at the treatment level of 200 mg (cohort 1) and 14 at the treatment level of 400 mg (cohort 2) twice daily (b.i.d.) for 28 days (cycle 1). One out of 13 patients in cohort 1 discontinued the study due to consent withdrawal after single‐dose administration. One out of 14 patients in cohort 2 dropped out of this study due to adverse events during cycle 1. Patient characteristics are shown in Table 1. The median number of cycles administered per patient was five (range, 1–13 cycles). None of the patients from the 200‐mg group reduced the dose of sorafenib, whereas two patients required dose reduction in the 400‐mg group.
Table 1.
Patient characteristics
| Characteristic | 200 mg bid (n = 13) | 400 mg bid (n = 14) | Total (n = 27) |
|---|---|---|---|
| Sex (n) | |||
| Male | 12 | 13 | 25 |
| Female | 1 | 1 | 2 |
| Median age (years) | 69 (range 48–77) | 70 (range 63–79) | 70 (range 48–79) |
| Eastern Cooperative Oncology Group performance status | |||
| 0 | 13 | 14 | 27 |
| Child–Pugh classification | |||
| A | 7 | 6 | 13 |
| B | 6 | 8 | 14 |
| Viral markers | |||
| HB antigen+, HCV antibody− | 3 | 1 | 4 |
| HB antigen−, HCV antibody+ | 9 | 11 | 20 |
| HB antigen−, HCV antibody− | 1 | 2 | 3 |
| Previous treatment | |||
| – | 1 | 3 | 4 |
| + | 12 | 11 | 23 |
| Tumor stage | |||
| II | 1 | 2 | 3 |
| III | 7 | 8 | 15 |
| IVa | 1 | 1 | 2 |
| IVb | 4 | 3 | 7 |
| Portal vein tumor thrombus | |||
| – | 12 | 13 | 25 |
| + | 1 | 1 | 2 |
| Metastasis | |||
| – | 9 | 11 | 20 |
| + | 4 | 3 | 7 |
| Lung | 3 | 1 | 4 |
| Lung + lymph node | 1 | 1 | 2 |
| Lymph node | 0 | 1 | 1 |
HB, hepatitis B; HCV, hepatitis C virus.
Evaluation of PK. Plasma drug concentrations were analyzed in 27 patients in the PK analysis. Plasma PK parameters of patients in the 200 and 400 mg bid groups are shown in 2, 3. There was a large interpatient variability in the PK of sorafenib. Geometric means of AUC, AUC0–12, and Cmax on day 1 of single‐dose administration were not statistically different between 200 and 400 mg bid or between Child–Pugh A and B. Dose‐dependent increases in AUC0–12 and Cmax were observed at steady state (day 14) in the 200‐mg bid and 400‐mg bid patients; however, these increases were not dose proportional. Geometric means of AUC0–12 and Cmax were slightly lower in the Child–Pugh B patients compared with the Child–Pugh A patients at steady state. The t1/2 after single dose was similar between the Child–Pugh A and B groups for both dose levels.
Table 2.
Pharmacokinetic parameters of sorafenib and metabolites M‐2, M‐4, and M‐5: sorafenib following single dose and multiple dose of 200 mg and 400 mg geometric mean (coefficient of variation)
| Sorafenib | Parameter | Unit | 200 mg bid | 400 mg bid | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Child–Pugh A | Child–Pugh B | Child–Pugh A | Child–Pugh B | |||||||
| Single | n | 7 | 6 | 6 | 8 | |||||
| Dose | AUC | mg*h/L | 28.29 | 190.29 ‡ | 18.64 | 74.1 | 20.33 | 90.31 | 26.87 | 96.97 |
| Day 1 | AUC0–12 | mg*h/L | 5.02 | 190.36 | 2.75 | 61.06 | 3.82 | 86.06 | 3.11 | 88.16 |
| Cmax | mg/L | 0.81 | 195.96 ‡ | 0.49 | 67.85 | 0.55 | 83.75 | 0.53 | 86.68 | |
| Tmax | h † | 7 | 3–12 ‡ | 18 | 4–24 | 8 | 6–24 | 24 | 4–24 | |
| T1/2 | H | 25.14 | 30.13 ‡ | 30.44 | 35.67 | 22.28 | 12.49 | 27.2 | 45.19 | |
| Cycle 1 | N | 6 | 6 | 6 | 6 | |||||
| Day 14 | AUC0–12 | mg*h/L | 25.52 | 75.04 | 15.28 | 55.26 | 33.47 | 60.13 | 29.45 | 59.44 § |
| Cmax | mg/L | 3.36 | 87.29 | 1.89 | 62.14 | 4.66 | 66.12 | 3.04 | 94.39 | |
| Cycle 1 | N | 6 | 6 | 6 | 5 | |||||
| Day 28 | AUC0–12 | mg*h/L | 31.63 | 101.64 | 20 | 73.4 | 28.91 | 86.79 | 20.71 | 72.06 |
| Cmax | mg/L | 4.22 | 92.32 | 3.32 | 78.65 | 3.32 | 113.47 | 4.01 | 79.12 | |
Median (range),
n = 6,
n = 5. AUC0–12, area under the concentration–time curve for 0–12 h.
