

Abstract
(Cancer Sci 2010; 101: 728–734)
Methotrexate (MTX) has been used to treat various hematological malignancies. Since MTX prevents tumor cells from proliferating by inhibiting dihydrofolate reductase (DHFR), DHFR expression is a key determinant of resistance to MTX in malignant hematological tumor cells. The antiproliferative effect of MTX was significantly enhanced by the knockdown of DHFR expression by siRNA in Jurkat cells. Therefore, a novel strategy down‐regulating DHFR expression seems promising for enhancing sensitivity to MTX. We found that SU9516, a cyclin‐dependent kinase inhibitor, reduced the expression of both DHFR mRNA and protein. Moreover, we found that DHFR promoter activity was attenuated by SU9516 dependent on the E2F site. Finally, pretreatment with SU9516 significantly enhanced sensitivity to MTX in a colony formation assay. We conclude that a combination of cyclin‐dependent kinase inhibitors and MTX may be useful for overcoming resistance to MTX.
Methotrexate (MTX), a classical anti‐folate drug, has been used in the treatment of leukemia, osteosarcoma, and breast cancer. Lower doses of MTX are also used as an immunosuppressant for a variety of other diseases including graft‐versus‐host disease, rheumatoid arthritis, and psoriasis.( 1 ) MTX was originally designed to inhibit dihydrofolate reductase (DHFR) by way of polyglutamated derivatives, which inhibit nucleotide synthesis, the cell cycle, and the proliferation of tumor cells.( 2 , 3 ) DHFR is a key enzyme for intracellular folate metabolism, and functions to regenerate tetrahydrofolate from dihydrofolate as a product of thymidylate synthase. Therefore, DHFR represents an important target for anticancer chemotherapy. As a consequence of DHFR’s inhibition, intracellular levels of tetrahydrofolate coenzymes are decreased, resulting in the inhibition of thymidylate and consequently, DNA biosynthesis.( 1 )
Drug resistance is occasionally a limiting factor in successful chemotherapy against cancer. Actually, drug resistance in the clinical use of MTX for chemotherapy is often observed. The mechanisms of resistance to MTX have been studied extensively. Five common mechanisms of resistance have been identified: (i) an increase in DHFR; (ii) decreased uptake due to impaired transport; (iii) decreased retention as a consequence of lack of polyglutamylation; (iv) an altered (mutated) DHFR that binds MTX less than the normal enzyme; and (v) an increased level of a lysosomal enzyme, γ‐glutamyl hydrolase, that hydrolyses MTX polyglutamates.( 4 , 5 , 6 )
We focused on the increase in DHFR expression in these resistant mechanisms. MTX is a tight‐binding inhibitor of DHFR, and the amount of MTX required to inhibit DHFR activity increases in proportion to the amount of DHFR in tumor cells. An important mechanism of resistance to MTX is an increase in DHFR activity due to high expression of the DHFR gene.( 7 , 8 , 9 ) Therefore, small compounds that repress DHFR expression may be useful as chemosensitizers in combination with MTX.
On the other hand, the expression of DHFR has been assumed to be controlled by the transcription factor E2F.( 10 ) Furthermore, retinoblastoma (RB) protein is known to bind and inhibit the E2F family.( 11 ) Actually, it has been reported that RB protein, which acts as a negative regulator of cell cycle transition, suppresses the expression of DHFR.( 12 ) When RB is phosphorylated by cyclin‐dependent kinase (CDK)‐cyclins, E2F is released from the complex and binds to a cis‐element in the promoter region of various genes involved in cell proliferation and DNA synthesis. In other words, CDK inhibitors such as p16INK4a and the p21Waf1 family can activate RB function and inhibit E2F function.( 13 , 14 )
Based on these findings, we investigated whether a CDK inhibitor acted as a suppressor of DHFR, and whether it could be used to enhance sensitivity to MTX. Our results demonstrated that SU9516, a small chemical CDK inhibitor,( 15 , 16 , 17 , 18 ) effectively reduced DHFR expression and enhanced sensitivity to MTX in human leukemic cells.
Materials and Methods
Reagents. SU9516, Purvalanol A, Cdk4/6 Inhibitor IV, and MTX were purchased from Calbiochem (San Diego, CA, USA). SU9516, Purvalanol A, and Cdk4/6 Inhibitor IV were dissolved in dimethyl sulfoxide (DMSO). DMSO alone was used as a control. The maximum volume (%) of DMSO in the assays was 0.1%. MTX was dissolved in phosphate‐buffered saline (PBS). PBS alone was used as a control.
Cell culture and cell growth. Human T‐cell leukemia Jurkat cells and CCRF‐CEM cells, and human erythroleukemia K562 cells were cultured in RPMI‐1640 medium (Nissui Pharmaceutical, Tokyo, Japan) supplemented with 10% heat‐inactivated fetal bovine serum (FBS), 1 mmol/L l‐glutamine, and antibiotics (penicillin and streptomycin). The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 and regularly passaged to maintain exponential growth. For the experiments described herein, the cells were exposed to designated concentrations of the CDK inhibitors and/or MTX. To measure the cell growth, the cells were seeded at a density of 1 × 104 cells in a 12‐well plate. SU9516 was added at various concentrations simultaneously. Cell growth was compared with a control culture with equivalent DMSO alone. From 24 to 72 h after the treatment, the numbers of viable cells were counted using a Trypan blue dye exclusion test. Following these treatments, cells or cell pellets were washed free of the drug(s) prior to the experiments.
