

Abstract
There are a number of kinase inhibitors that regulate components of the neovasculature. We previously reported the use of transforming growth factor (TGF)‐β inhibitor on neovasculature in stroma‐rich tumor models to increase the intratumoral distribution of nanoparticles. Here, we compared the effects of two other kinase inhibitors, imatinib and sorafenib, with TGF‐β inhibitor (LY364947) on extravasation of a modeled nanoparticle, 2 MDa dextran. We first used a mouse model of neoangiogenesis, the Matrigel plug assay, to compare neovasculature formed inside of and around Matrigel plugs (intraplug and periplug regions, respectively). Intraplug vasculature was more strongly pericyte covered, whereas periplug vasculature was less covered. In this model, TGF‐β inhibitor exhibited the most potent effect on intraplug vasculature in increasing the extravasation of dextran, whereas sorafenib had the strongest effect on periplug vasculature. Although imatinib and TGF‐β inhibitor each reduced pericyte coverage, imatinib also reduced the density of endothelium, resulting in a decrease in overall delivery of nanoparticles. These findings were confirmed in two tumor models, the CT26 colon cancer model and the BxPC3 pancreatic cancer model. The vasculature phenotype in the CT26 model resembled that in the periplug region, whereas the latter resembled that in the intraplug region. Consistent with this, sorafenib most potently enhanced the accumulation of nanoparticles in the CT26 model, whereas TGF‐β inhibitor did in the BxPC3 model. In conclusion, the appropriate strategy for optimization of tumor vasculature for nanoparticles may differ depending on tumor type, and in particular on the degree of pericyte coverage around the vasculature. (Cancer Sci 2009; 100: 173–180)
The effectiveness of drug delivery into tumor tissues is an important issue in the treatment of solid tumors, in addition to the efficacy of drugs in treating tumor cells. For example, gemcitabine, a first‐line anticancer agent for pancreatic adenocarcinoma, exhibited potent in vitro growth‐inhibitory effects on a cultured cell line derived from the human pancreatic adenocarcinoma line BxPC3.( 1 ) However, it exhibited only slight inhibitory effects on xenografted BxPC3 tumors in mice( 2 ) and slight elongation of survival time in tumor‐bearing patients, with significant effects only in the improvement of quality of life index in clinical trials.( 3 )
Many factors might potentially explain this discrepancy, particularly those related to tumor stroma.( 4 ) Among them, tumor vasculature plays an important role in the delivery of anticancer agents. Extravasation of drugs to tumor tissue constitutes an essential part of drug delivery to tumor tissues,( 5 ) whereas the molecular size of compounds is another important determinant of accumulation.( 6 ) We have recently shown that increased leakiness in tumor neovasculature improves the accumulation of nanoparticles in tumor tissues in animal models of pancreatic adenocarcinoma and diffuse‐type advanced gastric cancer.( 7 ) In that study, inhibition of transforming growth factor (TGF)‐β signaling reduced pericyte coverage and slightly increased endothelial area, resulting in an increase in vascular leakiness without loss of blood flow. However, numerous studies of tumor neovasculature have shown that it is leaky by nature, and that manipulation of vessels to make them less leaky, or induction of vascular normalization, may therefore benefit drug delivery to tumor tissues.( 8 ) This theory has been supported with the use of vascular endothelial growth factor (VEGF) inhibitors. There are a number of VEGF inhibitors available, including neutralizing anti‐VEGF antibodies such as bevacizumab (Avastin) and sorafenib (Nexavar). Sorafenib is a small molecular‐weight (SMW) compound inhibiting multiple tyrosine kinases, including VEGF receptor (VEGFR) 2.( 9 )
The roles of pericytes in neoangiogenesis have also been well investigated.( 10 ) Coverage of the neovasculature by pericytes stabilizes vascular structure.( 11 ) Genetic ablation of platelet‐derived growth factor (PDGF)‐B signaling, one of the major signaling pathways in induction of pericyte maturation and recruitment to the endothelium, results in a bleeding tendency of the neovasculature.( 11 , 12 , 13 ) PDGF‐B signaling can be inhibited by the SMW inhibitor (SMWI) imatinib (Gleevec or Glivec), which inhibits the receptor for PDGF‐B signaling, PDGF receptor (PDGFR) β, as well as PDGFRα and c‐kit.( 14 ) The use of imatinib along with VEGF inhibitors was shown to be effective in inhibiting tumor neovascularization in an animal model of spontaneous pancreatic islet tumor, the RIP‐Tag model, through disruption of both pericytes and endothelium.( 15 )
Here we investigated the changes in vascular leakiness induced by three of the SMWI mentioned above, TGF‐β inhibitor (LY364947), sorafenib, and imatinib, in the Matrigel plug assay as well as two animal cancer models. The Matrigel plug assay was carried out by mixing BD Matrigel Basement Membrane Matrix with VEGF‐A, fibroblast growth factor (FGF)‐2, and heparin as angiogenic molecules to form mature neovasculature inside the gel plug, according to our previous report.( 16 ) Of the two cancer models used in the present study, one was a well‐established hypervascular cancer model using the murine colon cancer cell line CT26, whereas the other was an interstitium‐rich cancer model using the human pancreatic cancer cell line BxPC3. With the latter model, we previously demonstrated therapeutic effects of combined use of TGF‐β inhibitor on nanoparticles.( 7 ) Using these models, we investigated the effects of SMWI on the distribution of 2 MDa dextran, a model of nanoparticles with an estimated hydrodynamic diameter of 50 nm.( 6 ) The Matrigel plug assay and tumor model experiments revealed that TGF‐β inhibitor increased extravasation of 2 MDa dextran in pericyte‐covered neovasculature, whereas sorafenib increased that in vasculature with less pericyte coverage. These findings are important for determination of the optimal choice of angiogenic regulators in combination with nanoparticles for chemotherapy of cancer in general.