Table 3.
Pharmacokinetic parameters of sorafenib and metabolites M‐2, M‐4, and M‐5: metabolites following multiple dose of 200 mg and 400 mg, measured at steady state (cycle 1, day 14) geometric mean (% coefficient of variation)
| Parameter | 200 mg bid | 400 mg bid | ||||
|---|---|---|---|---|---|---|
| M‐2 | M‐4 | M‐5 | M‐2 | M‐4 | M‐5 | |
| Child–Pugh A | ||||||
| n | 6 | 6 | 6 | 6 | 6 | 6 |
| AUC0–12 (mg × h/L) | 4.18 (126) | 0.92 (158) | 0.79 (167) | 6.18 (127) | 1.68 (159) | 1.22 (193) |
| Ratio † (%) | 13.08 (30) | 2.89 (60) | 2.48 (81) | 14.16 (39) | 3.85 (55) | 2.79 (85) |
| Child–Pugh B | ||||||
| n | 6 | 5 | 4 | 5 | 5 | 5 |
| AUC0–12 (mg × h/L) | 1.62 (173) | 0.36 (131) | 0.44 (351) | 5.67 (90) | 2.13 (142) | 1.25 (117) |
| Ratio † (%) | 9.05 (67) | 1.85 (42) | 1.95 (157) | 14.46 (36) | 5.44 (56) | 3.19 (47) |
Median ratio of each metabolite to sum of all analytes. BAY 43‐9006: M‐2, BAY 67‐3472; M‐4, BAY 43‐9007; and M‐5, BAY 68‐7769. AUC0–12, area under the concentration–time curve for 0–12 h.
Dose‐dependent increases in the AUC0–12 and Cmax of metabolites M‐2 (N‐oxide), M‐4 (N‐demethyl), and M‐5 (N‐oxide, desmethyl derivative) were observed. M‐2 was the main metabolite in plasma. Ratios of each metabolite to the sum of all analytes were similar between the 200‐mg bid and 400‐mg bid patients and for baseline Child–Pugh class (2, 3). M‐7 (glucuronide of sorafenib) and M‐8 (glucuronide of M‐2) were detected in urine though no unchanged substance or M‐2 was detected. There was no difference between the Child–Pugh A (1.21% for M‐7 and 0.02% for M‐8 at 400 mg) and B (1.18% and 0.02%, respectively, at 400 mg) groups in the urinary excretion rate of compounds at steady state. Interestingly, these PK results were similar to those obtained from the Japanese phase I study in non‐HCC tumors.( 18 )
Adverse events. Adverse events of all 27 patients are shown in Table 4. Twenty‐six out of 27 patients (96.3%) experienced an adverse event: 12 out of 13 patients (92.3%) in the 200‐mg group and 14 out of 14 patients (100%) in the 400‐mg group. The most common drug‐related adverse events were elevated lipase or amylase (88.9%), dermatological events (81.5%), and gastrointestinal events (70.4%). Common dermatological events were rash or desquamation (55.6%), and hand–foot skin reaction (44.4%). The incidence of adverse events in the 400‐mg dose level was higher than that in the 200‐mg dose level by ≥20%. These events fell under the categories of dermatology/skin (100.0 vs 61.5%), general cardiovascular (35.7 vs 7.7%), and renal/genitourinary (21.4 vs 0%).
Table 4.