Small‐interfering RNA‐mediated knockdown of endogenous DHFR. A synthetic ready‐to‐use siRNA (21 nucleotides) complementary to a region of a DHFR‐specific domain and Silencer Negative Control #1 siRNA were purchased from Ambion (Austin, TX, USA). For electroporation, 5 μmol of dsRNA was added to prechilled 0.4‐cm electrode gap cuvettes (Bio‐Rad, Hercules, CA, USA). Jurkat cells (1.5 × 107) were resuspended to 3 × 107 cells/mL in cold OPTI‐MEM, added to the cuvettes, mixed, and pulsed once at 280 V and 1000 μF with a Gene Pulser electroporator Xcell (Bio‐Rad). The cells were plated into six‐well culture plates containing 2 mL of complete medium and incubated for 48 h at 37°C in a humidified 5% CO2 chamber. The cells were then harvested for Western blotting.
Western blotting. The Western blot analysis was performed as described previously.( 19 ) Briefly, the cells were treated with the indicated concentrations of the CDK inhibitors for predetermined periods. The cultured cells were washed once with PBS, lysed with 100–150 μL of sodium dodecyl sulfate (SDS) buffer (50 mM Tris‐HCl [pH 7.5] 1% SDS, 0.5 mM PMSF, 2 μg/mL aprotinin, 2 μg/mL leupeptin, and 1 mM DTT) by incubation on ice, and clarified by centrifugation at 18 000g for 15 min at 4°C. The whole‐cell lysates were boiled for 5 min in the presence of SDS sample buffer, electrophoresed with 12% (for DHFR detection) or 7% (for RB detection) SDS‐polyacrylamide gels, and electrophoretically transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). A mouse monoclonal anti‐DHFR antibody (BD Biosciences, San Jose, CA, USA), a rabbit polyclonal anti‐phospho‐RB (Ser780) antibody (Cell Signaling, Danvers, MA, USA), and a mouse monoclonal anti‐glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) antibody (BD Biosciences) as a loading control were used as the primary antibodies. For the electroporation of siRNA, a mouse monoclonal anti‐β‐actin antibody (Oncogene Res. Prod., San Diego, CA, USA) was used. The signal was then developed with the enhanced chemiluminescence system (GE Healthcare, Buckinghamshire, UK) for detection.
Colony formation assay. To study the effect of DHFR siRNA, Jurkat cells were transfected with the DHFR siRNA or negative control siRNA in 10‐cm dishes and washed in RPMI‐1640 medium after 48 h. Next, they were treated with various concentrations of MTX, and 200 cells were inoculated into soft agarose in six‐well dishes containing RPMI‐1640 medium with 10% FBS in triplicate. The soft agarose was adjusted to 0.53% for the lower layer and 0.4% for the upper layer. The cells were incubated without a change of medium for 14–21 days, and colony numbers per dish were counted. To study the effect of SU9516, Purvalanol A, and Cdk4/6 Inhibitor IV, the Jurkat cells were treated with designated concentrations of the CDK inhibitors in 10‐cm dishes for 24 h, and washed in RPMI‐1640 medium. Next, they were treated with various concentrations of MTX, and seeded in soft agarose in six‐well dishes containing RPMI‐1640 medium with 10% FBS in triplicate. As mentioned above, cells were incubated in growth medium for 14–21 days, and the number of colonies per dish was counted. To study the effect on CCRF‐CEM cells and K562 cells of SU9516, these cells were also treated with designated concentrations of SU9516 in 10‐cm dishes for 24 h, and washed in RPMI‐1640 medium. In the same way as in the colony formation assay, the cells were incubated without a change of medium for 14–21 days, and the number of viable colonies was counted. The data are presented as a percentage compared to the untreated control cells.
Quantitative real‐time PCR analysis. Jurkat cells were treated with DMSO or various concentrations of SU9516 as for 24 h. Alternatively, the cells were treated with DMSO or 2 μM of SU9516 for the indicated hours. After the treatment, the cells were lysed, and total RNA was extracted using Sepasol‐RNA I (Nacalai Tesque, Kyoto, Japan), according to the manufacturer’s instructions. Reverse‐transcribed cDNA was constructed from 10 μg of total RNA using the SuperScript reverse transcriptase (Invitrogen, Carlsbad, CA, USA) and oligo (dT) primer (Toyobo, Osaka, Japan) according to the manufacturer’s instructions. For the analysis of mRNA expression, we performed a real‐time PCR. The reaction mixture was incubated at 42°C for 50 min, and then at 70°C for 15 min to stop the reaction. An equivalent volume (1 μL) of cDNA solution was used for the quantification of specific cDNAs by quantitative real‐time PCR. The Taqman probes and DHFR‐specific primers were purchased from Applied Biosystems (Foster, CA, USA). Quantitative real‐time PCR was carried out using the RT‐PCR system GeneAmp 5700 (Applied Biosystems). The relative mRNA expression data were normalized to GAPDH (Applied Biosystems) expression. Each sample was tested in triplicate, and the level of DHFR mRNA was normalized to that of GAPDH mRNA.
Preparation of plasmids. The human DHFR promoter–luciferase fusion plasmid, pDHFR‐luc, a kind gift from Dr. K. Ohtani (Tokyo Medical and Dental University, Tokyo, Japan), was constructed as previously described.( 20 ) A QuikChange site‐directed mutagenesis kit (Stratagene Cloning Systems, La Jolla, CA, USA) was used to generate point mutations at the E2F site of the pDHFR reporter plasmid. The primer used to mutate the E2F site in the region from −12 to −1 bp of pDHFR was 5′‐gc ggccacaatt tcgatccaaa cttgaccgcg c‐3′ (the mutated nucleotides are underlined). The vacant control luciferase plasmid pGVB2 was purchased from Nippon Gene (Tokyo, Japan).