Materials and Methods
Reagents and antibodies. TGF‐β inhibitor was purchased from Calbiochem (San Diego, CA, USA; LY364947, catalog no. 616451), imatinib was from Novartis Pharma (Tokyo, Japan), and sorafenib was from Bayer Healthcare (West Haven, CT, USA). These compounds were diluted in dimethyl sulfoxide to 5, 25, and 10 mg/mL, respectively, as stock solutions. Fluorescein isothiocyanate (FITC)‐conjugated dextran of 2 000 000 Da (2 MDa) was obtained from Sigma‐Aldrich (St Louis, MO, USA). The antibody to platelet endothelial cell adhesion molecule (PECAM)‐1 was from BD PharMingen (San Diego, CA, USA), that to NG2 was from Chemicon (Temecula, CA, USA), and that to smooth muscle α‐actin (SMA) (Cy3‐conjugated) was from Sigma‐Aldrich. AlexaFluor‐conjugated secondary antibodies were purchased from Invitrogen Molecular Probes (Eugene, OR, USA).
Cancer cell lines and animals. The BxPC3 human pancreatic adenocarcinoma cell line was obtained from the American Type Culture Collection (Manassas, VA, USA), and was grown in RPMI‐1640 medium supplemented with 10% fetal bovine serum. The murine colon adenocarcinoma CT26 cell line was from the National Cancer Center Research Institute, Japan, and was cultured in Dulbecco's modified Eagle's medium (Sigma‐Aldrich) containing 10% fetal bovine serum. BALB/c mice and BALB/c nude mice, 5–6 weeks of age, were obtained from Sankyo Laboratory (Tokyo, Japan) and Charles River Laboratories (Tokyo, Japan), respectively.
In vivo Matrigel plug assay and cancer models. Matrigel plugs were created by mixing 0.2 mg/mL recombinant human VEGF‐A (VEGF165; R & D Systems, Minneapolis, MN, USA), 1 mg/mL FGF‐2 (R & D Systems), and 0.1 mg/mL heparin (Aventis Pharma, Tokyo, Japan) by pipetting, in combination with regular Matrigel (catalogue no. 354234; BD Biosciences, Franklin Lakes, NJ, USA). Matrigel (400 µL per plug; one plug per mouse) was injected subcutaneously into the abdominal region of BALB/c mice. Each Matrigel plug was harvested on day 7 and frozen directly in dry‐iced acetone for immunohistochemistry. As cancer models, 5 × 106 BxPC3 cells or 1 × 106 CT26 cells were implanted by subcutaneous injection into the abdominal region of BALB/c nude and normal BALB/c mice and allowed to grow for 3 weeks and 1 week, respectively, until reaching the proliferative phase. For the in vivo permeability assay, TGF‐β inhibitor at 1 mg/kg, imatinib at 50 mg/kg, or sorafenib at 40 mg/kg was administered as one shot intraperitoneally 18 h before injection of dextran. Dextran was administered intravenously via lateral tail veins 6 h before harvesting of samples. For perfusion study in the tumor tissues, dextran of 2 MDa was administered intravenously, at 24 h after SMWI‐administration and 10 min before harvesting, and the excised samples were directly fixed in formalin. All experimental protocols were carried out in accordance with the policies of the Animal Ethics Committee at the University of Tokyo.