Adverse events
| Child–Pugh | Grade 3/4 | All grades | ||||||
|---|---|---|---|---|---|---|---|---|
| 200 mg bid | 400 mg bid | 200 mg bid | 400 mg bid | |||||
| A (n = 7) | B (n = 6) | A (n = 6) | B (n = 8) | A (n = 7) | B (n = 6) | A (n = 6) | B (n = 8) | |
| Hematological | ||||||||
| Leukocytopenia | 0 | 0 | 0 | 0 | 2 (29%) | 0 | 1 (17%) | 0 |
| Lymphopenia | 2 (29%) | 1 (17%) | 1 (17%) | 1 (13%) | 2 (29%) | 1 (17%) | 1 (17%) | 2 (25%) |
| Platelets | 0 | 0 | 1 (17%) | 1 (13%) | 0 | 1 (17%) | 2 (33%) | 3 (38%) |
| Non‐hematological | ||||||||
| Hypertension | 0 | 1 (17%) | 1 (17%) | 3 (38%) | 0 | 1 (17%) | 1 (17%) | 3 (38%) |
| Fatigue | 0 | 0 | 0 | 0 | 0 | 1 (17%) | 0 | 0 |
| Fever | 0 | 0 | 0 | 0 | 1 (14%) | 2 (33%) | 0 | 1 (13%) |
| Weight loss | 0 | 0 | 0 | 0 | 2 (29%) | 1 (17%) | 1 (17%) | 4 (50%) |
| Hand–foot skin reaction | 0 | 0 | 0 | 2 (27%) | 2 (29%) | 2 (33%) | 5 (83%) | 3 (38%) |
| Rash | 0 | 0 | 0 | 2 (27%) | 2 (29%) | 3 (50%) | 4 (67%) | 6 (75%) |
| Alopecia | 0 | 0 | 0 | 0 | 2 (29%) | 1 (17%) | 2 (33%) | 0 |
| Dry skin | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (38%) |
| Pruritus | 0 | 0 | 0 | 0 | 0 | 1 (17%) | 4 (67%) | 3 (38%) |
| Anorexia | 0 | 0 | 0 | 0 | 2 (29%) | 1 (17%) | 1 (17%) | 2 (25%) |
| Diarrhea | 0 | 0 | 1 (17%) | 0 | 4 (57%) | 4 (67%) | 2 (33%) | 5 (63%) |
| Stomatitis | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17%) | 2 (25%) |
| Lipase | 3 (43%) | 4 (67%) | 4 (67%) | 6 (75%) | 6 (86%) | 6 (100%) | 6 (100%) | 6 (75%) |
| Amylase | 1 (14%) | 1 (17%) | 1 (17%) | 1 (13%) | 4 (57%) | 3 (50%) | 4 (67%) | 5 (63%) |
Elevation of lipase and amylase was transient in most of the cases, and decreased gradually in all patients without treatment. One patient on 400 mg bid experienced acute pancreatitis that necessitated sorafenib withdrawal. The patient experienced abdominal pain 6 months after beginning treatment (cycle 6). Moreover, high lipase and amylase, as assessed by blood test, and swelling of the pancreas were observed. The patient's abdominal pain resolved 1 day after stopping sorafenib, and lipase and amylase normalized 2 days later. Sorafenib was restarted 20 days after resolution and continued over 122 days, without recurrence of pancreatitis.
Grade 3 or worse drug‐related adverse events were observed in 23 patients (85.2%), the majority of which were related to laboratory abnormalities: 10 patients in the 200‐mg group and 13 in the 400‐mg group. One patient with Child–Pugh B in the 400‐mg bid group experienced DLT of hand–foot skin reaction at the end of cycle 1. There were no drug‐related deaths in either of the groups.
There was no major difference in the incidence and grade of drug‐related adverse events between the Child–Pugh A and B groups. At the dose level of 200 mg, the drug‐related adverse event whose incidence was at least 20% higher in the Child–Pugh B group than in the Child–Pugh A group was rash or desquamation (50.0 vs 28.6%). The differences at the 400‐mg dose level were diarrhea (62.5 vs 33.3%), weight loss (50.0 vs 16.7%), hypertension (37.5 vs 16.7%), dry skin (37.5 vs 0%), and fatigue (25.0 vs 0%).
Tumor response and survival. Partial response was achieved in one of the 27 patients. No complete response was observed (Table 5; Fig. 1). The overall response rate was 3.7% (95% confidence interval, 0.1–14.0%). Stable disease was noted in 21 patients (77.8%) and the disease control rate (partial response + stable disease rate) was 81.5% in 27 patients. Progressive disease was noted in three patients (11.1%).