Transient DNA transfection and luciferase assay. Jurkat cells were seeded in six‐well plates, and 0.5 μg of pDHFR (wild type and mutant type), or vacant vector plasmid (pGVB2) was transfected by the DEAE dextran method using a CellPhect transfection kit (GE Healthcare). After 24 h of incubation, the cells were treated with or without SU9516, and 48 h after the transfection, they were collected for luciferase assays. Cells were washed twice with PBS and lysed by PGC 50 (Toyo Ink, Tokyo, Japan). The luciferase activity was measured as previously described.( 21 ) Levels of activity were normalized to the amount of protein in cell lysates, and measured using a Bio‐Rad protein assay kit (Bio‐Rad). All luciferase assays were carried out in triplicate, and each assay was done at least twice.
Statistical analysis. Values are expressed as the mean ± SD. All the data were analyzed using the two‐tailed Student’s t‐test. Differences were considered to be statistically significant compared with the control when P‐values were less than 0.05.
Results
The knockdown of DHFR expression contributed to the enhancement of sensitivity to MTX in Jurkat cells. First, we examined whether the knockdown of DHFR expression contributed to the enhancement of sensitivity to MTX in Jurkat cells. The expression of DHFR was efficiently reduced by siRNA transiently transfected using electroporation (Fig. 1a). We subsequently confirmed the sensitivity of Jurkat cells to MTX in vitro with colony formation assays. As shown in Figure 1(b), MTX inhibited the growth of colonies pretreated with DHFR siRNA more strongly than that of negative control siRNA‐treated colonies. Pretreatment with DHFR siRNA significantly enhanced sensitivity to MTX at a concentration of 0.05 μM MTX or more (*P < 0.05, **P < 0.01).
Figure 1.
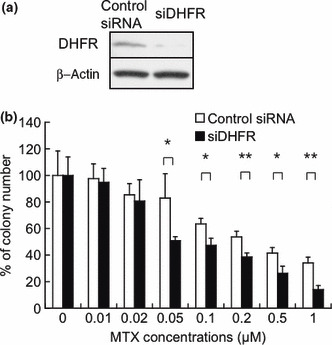
The knockdown of dihydrofolate reductase (DHFR) expression enhanced sensitivity to methotrexate (MTX) in Jurkat cells. (a) Jurkat cells were transfected with DHFR siRNA or negative control siRNA by electroporation. After 48 h, cells were subjected to a Western blot analysis using DHFR and β‐actin antibodies to verify the reduction in DHFR expression by siRNA. (b) Transfected cells were cultured with the indicated concentrations of MTX and subjected to colony formation assays. The data are presented as a percentage compared to the control with an equivalent amount of PBS, and the bars show SDs (*P < 0.05, **P < 0.01). PBS, phosphate‐buffered saline.
SU9516 inhibited growth and DHFR protein levels in Jurkat cells. The expression of DHFR is known to be up‐regulated by E2F. Furthermore, E2F is inactivated by RB protein. Therefore, we have examined whether the expression of DHFR is repressed by a CDK inhibitor, SU9516, which can unphosphorylate RB protein inactivating E2F. First, we examined the effect of SU9516 on the growth of Jurkat cells. Figure 2(a) shows the growth rate of Jurkat cells in the presence or absence of various concentrations of SU9516. A significant dose‐dependent inhibition of cell growth was observed (*P < 0.05, **P < 0.01). After 72 h, cell growth was inhibited to 56%, 31%, 17%, and 9% of controls by SU9516 at 1, 2, 5, and 10 μM, respectively. However, few dead cells were detected by counting the cells using a Trypan blue dye exclusion test in cell growth study and sub‐G1 population were hardly detected by flow cytometry in the concentration of 2 μM SU9516 (data not shown). Next, to confirm the effect of SU9516 on DHFR expression, we carried out a Western blot analysis. We found that the treatment of Jurkat cells with SU9516 repressed the levels of DHFR protein in a dose‐dependent manner (Fig. 2b). DHFR protein started to decrease after treatment with SU9516 at 2 μM or more. SU9516 treatment also inhibited the levels of phospho‐RB (Ser780) protein in a dose‐dependent manner, suggesting that SU9516 activates the RB pathway (Fig. 2c). A time‐course study showed that DHFR protein started to decrease 12 h after the treatment with 2 μM of SU9516 (Fig. 2d).
Figure 2.
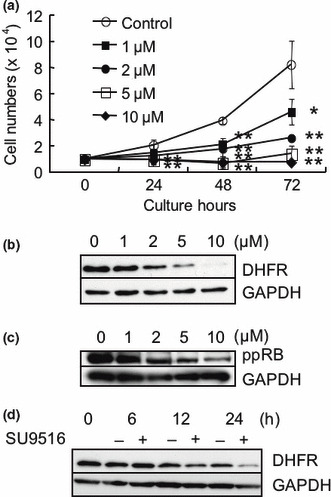
The effects of SU9516 on growth, dihydrofolate reductase (DHFR), and retinoblastoma (RB) proteins in Jurkat cells. (a) Numbers of cells measured every 24 h were compared in the presence or absence of various concentrations of SU9516 by counting the cells a Trypan blue dye exclusion test. The data represent means of triplicate and the bars show SDs (*P < 0.05, **P < 0.01). (b) Whole‐cell extracts from Jurkat cells treated with the indicated concentrations of SU9516 or vehicle control (dimethyl sulfoxide, DMSO) for 24 h were subjected to a Western blot analysis using DHFR and GAPDH antibodies. (c) Whole‐cell extracts from Jurkat cells treated with the indicated concentrations of SU9516 or DMSO for 24 h were subjected to a Western blot analysis using phosphorylated pRB (Ser780) and GAPDH antibodies. (d) Whole‐cell extracts from Jurkat cells treated with DMSO or 2 μM of SU9516 for the periods indicated were subjected to a Western blot analysis using DHFR and GAPDH antibodies. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
SU9516 attenuated DHFR mRNA levels. To ascertain the effect of SU9516 on DHFR mRNA in Jurkat cells, we carried out real‐time quantitative RT‐PCR. We found that DHFR mRNA expression was decreased by 24 h of exposure to SU9516 in a dose‐dependent manner (Fig. 3a). A time‐course study showed that DHFR mRNA expression started to decrease 6 h after the treatment with 2 μM of SU9516 (Fig. 3b).