Histology and immunohistochemistry. The excised samples were either directly frozen in dry‐iced acetone for immunohistochemistry, or fixed overnight in 4% paraformaldehyde and then paraffin embedded to prepare them for hematoxylin–eosin (HE) staining or perfusion study in the tumor tissues. Frozen samples were further sectioned at 10 µm thickness in a cryostat, briefly fixed with 10% formalin, and then incubated with primary and fluorescent secondary antibodies. Samples were observed with a LSM510 Meta confocal microscope (Zeiss, Thornwood, NY, USA) for immunohistochemistry, and with an AX80 microscope (Olympus, Tokyo, Japan) for HE staining.
Quantification. Areas in Matrigel plugs that were PECAM‐1‐positive, double‐positive for PECAM‐1 and SMA, and FITC–dextran‐positive in confocal micrographs (n = 15), or lengths of FITC–dextran‐positive structure in the tumor tissues (n = 12) were measured using Adobe Photoshop software (Adobe Systems, San Jose, CA, USA) and ImageJ software (freeware distributed by the National Institutes of Health, USA). Pericyte coverage was quantified as the ratio of PECAM‐1/SMA‐double‐positive areas to PECAM‐1‐positive areas, as described previously.( 7 ) Results were further analyzed statistically by Student's t‐test using Microsoft Excel software (Microsoft, Redmond WA, USA).
Results
We initially carried out the Matrigel plug assay in vivo, in which regular Matrigel was mixed with VEGF‐A, FGF‐2, and heparin( 16 ) to investigate the effects of three SMWI on the extravasation of 2 MDa dextran (Fig. 1). Marked induction of pericyte‐covered mature neovasculature was observed in the gel plug after a 7‐day incubation in mice, as we reported previously.( 16 ) Pericytes were determined to be SMA‐positive cells in a Matrigel plug assay. In this model, administration of TGF‐β inhibitor decreased pericyte coverage of the neovasculature and significantly enhanced the distribution of 2 MDa dextran. This observation was consistent with our previous study, in which we used animal models of pancreatic adenocarcinoma and diffuse‐type gastric cancer.( 7 ) Based on this result, we expected that a decrease in pericytes might induce more extravasation of 2 MDa dextran.
Figure 1.
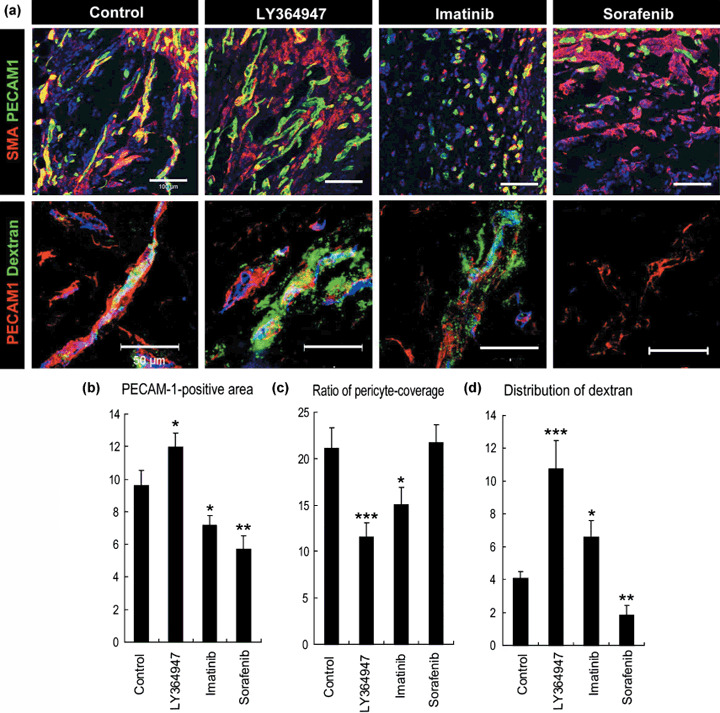
The effects of three types of kinase inhibitors on extravasation of dextran in the Matrigel plug assay. (a) Confocal microscopy analyses. Upper row: staining of platelet endothelial cell adhesion molecule (PECAM)‐1‐positive endothelium in green and smooth muscle α‐actin (SMA)‐positive pericytes in red. Scale bars = 100 µm. Lower row: distribution of 2 MDa dextran in green and PECAM‐1‐positive endothelium in red. Scale bars = 50 µm. (b–d) Results of quantification (n = 15) of areas of endothelium (b, in percentage in one microscopic view), ratio of pericyte‐covered endothelium (c, in percentage), and dextran distribution (d, in percentage in one microscopic view). Bars in the graphs represent standard errors. *P < 0.05; **P < 0.01; and ***P < 0.001.
