

Abstract
BCR–ABL tyrosine kinase, generated from the reciprocal chromosomal translocation t(9;22), causes chronic myeloid leukemia (CML). BCR–ABL is inhibited by imatinib; however, several mechanisms of imatinib resistance have been proposed that account for loss of imatinib efficacy in patients with CML. Previously, we showed that overexpression of the efflux drug transporter P‐glycoprotein partially contributed to imatinib resistance in imatinib‐resistant K562 CML cells having no BCR–ABL mutations. To explain an additional mechanism of drug resistance, we established a subclone (K562/R) of the cells and examined the BCR–ABL signaling pathway in these and wild‐type K562 (K562/W) cells. We found the K562/R cells were 15 times more resistant to imatinib than their wild‐type counterparts. In both cell lines, BCR–ABL and its downstream signaling molecules, such as ERK1/2, ERK5, STAT5, and AKT, were phosphorylated in the absence of imatinib. In both cell lines, imatinib effectively reduced the phosphorylation of all the above, except ERK1/2, whose phosphorylation was, interestingly, only inhibited in the wild‐type cells. We then observed that phospho‐ERK1/2 levels decreased in the presence of siRNA targeting BCR–ABL, again, only in the K562/W cells. However, using an ERK1/2 inhibitor, U0126, we found that we could reduce phospho‐ERK1/2 levels in K562/R cells and restore their sensitivity to imatinib. Taken together, we conclude that the BCR–ABL‐independent activation of ERK1/2 contributes to imatinib resistance in K562/R cells, and that ERK1/2 could be a target for the treatment of CML patients whose imatinib resistance is due to this mechanism. (Cancer Sci 2009)
Chronic myeloid leukemia, a hematopoietic stem cell disorder, is characterized by the expression of the chimeric BCR–ABL oncoprotein, caused by the reciprocal chromosomal translocation t(9;22) (q34;q11), which generates a shortened chromosome 22 or Philadelphia chromosome.( 1 ) BCR–ABL is a cytoplasmic protein with constitutive tyrosine kinase activity responsible for transformation and leukemogenic effects. BCR–ABL is the target of the tyrosine kinase inhibitor imatinib, which also inhibits c‐kit protooncogene/CD117 (c‐KIT) and plate‐derived growth factor receptor (PDGFR).( 2 ) More than 90% of chronic‐phase CML patients respond to imatinib, at least initially, and a high percentage of them achieve cytogenic complete responses.( 3 ) However, some patients fail to respond to treatment with imatinib in front‐line therapy (primary resistance), while others stop responding after an initial response (acquired resistance).( 4 ) Frequent clinical relevancies to imatinib resistance include point mutations in the ABL gene( 5 , 6 ) and amplification of the BCR–ABL fusion gene.( 7 ) In addition to these BCR–ABL‐dependent mechanisms, BCR–ABL‐independent mechanisms of imatinib resistance have been proposed, which involve the drug transporter P‐gp (MDR1, ATP‐binding cassette subfamily B1 (ABCB1)),( 8 , 9 , 10 ) the drug carrier serum α1 acid glycoprotein,( 11 ) and signal cascades via LYN kinase.( 12 , 13 ) Although some mechanisms of imatinib resistance are understood, resistance to this drug is still an important challenge confronting the effective treatment of CML.
We previously focused on drug transporters and reported the potential contribution to imatinib resistance of P‐gp, a multidrug efflux transporter, using K562 cells, which are known as a representative human CML cell line.( 9 ) In a previous study, we established imatinib‐resistant K562 (prevK562/R) cells, which had wild‐type BCR–ABL and overexpressed P‐gp. In prevK562/R cells, intracellular imatinib levels were 42% less than those in wild‐type K562 (K562/W) cells. Intracellular imatinib accumulation was restored completely by CysA, a P‐gp inhibitor; however, CysA did not completely overcome prevK562/R cell sensitivity to imatinib. Therefore, we assumed that another mechanism was also involved in imatinib resistance in K562 cells. To identify this unknown mechanism, we focused on signal transduction pathways that were downstream of BCR–ABL, and analyzed imatinib resistance factors, other than overexpression of P‐gp, using a new imatinib‐resistant subclone of K562 (K562/R) cells, which was cloned from prevK562/R cells.
BCR–ABL has multiple downstream survival pathways, including ERK1/2, ERK5, AKT, JAK/STAT, nuclear factor kappa beta (NF‐κB), and BCL‐xL.( 14 , 15 , 16 , 17 , 18 ) Phosphorylation of tyrosine 177 of BCR–ABL is necessary for binding of the adaptor growth factor receptor‐bound protein (GRB2) to BCR–ABL, which involves the recruitment of son of sevenless (SOS), the nucleotidic exchange factor of RAS.( 19 ) RAS activates both the RAF–MEK–ERK1/2 and PI3K–AKT pathways, which are engaged in cell survival and anti‐apoptosis.( 18 , 20 ) ERK5, like ERK1/2, is a member of the mitogen‐activated protein kinase family, is modulated by BCR–ABL, and contributes to the survival of leukemia cells.( 17 , 21 ) In the present study, we investigated the contributions of these downstream factors to imatinib resistance in K562/R cells.