Figure 3.
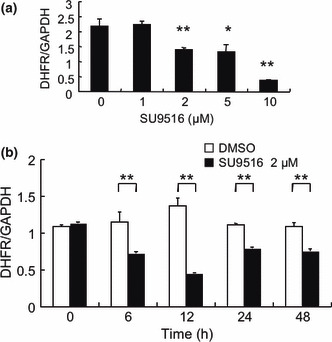
SU9516 decreased dihydrofolate reductase (DHFR) mRNA expression in Jurkat cells. (a) Jurkat cells were treated with the indicated concentrations of SU9516 or dimethyl sulfoxide (DMSO) for 24 h, and DHFR mRNA expression was evaluated using real‐time RT‐PCR as described. Total RNA (10 mg) was probed with DHFR cDNA. The quantity of DHFR mRNA obtained was normalized to the quantity of GAPDH mRNA (*P < 0.05, **P < 0.01). (b) Jurkat cells were treated with DMSO or 2 μM of SU9516 for the periods indicated, and DHFR mRNA was determined using real‐time RT‐PCR as described. Total RNA (10 μg) was probed with DHFR cDNA. The quantity of DHFR mRNA obtained was normalized to the quantity of GAPDH mRNA (*P < 0.05, **P < 0.01). GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
DHFR promoter activity was attenuated by SU9516 and mediated through an E2F site in Jurkat cells. We investigated the effect of SU9516 on DHFR promoter activity in Jurkat cells using a DHFR promoter–luciferase reporter plasmid with a transient luciferase assay. As shown in Figure 4(a), SU9516 inhibited the activity in a dose‐dependent manner. The results suggested that SU9516 inhibited DHFR expression at the promoter level. Moreover, to determine whether an E2F‐binding site of the DHFR promoter is responsible for the repression by SU9516, we generated point mutations at the E2F site of the DHFR promoter–luciferase reporter plasmid. We then studied the effect of SU9516 on the DHFR promoter activity in Jurkat cells using the DHFR promoter–luciferase reporter plasmids with the wild‐type or mutant E2F site. Notably, as compared to the effect on the mutant DHFR promoter, the wild‐type DHFR promoter activity was more strongly inhibited by SU9516 in Jurkat cells, suggesting that SU9516 inhibited DHFR promoter activity through the E2F site (Fig. 4b).
Figure 4.
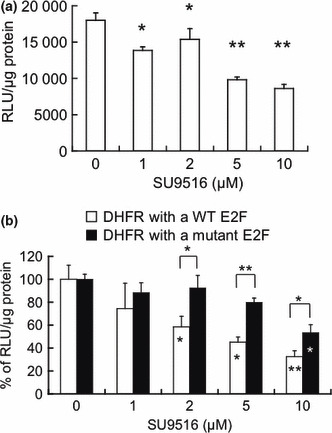
Dihydrofolate reductase (DHFR) promoter activity was inhibited by SU9516 in Jurkat cells. (a) Jurkat cells were transiently transfected with a DHFR promoter–luciferase reporter plasmid or vacant vector for a luciferase assay. Relative luciferase activity: raw light units (RLU) in cell lysates per μg of protein. Data are presented as means ± SD (n = 3; *P < 0.05, **P < 0.01). (b) Jurkat cells were transiently transfected with a vacant vector or a DHFR promoter–luciferase reporter plasmid with a wild‐type (□) or mutant E2F site () for a luciferase assay. Relative luciferase activity: RLU in cell lysates per μg of protein. The data are presented as a percentage compared to the control, and the bars show SDs (*P < 0.05, **P < 0.01).
SU9516 enhanced sensitivity to MTX in Jurkat cells. Next, to assess whether the repression of DHFR expression by the CDK inhibitor contributes to the enhancement of sensitivity to MTX in Jurkat cells, we investigated the sensitivity to MTX in vitro by conducting a colony formation assay after the treatment with DMSO or SU9516. As shown in Figure 5(a), MTX inhibited the growth of colonies pretreated with SU9516 more strongly than that of colonies pretreated with DMSO. Pretreatment of SU9516 significantly enhanced sensitivity to MTX at a concentration of 0.1 μM MTX or more (*P <0.05, **P <0.01).
Figure 5.