To confirm this, we compared the effects of imatinib administration, which inhibits PDGF signaling and may therefore decrease pericyte coverage. However, administration of imatinib decreased the total accumulation of 2 MDa dextran compared with TGF‐β inhibitor (Fig. 1). Although imatinib actually decreased pericyte coverage to the same level of TGF‐β inhibitor, it also decreased PECAM‐1‐positive endothelium together with pericyte coverage. These findings of morphological analysis were consistent with those noted in a previous report.( 17 ) TGF‐β inhibitor maintained the area of PECAM‐1‐positive endothelium and may therefore be superior to imatinib. In addition, although VEGF inhibition was expected to increase drug delivery, based on the results of previous studies,( 8 ) sorafenib nearly eliminated the influx of 2 MDa dextran and resulted in far less accumulation of it. This result can be explained by the potent reduction of PECAM‐1‐positive endothelium and increase in pericytes as sleeves. These morphological changes induced by VEGF inhibition were also consistent with previous reports.( 18 )
Although the neovasculature inside the gel plugs was as described above, the vasculature in the regions surrounding the gel plugs, or sites of acute inflammation in reaction to the plugs as foreign bodies (Fig. 2), exhibited different patterns. Compared to the vasculature inside the gel plug, that in regions around the plugs was denser and more tortuous, and was accompanied by pericytes to a smaller extent. These phenotypes resembled those of the vasculature in conventional animal models of tumors, such as the CT26 model, as we describe later in this report. We termed these two regions the ‘intraplug’ and ‘periplug’ regions, respectively, after the established terminology in oncology, ‘intratumoral’ and ‘peritumoral’.
Figure 2.
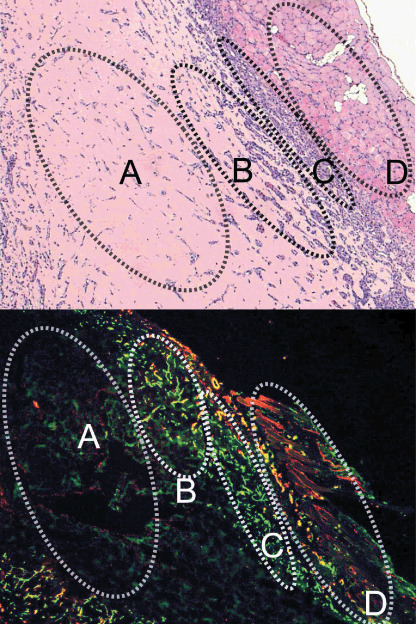
Low‐magnification view of the Matrigel plug and surrounding regions in sections with hematoxyline‐eosin (HE) staining (upper) and with immunohistochemistry (lower). (a) Avascular area, (b) vascularized intraplug region, (c) periplug region, and (d) normal tissue. Green, platelet endothelial cell adhesion molecule‐1; red, smooth muscle α‐actin.
Functionally, the vasculature in periplug regions was leaky to 2 MDa dextran in the control condition, that is, without any modulation by SMWI (Fig. 3). Surprisingly, the effects of SMWI on neovasculature in the periplug regions were quite different from those in the intraplug regions. In the periplug regions, pericyte coverage of the neovasculature was far less than in the intraplug region, even in the control condition. In this periplug region, neither TGF‐β inhibitor nor imatinib significantly altered pericyte coverage. Consequently, these compounds did not alter the accumulation of 2 MDa dextran. Sorafenib, on the other hand, did increase pericyte coverage, and increased the accumulation of 2 MDa dextran. This increase in extravasation was consistent with previous reports on the effects of VEGF inhibition.( 8 )
Figure 3.