Table 5.
Tumor response
| Response | 200 mg bid (n = 13) | 400 mg bid (n = 14) | Total (n = 27) |
|---|---|---|---|
| Partial response | 1 | 0 | 1 (3.7%) |
| Stable disease | 10 | 11 | 21 (77.8%) |
| Progressive disease | 1 | 2 | 3 (11.1%) |
| NA | 1 | 1 | 2 (7.4%) |
NA, not assessed because these patients did not complete cycle 1.
Figure 1.
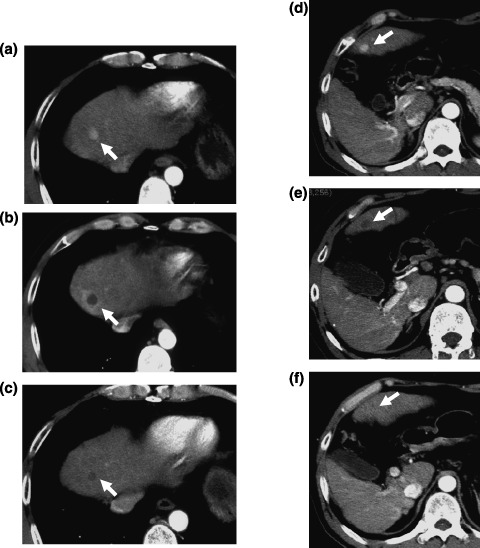
A 48‐year‐old man with multiple tumors of hepatocellular carcinoma (HCC) after hepatectomy, percutaneous ethanol injection, and transcatheter arterial embolization. (a) Hypervascular HCC lesion, 1 cm in diameter, was revealed at the early phase of dynamic computed tomography (CT) before administration of sorafenib at the anterior superior segment of the liver (arrow). (b) The vascularity of this tumor disappeared 1 month after the administration of sorafenib. (c) The tumor was reduced 3 months after the administration of sorafenib. (d) Another hypervascular HCC lesion, 1 cm in diameter, was revealed at the early phase of dynamic CT before administration of sorafenib in the left lobe of the liver (arrow). (e) The vascularity of this tumor disappeared 8 months after the administration of sorafenib. (f) The tumor almost completely disappeared 10 months after the administration of sorafenib.
Disease progression or death was observed in all patients. Sixteen of the 27 patients died of disease progression, and two died of cerebral infarction or myocardial infarction. Of the 27 patients, the median TTP was 4.9 months, and the median overall survival (OS) was 15.6 months (Fig. 2). The 6‐month progression‐free rate based on TTP was 46.2%, and 1‐ and 2‐year OS were 59.3 and 30.9%, respectively.
Figure 2.
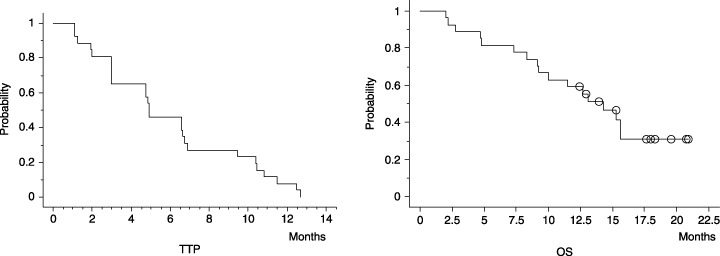
Time to progression (TTP) in all 27 patients treated with sorafenib. The median TTP was 4.9 months, and the 6‐month survival rate was 46.2%. Overall survival (OS) in the 27 patients treated with sorafenib. The median OS period was 15.6 months, and the 1‐year survival rate was 59.3%.
Discussion
The PK, safety, and tolerability of sorafenib were investigated in Japanese patients with HCC treated with doses of 200 mg bid or 400 mg bid.
Most of the HCC patients had hepatitis or cirrhosis with underlying liver disorder and a reduction in hepatic blood flow to various degrees. Liver dysfunction in patients with HCC may affect the PK of sorafenib. When comparing the PK by Child–Pugh classification, geometric means of AUC0–12 and Cmax at steady state were lower in the Child–Pugh B group than in the Child–Pugh A group, whereas after multiple doses of sorafenib, the mean plasma concentrations were highly variable and showed no clear dose dependency. Although the numerical differences in geometric means for PK parameters such as AUC, Cmax, and t1/2 were observed between Child–Pugh classifications, these differences were considered not to be clinically relevant in consideration of their large intersubject variability. No significant difference in clinical findings between these two groups was observed. There was also no major difference (i.e. over 20%) in the incidence of adverse events between Child–Pugh A and B groups. However, geometric means of AUC0–12 and Cmax at steady state were slightly lower in the Child–Pugh B patients compared with the Child–Pugh A patients.