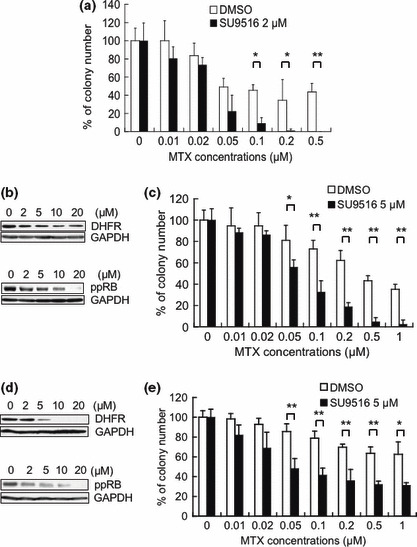
Effect of SU9516 on the sensitivity of Jurkat cells to methotrexate (MTX). (a) Cells were treated with dimethyl sulfoxide (DMSO) or SU9516 (2 μM) for 24 h. Then the cells were cultured with the indicated concentrations of MTX, and subjected to a colony formation assay. The data are presented as a percentage compared to the control with vehicle only, and the bars show SDs (*P < 0.05, **P < 0.01). (b) Whole‐cell extracts from CCRF‐CEM cells treated with the indicated concentrations of SU9516 or DMSO for 24 h were subjected to a Western blot analysis using DHFR, phosphorylated pRB (Ser780), and GAPDH antibodies. (c) CCRF‐CEM cells were treated with DMSO or SU9516 (5 μM) for 24 h. Then the cells were cultured with the indicated concentrations of MTX, and subjected to a colony formation assay. The data are presented as a percentage compared to the control with phosphate‐buffered saline (PBS), and the bars show SDs (*P < 0.05, **P < 0.01). (d) Whole‐cell extracts from K562 cells treated with the indicated concentrations of SU9516 or DMSO for 24 h were subjected to a Western blot analysis using DHFR, phosphorylated pRB (Ser780) and GAPDH antibodies. (e) K562 cells were treated with DMSO or SU9516 (5 μM) for 24 h. Then the cells were cultured with the indicated concentrations of MTX, and subjected to a colony formation assay. The data are presented as a percentage compared to the control with PBS, and the bars show SDs (*P < 0.05, **P < 0.01).
SU9516 attenuated DHFR protein levels and enhanced sensitivity to MTX in CCRF‐CEM cells and K562 cells. Furthermore, it was also observed that treatment with SU9516 repressed the levels of DHFR protein and the levels of phospho‐RB (Ser780) in human T‐cell leukemia CCRF‐CEM cells and human erythroleukemia K562 cells (Fig. 5b,d). Correspondingly, we investigated the sensitivity to MTX in vitro by conducting a colony formation assay after the treatment with DMSO or SU9516 in CCRF‐CEM cells and K562 cells. As shown in Figure 5(c), MTX inhibited the growth of colonies pretreated with SU9516 more strongly than that of colonies pretreated with DMSO in CCEF‐CEM cells. Pretreatment of SU9516 significantly enhanced sensitivity to MTX at a concentration of 0.05 μM MTX or more (*P < 0.05, **P < 0.01). As shown in Figure 5(e), MTX also showed strong growth inhibition as a pretreatment in SU9516 compared with the use of DMSO as a pretreatment in K562 cells. Pretreatment of SU9516 significantly enhanced sensitivity to MTX at a concentration of 0.05 μM MTX or more (*P < 0.05, **P < 0.01). These results might indicate that pretreatment of SU9516 is effective to enhance sensitivity to MTX in not only the T‐cell leukemia cell lines but also erythroleukemia cell lines such as K562 cells.
Other CDK inhibitors, Purvalanol A and Cdk4/6 Inhibitor IV, attenuated DHFR protein levels and enhanced sensitivity to MTX in Jurkat cells. Finally, to assess whether other CDK inhibitors contribute to the enhancement of sensitivity to MTX in Jurkat cells, we investigated the expression of DHFR and the sensitivity to MTX in vitro by conducting a colony formation assay after the treatment with DMSO, Purvalanol A, or Cdk4/6 Inhibitor IV. As shown in Figure 6(a), it was observed that treatment with Purvalanol A or Cdk4/6 Inhibitor IV repressed the levels of DHFR protein and the levels of phospho‐RB (Ser780) in Jurkat cells. Moreover, as shown in Figure 6(b,c), MTX inhibited the growth of colonies pretreated with Purvalanol A or Cdk4/6 Inhibitor IV more strongly than that of colonies pretreated with DMSO. Pretreatment of Purvalanol A significantly enhanced sensitivity to MTX at a concentration of 0.1 μM MTX or more (*P < 0.05, **P < 0.01). Pretreatment of Cdk4/6 Inhibitor IV significantly enhanced sensitivity to MTX at a concentration of 0.05 μM MTX or more (*P < 0.05, **P < 0.01).
Figure 6.
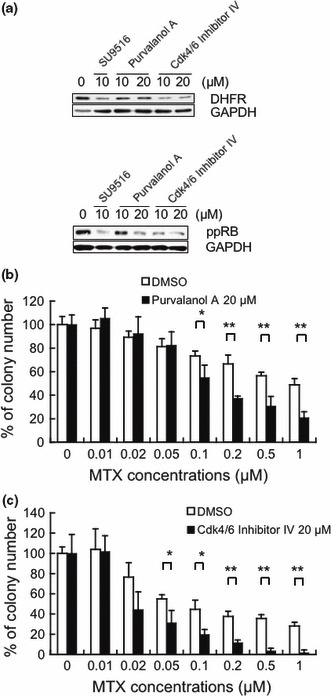
Effect of Purvalanol A and Cdk4/6 Inhibitor IV on the sensitivity of Jurkat cells to methotrexate (MTX). (a) Whole‐cell extracts from Jurkat cells treated with the indicated concentrations of SU9516, Purvalanol A, Cdk4/6 Inhibitor IV, or dimethyl sulfoxide (DMSO) for 24 h were subjected to a Western blot analysis using dihydrofolate reductase (DHFR), phosphorylated pRB (Ser780), and GAPDH antibodies. (b) The cells were treated with DMSO or Purvalanol A (20 μM) for 24 h. Then the cells were cultured with the indicated concentrations of MTX, and subjected to a colony formation assay. The data are presented as a percentage compared to the control with PBS only, and the bars show SDs (*P < 0.05, **P < 0.01). (c) The cells were treated with DMSO or Cdk4/6 Inhibitor IV (20 μM) for 24 h. Then the cells were cultured with the indicated concentrations of MTX, and subjected to a colony formation assay. The data are presented as a percentage compared to the control with phosphate‐buffered saline (PBS) only, and the bars show SDs (*P < 0.05, **P < 0.01).