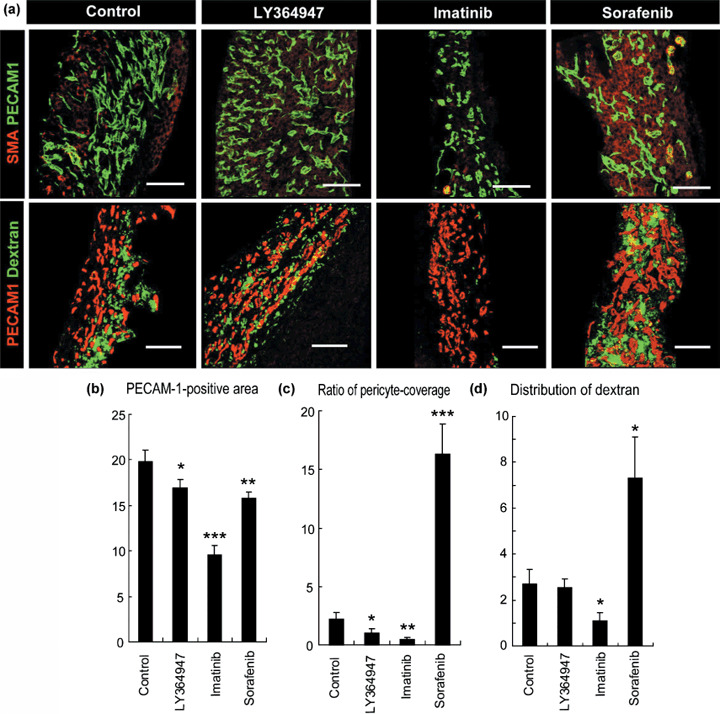
Effects of three types of kinase inhibitors on extravasation of dextran from vasculature in the periplug region. (a) Confocal microscopy analyses. Upper row: staining of platelet endothelial cell adhesion molecule (PECAM)‐1‐positive endothelium in green and smooth muscle α‐actin (SMA)‐positive pericytes in red. Lower row: distribution of 2 MDa dextran in green and PECAM‐1‐positive endothelium in red. Scale bars = 100 µm. (b–d) Results of quantification (n = 15) of areas of endothelium (b, in percentage in one microscopic view), ratio of pericyte‐covered endothelium (c, in percentage), and dextran distribution (d, in percentage in one microscopic view). Bars in the graphs represent standard errors. *P < 0.05; **P < 0.01; and ***P < 0.001.
We subsequently compared these findings in the Matrigel plug assay with those in two subcutaneous tumor xenograft models. We used the CT26 cell line derived from murine colon cancer and the BxPC3 cell line derived from human pancreatic adenocarcinoma (Fig. 4). HE staining of CT26 xenografts revealed a well‐vascularized medullary histological pattern with little tumor stroma, whereas that of BxPC3 xenografts revealed a stroma‐rich histology. Immunostaining of PECAM‐1 and SMA confirmed this stroma‐rich characteristic of the BxPC3 model. Although the BxPC3 model grew more slowly than the CT26 model, the BxPC3 model also reached the proliferative phase. Compared to the BxPC3 model, the CT26 model required one‐fifth of the number of inoculating cells and one‐third of the duration to reach the proliferative phase, which was 1 week for the CT26 model and 3 weeks for the BxPC3 model (data not shown). These differences may well be due to the differences in requirements for induction of stromal components from host animals, as well as rates of proliferation of tumor cell lines.
Figure 4.
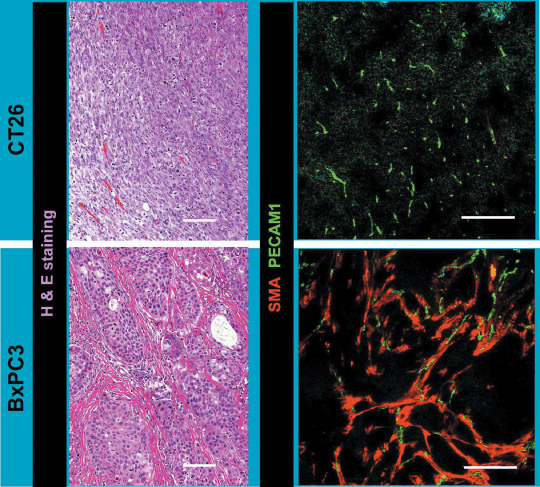
Two animal tumor models using CT26 and BxPC3 cell lines. Histological examination of tumor models by hematoxylin–eosin staining and immunohistochemistry with platelet endothelial cell adhesion molecule (PECAM)‐1 in green and smooth muscle α‐actin (SMA) in red. Scale bars = 100 µm.
We then tested the alterations in vascular phenotypes as well as accumulation of 2 MDa dextran with or without SMWI in these tumor models (Fig. 5). We here used NG2 as the pericyte marker (Fig. 5a), because SMA‐positive cells (i.e. myofibroblasts) are abundant especially in the stroma of BxPC3 tumor (Fig. 4). In the CT26 model, sorafenib did increase the pericyte‐covered vasculature, whereas other SMWI did not increase the pericytes. Imatinib decreased endothelial cells. These observations in the CT26 tumor model were consistent with those in the periplug region of the Matrigel plug. In the BxPC3 model, pericyte coverage was less with LY364947 and imatinib, and endothelial cells were decreased with imatinib and sorafenib. These findings in the BxPC3 tumor model were consistent with those in the intraplug region. Accordingly, 2 MDa dextran was diffusely distributed in tumor tissue without any treatment in the CT26 model, whereas almost no leakage of dextran was observed in the BxPC3 model (Fig. 5b). Sorafenib exhibited the best effect in the CT26 model, whereas TGF‐β inhibitor did in the BxPC3 model. The latter result was consistent with the findings of our previous work using nanoparticles including PEGylated liposomes incorporating doxorubicin (Doxil) of approximately 100 nm in diameter, which exhibited antitumor effects in the BxPC3 model only when combined with TGF‐β inhibitor.( 7 ) We also tested the effects of Doxil with or without TGF‐β inhibitor in the CT26 model. Monotherapy with Doxil at 8 mg/kg almost completely inhibited tumor growth, and combined administration of TGF‐β inhibitor did not yield any significant additional effects (data not shown). These findings were consistent with those observed in the Matrigel plug assay. The effects of combined use of imatinib were also consistent with those in the Matrigel plug assay.