There were no remarkable differences in the overall incidence of adverse events for each dose level (92% for the 200‐mg group and 100% for the 400‐mg group). For a few drug‐related adverse events, the incidences were at least 20% higher in the 400‐mg group than in the 200‐mg group, including rash or desquamation (71.4 vs 38.5%), hand–foot skin reaction (57.1 vs 30.8%), pruritus (50.0 vs 7.7%), decrease of platelets (35.7 vs 7.7%), hypertension (28.6 vs 7.7%), dry skin (21.4 vs 0%), and stomatitis or pharyngitis (21.4 vs 0%). DLT of hand–foot skin reaction was observed in a patient with Child–Pugh B at the end of cycle 1 with 400 mg bid, whereas no DLT was observed in the 200‐mg bid group.
The most common drug‐related adverse events were elevated lipase (88.9%) and amylase (59.3%). Twenty‐four (88.9%) of the 27 patients showed high values of grade 3 or worse. Most of the patients were asymptomatic and only one patient had abdominal pain with findings to indicate pancreatitis on ultrasonography during cycle 6. His pancreatitis resolved shortly after discontinuation of sorafenib, and the patient restarted and continued with a reduced dose of sorafenib after recovery.
A separate phase I clinical study was carried out to evaluate the safety of sorafenib in patients with solid tumor, excluding HCC, at doses of 100, 200, 400, and 600 mg bid.( 18 ) In that study, the most common type of adverse events included skin reaction, elevation of pancreatic enzyme, and gastrointestinal (GI) toxicity such as diarrhea. In the current study, a similar pattern of adverse events was observed. These results suggest that ‘gastrointestinal’ and ‘dermatology/skin’ are common adverse events regardless of cancer type and liver function status. One finding to note is that the incidence of elevation (grade 3/4) of lipase (63.0%) or amylase (14.8%) in the present study in HCC patients was higher than that observed in non‐HCC patients (lipase 23% and amylase 10%).( 18 )
In summary, the present study showed no clinically significant difference in PK, safety, tolerability, or efficacy by Child–Pugh status or between HCC patients and non‐HCC patients, whereas some dose dependency in adverse events was observed.
Investigations into cytotoxic agents for HCC have been conducted.( 20 , 21 ) However, no standard chemotherapy has been established. Recently, a number of agents targeting growth factors were investigated in HCC. Through these investigations, it was indicated that epidermal growth factor receptor/human epidermal growth factor receptor 1 (EGFR/HER1) is actively expressed in human hepatoma cells.( 22 , 23 ) Erlotinib, which is an EGFR/HER1 tyrosine kinase inhibitor, and lapatinib, which is an EGFR/HER1 and ErbB‐2 (Her2/neu) dual tyrosine kinase inhibitor, have been investigated in phase II studies in HCC patients.( 24 , 25 , 26 ) For erlotinib, the response rate was 4–9%, the median TTP was 2.1–3.2 months, and the OS was 5.8–13 months,( 24 , 25 ) whereas for lapatinib, the response rate was 0%, and the median progression‐free survival time was 1.8 months.( 26 )
Hepatocellular carcinoma, given its hypervascular characteristics, may be sensitive to antiangiogenic agents.( 9 ) It is known that VEGF augments the development and metastasis of HCC. Bevacizumab, a monoclonal antibody against VEGF, has been investigated in phase II studies.( 27 ) The response rate with bevacizumab was 10% and the disease control rate was 80%. A combination of gemox (gemcitabine plus oxaliplatin) and bevacizumab showed a better response rate of 20%.( 28 )
Sorafenib, an orally active multikinase inhibitor, blocks tumor‐cell proliferation by targeting Raf/MEK/ERK signaling at the level of Raf kinase, and exerts an antiangiogenic effect by targeting VEGFR‐2, VEGFR‐3, and PDGFR‐β tyrosine kinases. In phase II studies in non‐Japanese and Japanese HCC patients, comparable median TTP of 4.2 and 4.9 months, respectively, and response rates of 2 and 4%, respectively, were shown.( 15 ) However, OS in the two studies were different: 9.2 months in the non‐Japanese study and 15.6 months in the Japanese study. Difference in backgrounds such as liver function or treatment after progression may play a role in this discrepancy in survival time.