Discussion
Drug resistance is occasionally a limiting factor in successful anticancer chemotherapy. Mechanisms of resistance to MTX have already been investigated extensively. Most of the mechanisms are supposed to be positively related to the quantity and quality of DHFR, such as an increase in DHFR or mutated DHFR not binding MTX.( 22 , 23 , 24 ) We focused on the high expression of the target enzyme DHFR in these MTX‐resistant mechanisms. In the present study, we found that knockdown of the target enzyme DHFR using siRNA enhanced sensitivity to MTX. Although the human osteosarcoma cell line Saos2 lacking pRB is intrinsically resistant to MTX, reintroducing cDNA encoding pRB into this cell line restored sensitivity to MTX.( 25 ) Therefore, we also considered that the lack of a functional retinoblastoma protein (pRB) might lead to resistance to MTX as a consequence of an increase in DHFR mRNA expression. Since E2F regulates the expression of DHFR, this increase of DHFR has been linked to an increase in free and active E2F, a transcription factor that is bound and inactivated by hypophosphorylated RB protein.
Therefore, we speculated that CDK inhibitors might act as a DHFR repressor through the inhibition of E2F caused by dephosphorylated and activated RB protein. Concretely, we found that the CDK inhibitor SU9516 decreased the expression of DHFR protein and mRNA in a dose‐ and time‐dependent manner as shown in 2, 3. Furthermore, the inhibition of DHFR expression by SU9516 was associated with dephosphorylation of pRB (Fig. 2c). These results may reflect that the inhibition of DHFR expression occurred through the inhibition of CDK and activating the RB pathway.
In the present study, we showed that SU9516 down‐regulates DHFR protein expression, resulting in increased sensitivity to MTX. Regarding the mechanism down‐regulating DHFR expression, we observed that the expression was reduced at the promoter level through the E2F site on treatment with SU9516. These findings suggested that the E2F–RB pathway is important in regulating sensitivity to MTX.
Many investigators have been evaluating the role of CDK’s inhibition in cancer therapeutic strategy.( 26 , 27 ) In recent years, various CDK inhibitors have been developed. Flavopiridol is a CDK inhibitor that has completed phase I and II clinical trials. However, despite promising preclinical results, clinical activity observed with flavopiridol has been limited in solid and hematologic malignancies.( 28 , 29 , 30 , 31 , 32 , 33 , 34 ) A recent phase I study of a pharmacokinetically new schedule of administering flavopiridol for refractory chronic lymphocytic leukemia showed an improvement in clinical outcome.( 35 ) Another CDK inhibitor, AZD5438, a novel and orally bioavailable CDK inhibitor, is also under phase I study.( 36 )
We previously proposed a strategy for chemotherapy against malignancies with inactivated p53, which we called “gene‐regulating chemotherapy,” to reactivate gene targets of p53, such as p21/WAF1/Cip1, gadd45, and DR5 genes.( 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 ) Since the expression of DHFR affects sensitivity to MTX, we consider that the expression of DHFR is also a target for gene‐regulating chemotherapy to overcome MTX resistance. Similarly, we have previously reported that a CDK inhibitor enhanced sensitivity to 5‐FU through repression of the thymidylate synthase gene.( 46 )
In most malignant tumors, function of RB protein is inactivated by the hyperactivation of CDKs. Against such malignancies, CDK inhibitors can reactivate pRB, resulting in G1 arrest through suppression of E2F. The CDK inhibitors SU9516, Purvalanol A, and Cdk4/6 Inhibitor IV are potential drugs in the treatment of some malignancies with MTX as suppressors of DHFR. The CDK inhibitors significantly enhance sensitivity to MTX by inhibiting the expression of DHFR. This finding implicates that the CDK inhibitors are promising for combined use with MTX in cancer therapy.
In conclusion, we showed for the first time that SU9516 significantly enhanced sensitivity to MTX through the inhibition of DHFR expression in human T‐cell leukemia Jurkat cells and CCRF‐CEM cells, and human erythroleukemia K562 cells. Although further study is needed, these results raise the possibility that a combination of CDK inhibitors and MTX might be a suitable chemotherapeutic option to overcome resistance to MTX when treating hematological malignancies.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
We thank Dr. K. Ohtani (Tokyo Medical and Dental University) for the DHFR promoter–luciferase fusion plasmids.