Figure 5.
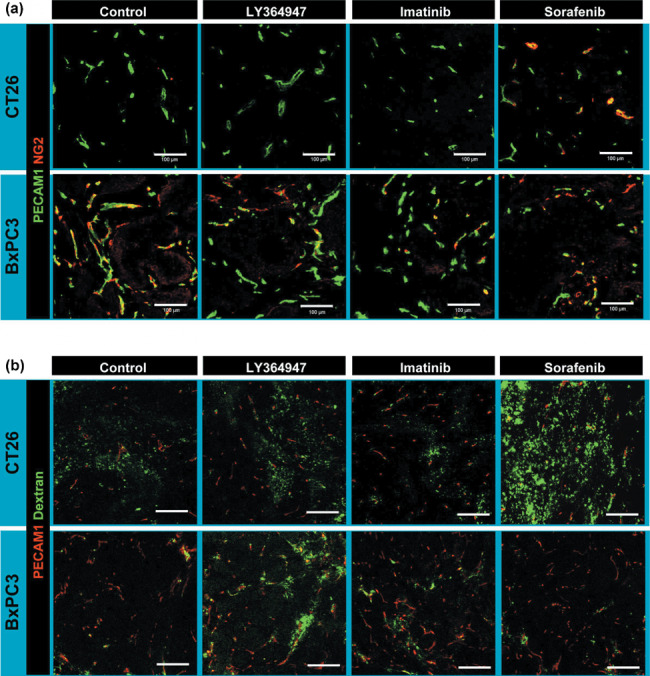
Effects of three kinase inhibitors in the tumor models. (a) Vascular phenotypes revealed by immunohistochemistry. Green, platelet endothelial cell adhesion molecule (PECAM)‐1; red, NG2. (b) Extravasation of 2 MDa dextran from vasculature. Dextran in green and PECAM‐1 in red. Scale bars = 100 µm.
Increased accumulation of dextran in these tumor models at 7 h after injection, by sorafenib in the CT26 tumor and by LY364947 in the BxPC3 tumor, can also be explained by an increase in the amount of vasculature with perfusion, not only by an increase in leakage. To test this possibility, we examined changes in perfusion by intravascular existence of dextran at only 10 min after administration, because dextran of 2 MDa should basically remain inside vasculature at that time after injection.( 6 ) As shown in Figure 6, the lengths of vasculature with blood flow were not altered in the conditions exhibiting increased accumulation of dextran, that is, by sorafenib treatment in the CT26 tumor and by LY364947 treatment in the BxPC3 tumor. Therefore, the increased accumulation of dextran in these conditions may largely be due to an increase in vascular leakiness.
Figure 6.
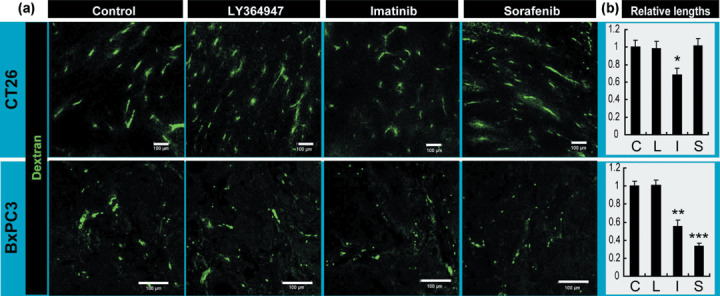
Perfusion study in the tumor models. (a) Tumor vessels with perfusion were determined by the existence of dextran in green, administrated at 10 min before harvesting. Scale bars = 100 µm. (b) Relative lengths of vessels with perfusion. C, control; I, imatinib; L, LY364947; S, sorafenib. Bars represent standard errors. *P < 0.05; **P < 0.01; and ***P < 0.001.
Discussion
We have previously shown that use of short‐acting SMW TGF‐β inhibitor can increase the distribution of nanoparticles in stroma‐rich tumors by increasing the leakiness of the tumor neovasculature.( 7 ) By virtue of the brief duration of SMWI effects, potential side effects can be decreased due to long‐term suppression of essential signaling pathways. There are still a number of SMWI that can be used for manipulation of tumor neovasculature via their effects on pericytes or endothelium. We therefore compared the effects of two of these SMWI, imatinib and sorafenib.