In the current study, one patient achieved partial response (Fig. 1). The patient had several small viable HCC lesions after hepatectomy, percutaneous ethanol injection, and TACE. Following administration of sorafenib, tumor vascularity decreased dramatically preceding a gradual tumor reduction. Time to tumor shrinking varied across lesions, ranging from 1 to 8 months after initiation of treatment with sorafenib. It is likely that, with anti‐VEGF agents such as sorafenib, it may take time to achieve tumor reduction to meet partial response by RECIST, whereas the duration of stable disease may persist due to its tumor stabilization activity.
With the relatively long TTP of VEGF pathway‐targeting agents such as bevacizumab or sorafenib, these agents may have antitumor effects on HCC and prolong survival. With its profile of tumor stabilization and tolerability, sorafenib may be applicable not only for advanced HCC but also for the adjuvant setting after curative treatment, such as surgery or radiofrequency ablation therapy.
In conclusion, in the present phase I study, sorafenib demonstrated favorable safety and tolerability, and promising preliminary antitumor activity in Japanese HCC patients. Considering that DLT was observed in one of 14 patients treated with 400 mg bid, 400 mg bid could also be recommended for future studies in Japanese HCC patients, as well as non‐HCC Japanese and Caucasian patients. However, as the number of patients was limited in this phase I study, a confirmatory study will be required with a larger number of patients.
Acknowledgments
This study was supported by Bayer Yakuhin.
References
- 1. Ryu M, Shimamura Y, Kinoshita T et al . Therapeutic results of resection, transcatheter arterial embolization and percutaneous transhepatic ethanol injection in 3225 patients with hepatocellular carcinoma: a retrospective multicenter study. Jpn J Clin Oncol 1997; 27: 251–7. [DOI] [PubMed] [Google Scholar]
- 2. Okuda K, Mitchell DG, Itai Y et al . Hepatobiliary disease. Primary Malignant Tumors of the Liver. London: Blackwell Science, 2001: 343–89. [Google Scholar]
- 3. Bruix J, Sherman M, Llovet JM et al . Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona‐2000 EASL conference. European Association for the Study of the Liver. J Hepatol 2001; 35: 421–30. [DOI] [PubMed] [Google Scholar]
- 4. Llovet JM, Real MI, Montana X et al . Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet 2002; 18: 1734–9. [DOI] [PubMed] [Google Scholar]
- 5. Takayasu K, Arii S, Ikai I et al . Prospective cohort study of transarterial chemoembolization for unresectable hepatocellular carcinoma in 8510 patients. Gastroenterology 2006; 131: 461–9. [DOI] [PubMed] [Google Scholar]
- 6. Wilhelm S, Chien DS. BAY 43‐9006: preclinical data. Curr Pharm Des 2002; 8: 2255–7. [DOI] [PubMed] [Google Scholar]
- 7. Hotte SJ, Hirte HW. BAY 43‐9006: early clinical data in patients with advanced solid malignancies. Curr Pharm Des 2002; 8: 2249–53. [DOI] [PubMed] [Google Scholar]
- 8. Huynh H, Nguyen TT, Chow KH et al . Overexpression of the mitogen‐activated protein kinase (MAPK) kinase (MEK)‐MAPK in hepatocellular carcinoma: Its role in tumor progression and apoptosis. BMC Gastroenterol 2003; 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pang R, Poon RTP. Angiogenesis and antiangiogenic therapy in hepatocellular carcinoma. Cancer Lett 2006; 242: 151–67. [DOI] [PubMed] [Google Scholar]
- 10. Strumberg D, Richly H, Hilger RA et al . Phase I clinical and pharmacokinetic study of the novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43‐9006 in patients with advanced refractory solid tumors. J Clin Oncol 2005; 23: 965–72. [DOI] [PubMed] [Google Scholar]
- 11. Moore M, Hirte HW, Siu L et al . Phase I study to determine the safety and pharmacokinetics of the novel Raf kinase and VEGFR inhibitor BAY 43‐9006, administered for 28 days on/7 days off in patients with advanced, refractory solid tumors. Ann Oncol 2005; 16: 1688–94. [DOI] [PubMed] [Google Scholar]