References
- 1. Chu E, Allegra CJ. Antifolates. In: Chabner BA, Longo DL, eds. Cancer Chemotherapy and Biotherapy, 2nd edn. Philadelphia: Lippincott‐Raven Publishers, 1996. 109–48. [Google Scholar]
- 2. Chabner BA, Allegra CJ, Curt GA et al. Polyglutamation of methotrexate. Is methotrexate a prodrug? J Clin Invest 1985; 76: 907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fairbanks LD, Rückemann K, Qui Y et al. Methotrexate inhibits the first committed step of purine biosynthesis in mitogen‐stimulated human T‐lymphocytes: a metabolic basis for efficacy in rheumatoid arthritis? Biochem J 1999; 342: 143–52. [PMC free article] [PubMed] [Google Scholar]
- 4. Bertino JR, Göker E, Gorlick R, Li WW, Banerjee D. Resistance mechanisms to methotrexate in tumors. Stem Cells 1996; 14: 5–9. [DOI] [PubMed] [Google Scholar]
- 5. Rhee MS, Wang Y, Nair MG, Galivan J. Acquisition of resistance to antifolates caused by enhanced γ‐glutamyl hydrolase activity. Cancer Res 1993; 53: 2227–30. [PubMed] [Google Scholar]
- 6. Chave KJ, Ryan TJ, Chmura SE, Galivan J. Identification of single nucleotide polymorphisms in the human γ‐glutamyl hydrolase gene and characterization of promoter polymorphisms. Gene 2003; 319: 167–75. [DOI] [PubMed] [Google Scholar]
- 7. Schimke RT. Gene amplification in cultured cells. J Biol Chem 1988; 263: 5989–92. [PubMed] [Google Scholar]
- 8. Göker E, Waltham M, Kheradpour A et al. Amplification of the dihydrofolate reductase gene is a mechanism of acquired resistance to methotrexate in patients with acute lymphoblastic leukemia and is correlated with p53 gene mutations. Blood 1995; 86: 677–84. [PubMed] [Google Scholar]
- 9. Banerjee D, Mayer‐Kuckuk P, Capiaux G, Budak‐Alpdogan T, Gorlick R, Bertino JR. Novel aspects of resistance to drugs targeted to dihydrofolate reductase and thymidylate synthase. Biochim Biophys Acta 2002; 1587: 164–73. [DOI] [PubMed] [Google Scholar]
- 10. Blake MC, Azizkhan JC. Transcription factor E2F is required for efficient expression of the hamster dihydrofolate reductase gene in vitro and in vivo . Mol Cell Biol 1989; 9: 4994–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dyson N. The regulation of E2F by pRB‐family proteins. Genes Dev 1998; 12: 2245–62. [DOI] [PubMed] [Google Scholar]
- 12. Angus SP, Wheeler LJ, Ranmal SA et al. Retinoblastoma tumor suppressor targets dNTP metabolism to regulate DNA replication. J Biol Chem 2002; 277: 44376–84. [DOI] [PubMed] [Google Scholar]
- 13. Lees JA, Weinberg RA. Tossing monkey wrenches into the clock: new ways of treating cancer. Proc Natl Acad Sci USA 1999; 96: 4221–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pines J. Cyclins and cyclin‐dependent kinases: a biochemical view. Biochem J 1995; 308: 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li X, Huang P, Cui JJ, Zhang J, Tang C. Novel pyrrolyllactone and pyrrolyllactam indolinones as potent cyclin‐dependent kinase 2 inhibitors. Bioorg Med Chem Lett 2003; 13: 1939–42. [DOI] [PubMed] [Google Scholar]
- 16. Mohammadi M, McMahon G, Sun L et al. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997; 276: 955–60. [DOI] [PubMed] [Google Scholar]
- 17. Lane ME, Yu B, Rice A et al. A Novel cdk2‐selective inhibitor, SU9516, induces apoptosis in colon carcinoma cells. Cancer Res 2001; 61: 6170–7. [PubMed] [Google Scholar]
- 18. Moshinsky DJ, Bellamacina CR, Boisvert DC et al. SU9516: biochemical analysis of cdk inhibition and crystal structure in complex with cdk2. Biochem Biophys Res Commun 2003; 310: 1026–31. [DOI] [PubMed] [Google Scholar]
- 19. Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL‐R2 and sensitize apoptosis induced by TRAIL/APO2‐L in human malignant tumor cells. Oncogene 2004; 23: 6261–71. [DOI] [PubMed] [Google Scholar]
- 20. Iwanaga R, Komori H, Ohtani K. Differential regulation of expression of the mammalian DNA repair genes by growth stimulation. Oncogene 2004; 23: 8581–90. [DOI] [PubMed] [Google Scholar]
- 21. Brasier AR, Tate JE, Habener JF. Optimized use of the firefly luciferase assay as a reporter gene in mammalian cell lines. BioTechniques 1989; 7: 1116–22. [PubMed] [Google Scholar]
- 22. Alt FW, Kellems RE, Bertino JR, Schimke RT. Selective multiplication of dihydrofolate reductase genes in methotrexate resistant variants of cultured murine cells. J Biol Chem 1978; 253: 1357–70. [PubMed] [Google Scholar]
- 23. Srimatkandada S, Schweitzer BI, Moroson BA, Dube S, Bertino JR. Amplification of a polymorphic dihydrofolate reductase gene expressing an enzyme with decreased binding to methotrexate in a human colon carcinoma cell line, HCT‐8R4, resistant to this drug. J Biol Chem 1989; 264: 3524–8. [PubMed] [Google Scholar]
- 24. Haber DA, Beverly SM, Kiely ML, Schimke RT. Properties of an altered dihydrofolate reductase encoded by amplified genes in cultured mouse fibroblasts. J Biol Chem 1981; 256: 9501–10. [PubMed] [Google Scholar]