Combined use of VEGF inhibition has been reported to have potent effects on drug delivery into tumor tissues.( 19 ) The underlying mechanism for this has been explained by the vascular ‘normalization’ theory,( 8 ) or decreased interstitial fluid pressure by decreased leakiness of tumor vasculature, via a decrease in endothelial cells and increase in pericyte coverage. Consistent with this, VEGF inhibition by sorafenib had significant effects on the retention of 2 MDa dextran in the periplug regions and in CT26 tumor, where vasculature showed less pericyte coverage and denser endothelium than in normal tissues. However, VEGF inhibition significantly decreased retention of the same dextran in adjacent areas, the intraplug region, and in BxPC3 tumor. Vascular phenotypes in these regions were characterized by more pericyte coverage and sparser endothelium, that is, they were more ‘normal’ than those in the periplug region and in CT26 tumor.
One of the differences between these two kinds of vasculature was the blood flow in the vasculature after sorafenib treatment. In CT26 tumor after sorafenib treatment, blood flow was maintained, whereas the flow ceased in sorafenib‐treated BxPC3 tumor. These differences may partially be because of differences in sensitivity of the endothelium to the change in VEGF signaling, known to be at least due to differences in expression levels of VEGFR2.( 18 )
Another apparent difference in these tumor models was pericyte coverage before drug administration. Less pericyte coverage has been reported to result in more leakiness.( 11 , 12 , 13 ) The degrees of dextran accumulation in all control conditions (i.e. without modification by SMWI) are consistent with the degrees of pericyte coverage. Increased dextran accumulation in LY364947‐treated BxPC3 tumors can also be explained by decreased pericyte coverage, not by normalization. Both blood perfusion (Fig. 6) and interstitial fluid pressure, which we previously reported,( 7 ) did not differ with or without LY364947 treatment in BxPC3 tumor. These findings suggest that we may need different approaches, such as the use of TGF‐β inhibitor to increase drug delivery (at least for nanoparticles), to develop effective treatment for tumors with originally ‘more normal’ tumor vasculature. Note, however, that these more normal vessels in tumors might not be completely normal, because TGF‐β inhibitor did not alter the accumulation of nanoparticles in true normal tissues, as we previously reported.( 7 )
Regarding the degree of original pericyte coverage in tumor vasculature, an increase in the amount of stromal components in tumor tissue may result in an increase in pericyte coverage. In a previous study, we found that the presence of FGF‐2 together with VEGF‐A enhances mature neovascularization compared with VEGF‐A alone.( 16 ) In addition to FGF‐2, a set of signaling molecules is needed to recruit and to induce proliferation of pericytes. These include PDGF‐BB( 12 , 13 ) (homodimer of PDGF‐B chain) and TGF‐β.( 11 , 20 ) These signaling molecules are reported to be secreted from components of the tumor stroma and, above all, cancer‐associated fibroblasts( 21 , 22 , 23 ) and macrophages.( 24 ) Tumors with more stroma, including fibroblasts and immune cells, have more pericyte coverage of the vasculature with greater maturity and less leakiness. Although chemoresistance of tumors has been largely investigated from the aspect of drug sensitivity of tumor cells per se, it is possible that the histological pattern of the tumor tissues may also constitute a reason for chemoresistance, because of insufficient drug delivery to the tumor cells.
The Tie2–angiopoietin signaling pathway is also known to be involved in vessel maturation and to affect pericytes.( 25 ) Because there are no SMW compounds available to inhibit this signaling pathway, we tested the effects of one‐shot Tie2‐Fc chimeric protein at 50 mg/kg bodyweight with 2 MDa dextran in the Matrigel plug assay, but no significant effects on accumulation of 2 MDa dextran were observed (M.R. Kano, unpublished data, 2008). There are two possible explanations for this observation: spatial and temporal. According to the spatial explanation, because Tie2–angiopoietin signaling occurs between the endothelium and pericytes and thus outside the vessel lumen, the Fc chimera, which is of fairly large molecular size and may therefore be retained inside vessel lumens, is not able to affect signaling. The other drugs used in the present study were all SMWI, which may easily exit the vessel lumen and penetrate the perivascular tissues. The second explanation is temporal. Although this signaling pathway is known to be deeply involved in development, whether it is also involved in the maintenance of endothelial‐mural structure is not known. Because we observed the effects of drugs only at 24 h after administration, it is possible that other inhibitors inhibited only the maintenance functions of the signaling pathways, and not functions related to development. Administration of TGF‐β inhibitor did not significantly affect the vasculature in normal organs,( 7 ) so it is possible that the neovasculature requires larger amounts of signaling molecules to maintain structure than do established vessels.