- 12. Awada A, Hendlisz A, Gil T et al . Phase I safety and pharmacokinetics of BAY 43‐9006 administered for 21 days on/7 days off in patients with advanced, refractory solid tumors. Br J Cancer 2005; 92: 1855–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tong FK, Chow S, Hedley D. Pharmacodynamic monitoring of BAY 43‐9006 (Sorafenib) in phase I clinical trials involving solid tumor and AML/MDS patients, using flow cytometry to monitor activation of the ERK pathway in peripheral blood cells. Cytometry B Clin Cytom 2006; 70: 107–14. [DOI] [PubMed] [Google Scholar]
- 14. Ratain MJ, Eisen T, Stadler WM et al . Phase II placebo‐controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 2006; 24: 2505–12. [DOI] [PubMed] [Google Scholar]
- 15. Abou‐Alfa GK, Schwartz L, Ricci S et al . Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol 2006; 24: 4293–300. [DOI] [PubMed] [Google Scholar]
- 16. The Liver Cancer Study Group of Japan . Primary liver cancer in Japan: Clinicopathologic features and results of surgical treatments. Ann Surg 1990; 211: 277–87. [PMC free article] [PubMed] [Google Scholar]
- 17. Bruix J, Llovet JM. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology 2002; 35: 519–24. [DOI] [PubMed] [Google Scholar]
- 18. Minami H, Kawada K, Ebi H et al . A phase I study of BAY 43‐9006, a dual inhibitor of Raf and VEGFR kinases, in Japanese patients with solid cancers. J. Clin. Oncol. 2005 Proc. Am. Soc. Clin. Oncol. 2005; 23 (Suppl.): 207s (abstract 3062). [Google Scholar]
- 19. Therasse P, Arbuck SG, Eisenhauer EA et al . New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92: 205–16. [DOI] [PubMed] [Google Scholar]
- 20. Lai CL, Wu PC, Chan GC, Lok AS, Lin HJ. Doxorubicin versus no antitumor therapy in inoperable hepatocellular carcinoma: A prospective randomized trial. Cancer 1988; 62: 479–83. [DOI] [PubMed] [Google Scholar]
- 21. Yeo W, Mok TS, Zee B et al . A randomized phase III study of doxorubicin versus cisplatin/interferon α‐2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst 2005; 97: 1532–8. [DOI] [PubMed] [Google Scholar]
- 22. Kaneko Y, Shibuya M, Nakayama T et al . Hypomethylation of c‐myc and epidermal growth factor receptor genes in human hepatocellular carcinoma and fetal liver. Jpn J Cancer Res 1985; 76: 1136–40. [PubMed] [Google Scholar]
- 23. Hung WC, Chuang LY, Tsai JH et al . Effects of epidermal growth factor on growth control and signal transduction pathways in different human hepatoma cell lines. Biochem Mol Biol Int 1993; 30: 319–28. [PubMed] [Google Scholar]
- 24. Thoma MB, Dutta A, Brown T et al . A phase II open‐label study of OSI‐774 (NSC 718781) in unresectable hepatocellular carcinoma. Proc Am Soc Clin Oncol 2005; 23: 317S. [Google Scholar]
- 25. Philip PA, Mahoney MR, Allmer C et al . Phase II study of Erlotinib (OSI‐774) in patients with advanced hepatocellular cancer. J Clin Oncol 2005; 23: 6657–63. [DOI] [PubMed] [Google Scholar]
- 26. Ramanathan RK, Belani CP, Singh DA et al . Phase II study of lapatinib, a dual inhibitor of epidermal growth factor receptor (EGFR) tyrosine kinase 1 and 2 (Her2/Neu) in patients (pts) with advanced biliary tree cancer (BTC) or hepatocellular cancer (HCC): A California Consortium (CCC‐P) Trial. Proc Am Soc Clin Oncol 2006; 24: 181S. [Google Scholar]
- 27. Schwartz JD, Schwartz M, Lehrer D et al . Bevacizumab in unresectable hepatocellular carcinoma (HCC) for patients without metastasis and without invasion of the portal vein. Proc Am Soc Clin Oncol 2006; 24: 213S. [Google Scholar]
- 28. Zhu AX, Blaszkowsky LS, Ryan DP et al . Phase II study of gemcitabine and oxaliplatin in combination with bevacizumab in patients with advanced hepatocellular carcinoma. J Clin Oncol 2006; 24: 1898–903. [DOI] [PubMed] [Google Scholar]