- 25. Li W, Fan J, Hochhauser D et al. Lack of functional retinoblastoma protein mediates intrinsic resistance to antimetabolites in sarcoma cell lines. Proc Natl Acad Sci USA 1995; 92: 10436–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mani S, Wang C, Wu K, Francis R, Pestell R. Cyclin‐dependent kinase inhibitors: novel anticancer agents. Exp Opin Investig Drugs 2000; 9: 1–21. [DOI] [PubMed] [Google Scholar]
- 27. Collins I, Garrett MD. Targeting the cell division cycle in cancer: CDK and cell cycle checkpoint kinase inhibitors. Curr Opin Pharmacol 2005; 5: 366–73. [DOI] [PubMed] [Google Scholar]
- 28. Senderowicz AM, Headlee D, Stinson SF et al. Phase I trial of continuous infusion flavopiridol, a novel cyclin‐dependent kinase inhibitor, in patients with refractory neoplasms. J Clin Oncol 1998; 16: 2986–99. [DOI] [PubMed] [Google Scholar]
- 29. Senderowicz AM. Inhibitors of cyclin‐dependent kinase modulators for cancer therapy. Prog Drug Res 2005; 63: 183–206. [DOI] [PubMed] [Google Scholar]
- 30. Kouroukis CT, Belch A, Crump M et al. Flavopiridol in untreated or relapsed mantle‐cell lymphoma: results of a phase II study of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2003; 21: 1740–5. [DOI] [PubMed] [Google Scholar]
- 31. Aklilu M, Kindler HL, Donehower RC, Mani S, Vokes EE. Phase II study of flavopiridol in patients with advanced colorectal cancer. Ann Oncol 2003; 14: 1270–3. [DOI] [PubMed] [Google Scholar]
- 32. Burdette‐Radoux S, Tozer RG, Lohmann RC et al. Phase II trial of flavopiridol, a cyclin dependent kinase inhibitor, in untreated metastatic malignant melanoma. Invest New Drugs 2004; 22: 315–22. [DOI] [PubMed] [Google Scholar]
- 33. Liu G, Gandara DR, Lara PN Jr et al. A phase II trial of flavopiridol (NSC #649890) in patients with previously untreated metastatic androgen‐independent prostate cancer. Clin Cancer Res 2004; 10: 924–8. [DOI] [PubMed] [Google Scholar]
- 34. Schwartz GK, Ilson D, Saltz L et al. Phase II study of the cyclin‐dependent kinase inhibitor flavopiridol administered to patients with advanced gastric carcinoma. J Clin Oncol 2001; 19:1985–92. [DOI] [PubMed] [Google Scholar]
- 35. Phelps MA, Lin TS, Johnson AJ et al. Clinical response and pharmacokinetics from a phase I study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood 2009; 113: 2637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Camidge DR, Pemberton M, Growcott J et al. A phase I pharmacodynamic study of the effects of the cyclin‐dependent kinase‐inhibitor AZD5438 on cell cycle markers within the buccal mucosa, plucked scalp hairs and peripheral blood mononucleocytes of healthy volunteers. Cancer Chemother Pahrmacol 2007; 60: 479–88. [DOI] [PubMed] [Google Scholar]
- 37. Matsui TA, Sowa Y, Yoshida T et al. Sulforaphane enhances TRAIL‐induced apoptosis through the induction of DR5 expression in human osteosarcoma cells. Carcinogenesis 2006; 27: 1768–77. [DOI] [PubMed] [Google Scholar]
- 38. Sowa Y, Orita T, Minamikawa S et al. Histone deacetylase inhibitor activates the WAF1/Cip1 gene promoter through the Sp1 sites. Biochem Biophys Res Commun 1997; 241: 142–50. [DOI] [PubMed] [Google Scholar]
- 39. Nakano K, Mizuno T, Sowa Y et al. Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53‐negative human colon cancer cell line. J Biol Chem 1997; 272: 22199–206. [DOI] [PubMed] [Google Scholar]
- 40. Hirose T, Sowa Y, Takahashi S et al. p53‐independent induction of Gadd45 by histone deacetylase inhibitor: coordinate regulation by transcription factors Oct‐1 and NF‐Y. Oncogene 2003; 22: 7762–73. [DOI] [PubMed] [Google Scholar]
- 41. Oki T, Sowa Y, Hirose T et al. Genistein induces Gadd45 gene and G2/M cell cycle arrest in the DU145 human prostate cancer cell line. FEBS Lett 2004; 577: 55–9. [DOI] [PubMed] [Google Scholar]
- 42. Kouhara J, Yoshida T, Nakata S et al. Fenretinide up‐regulates DR5/TRAIL‐R2 expression via the induction of the transcription factor CHOP and combined treatment with fenretinide and TRAIL induces synergistic apoptosis in colon cancer cell lines. Int J Oncol 2007; 30: 679–87. [PubMed] [Google Scholar]
- 43. Horinaka M, Yoshida T, Shiraishi T et al. Luteolin induces apoptosis via death receptor 5 upregulation in human malignant tumor cells. Oncogene 2005; 24: 7180–9. [DOI] [PubMed] [Google Scholar]
- 44. Yoshida T, Shiraishi T, Horinaka M et al. Lipoxygenase inhibitors induce death receptor 5/TRAIL‐R2 expression and sensitize malignant tumor cells to TRAIL‐induced apoptosis. Cancer Sci 2007; 98: 1417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakata S, Yoshida T, Shiraishi T et al. 15‐Deoxy‐Δ12,14‐prostaglandin J2 induces death receptor 5 expression through mRNA stabilization independently of PPARg and potentiates TRAIL‐induced apoptosis. Mol Cancer Ther 2006; 5: 1827–35. [DOI] [PubMed] [Google Scholar]
- 46. Takagi K, Sowa Y, Cevik OM, Nakanishi R, Sakai T. CDK inhibitor enhances the sensitivity to 5‐fluorouracil in colorectal cancer cells. Int J Oncol 2008; 32: 1105–10. [PubMed] [Google Scholar]