In conclusion, the appropriate strategy for optimization of tumor vasculature for drug delivery system using nanoparticles may not be uniform, and may depend on tumor type, including differences in the degree of pericyte coverage of tumor vasculature.
Acknowledgments
We thank Dr Peter Baluk, Cardiovascular Research Institute, University of California, San Francisco (UCSF), for advice about perfusion. This work was supported by Grant‐in‐Aid for Scientific Research (KAKENHI 19790282 and 17016011) from the Ministry of Education, Culture, Sports, and Technology of Japan (MEXT), and a Health Labor Sciences Research Grant from the Ministry of Health, Labor, and Welfare of Japan.
References
- 1. Giroux V, Malicet C, Barthet M et al . p8 is a new target of gemcitabine in pancreatic cancer cells. Clin Cancer Res 2006; 12: 235–41. [DOI] [PubMed] [Google Scholar]
- 2. Merriman RL, Hertel LW, Schultz RM et al . Comparison of the antitumor activity of gemcitabine and ara‐C in a panel of human breast, colon, lung and pancreatic xenograft models. Invest New Drugs 1996; 14: 243–7. [DOI] [PubMed] [Google Scholar]
- 3. Rothenberg ML, Moore MJ, Cripps MC et al . A phase II trial of gemcitabine in patients with 5‐FU‐refractory pancreas cancer. Ann Oncol 1996; 7: 347–53. [DOI] [PubMed] [Google Scholar]
- 4. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 5. Jain RK. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res 1990; 50: 814S–19S. [PubMed] [Google Scholar]
- 6. Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst 2006; 98: 335–44. [DOI] [PubMed] [Google Scholar]
- 7. Kano MR, Bae Y, Iwata C et al . Improvement of cancer‐targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF‐β signaling. Proc Natl Acad Sci USA 2007; 104: 3460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005; 307: 58–62. [DOI] [PubMed] [Google Scholar]
- 9. Wilhelm S, Carter C, Lynch M et al . Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006; 5: 835–44. [DOI] [PubMed] [Google Scholar]
- 10. Bergers G, Song S. The role of pericytes in blood‐vessel formation and maintenance. Neuro Oncol 2005; 7: 452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Von Tell D, Armulik A, Betsholtz C. Pericytes and vascular stability. Exp Cell Res 2006; 312: 623–9. [DOI] [PubMed] [Google Scholar]
- 12. Abramsson A, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF‐B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest 2003; 112: 1142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lindblom P, Gerhardt H, Liebner S et al . Endothelial PDGF‐B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev 2003; 17: 1835–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buchdunger E, Zimmermann J, Mett H et al . Inhibition of the Abl protein‐tyrosine kinase in vitro and in vivo by a 2‐phenylaminopyrimidine derivative. Cancer Res 1996; 56: 100–4. [PubMed] [Google Scholar]
- 15. Bergers G, Song S, Meyer‐Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest 2003; 111: 1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kano MR, Morishita Y, Iwata C et al . VEGF‐A and FGF‐2 synergistically promote neoangiogenesis through enhancement of endogenous PDGF‐B–PDGFRβ signaling. J Cell Sci 2005; 118: 3759–68. [DOI] [PubMed] [Google Scholar]
- 17. Vlahovic G, Rabbani ZN, Herndon JE, Dewhirst MW, Vujaskovic Z. Treatment with Imatinib in NSCLC is associated with decrease of phosphorylated PDGFR‐β and VEGF expression, decrease in interstitial fluid pressure and improvement of oxygenation. Br J Cancer 2006; 95: 1013–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mancuso MR, Davis R, Norberg SM et al . Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest 2006; 116: 2610–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hurwitz H, Fehrenbacher L, Novotny W et al . Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–42. [DOI] [PubMed] [Google Scholar]
- 20. Hirschi KK, Rohovsky SA, D’Amore PA. PDGF, TGF‐β, and heterotypic cell–cell interactions mediate endothelial cell‐induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol 1998; 141: 805–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med 2008; 5: e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Micke P, Ostman A. Tumour–stroma interaction: cancer‐associated fibroblasts as novel targets in anti‐cancer therapy? Lung Cancer 2004; 45: S163–75. [DOI] [PubMed] [Google Scholar]
- 23. Bhowmick NA, Moses HL. Tumor–stroma interactions. Curr Opin Genet Dev 2005; 15: 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res 2006; 66: 605–12. [DOI] [PubMed] [Google Scholar]
- 25. Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res 2005; 97: 512–23. [DOI] [PubMed] [Google Scholar]