Here, we demonstrate that BCR–ABL‐independent activation of ERK1/2 may contribute to imatinib resistance in certain CML cell lines. This resistance can be overcome by co‐treatment with the specific ERK1/2 inhibitor and imatinib, indicating that this co‐treatment may be effective for imatinib‐resistant CML patients.
Materials and Methods
Cell culture and cloning of imatinib‐resistant K562 cells. K562/W cells were cultured in RPMI‐1640 medium supplemented with 10% FBS under an atmosphere of 5% CO2–95% air at 37°C. prevK562/R cells were established by exposing gradually increasing concentrations of imatinib (from 0.3 to 10 μm) from K562/W cells.( 9 ) The new clonal cell line K562/R was established by limiting dilution from prevK562/R cells. K562/R cells were maintained under the same culture conditions in the presence of 1 μm imatinib.
mRNA isolation and cDNA synthesis. For mRNA extraction, MagNA Pure LC mRNA Isolation Kit II (Roche Diagnostics, Basel, Switzerland) was used as per the instruction manual. cDNA was synthesized by reverse transcription using the High‐Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). Each prepared cellular cDNA was stored at –30°C.
Mutation analysis of the BCR–ABL kinase domain and KRAS. The kinase domain of BCR–ABL was amplified by PCR using each cellular cDNA.( 9 ) For the primary PCR, we used the forward primer 5′‐CCAGACTGTCCACAGCATTC‐3′ and the reverse primer 5′‐ATGGTCCAGAGGATCGCTCTCT‐3′, and for the secondary PCR, the forward primer 5′‐GGGAGGGTGTACCATTACAGG‐3′ and the reverse primer, 5′‐GCTGTGTAGGTGTCCCCTGT‐3′, or the forward primer 5′‐CCACTTGGTGAAGGTAGCTG‐3′ and the reverse primer, 5′‐CCTGCAGCAAGGTAGTCACA‐3′, were used. The analysis was carried out by DNA sequencing using an Applied Biosystems 3130 Genetic Analyzer. For the mutation analysis of KRAS ORF, the forward primer 5′‐CGGGAGAGAGGCCTGCTG‐3′ and the reverse primer 5′‐CCACTTGTACTAGTATGCCT‐3′ were used in PCR amplification. The sequence of the KRAS gene was analyzed by the Sigma‐Aldrich DNA sequencing Service.
Cytotoxicity assay. The individual or combined cytotoxicities of imatinib and U0126 were determined by the Alamar Blue assay as described previously.( 9 , 22 ) Absorbance in cells without drug treatment was 100%.
Western blot analysis. K562/W and K562/R cells were homogenized in a solution containing 7 m urea, 2 m thiourea, 4% CHAPS, protease inhibitor cocktail (P8430; Sigma‐Aldrich, St Louis, MO, USA), 2 mm Na3VO4, 10 mm NaF, 1 μm okadaic acid, and 1 mm DTT, using Micropestle (Eppendorf, Westbury, NY, USA). Crude membrane fraction selection and Western blotting conditions were as described previously.( 9 ) The following primary antibodies were used: LYN, phospho‐STAT5 (Tyr694), STAT5 (BD Transduction Laboratories, Lexington, KY, USA), β‐actin (Sigma‐Aldrich), 4G10, Na+/K+ ATPase α‐1 (Upstate Biotechnology, Lake Placid, NY, USA), c‐ABL (Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho‐ERK1/2, ERK1/2, phospho‐AKT (Thr308), phospho‐AKT (Ser473), AKT, ERK5 (Cell Signaling Technology, Beverly, MA, USA), and P‐gp (C219 monoclonal antibody; Signet Laboratories, Dedham, MA, USA).
Two‐dimensional Western blotting. Samples were desalted using the 2‐D Clean‐Up kit (GE Healthcare, Amersham Place, UK) and resolved with sample buffer (7 m urea, 2 m thiourea, 4% CHAPS, 0.5% IPG Buffer pH 3–10, and Destreaking buffer). The first‐dimensional isoelectric focusing (pH 3–10, 7 cm) was carried out using the Ettan IPGPhor Cup Loading Manifold electrophoresis system (GE Healthcare) as per manufacturer recommendations. After reduction and alkylation of disulfide bonds with 10 mg/mL DTT and 25 mg/mL iodoacetamide, respectively, the second‐dimensional separation was carried out by 12% SDS‐PAGE. The 2D gel was immunoblotted as indicated above.
Real‐time RT‐PCR analysis. To determine the expression levels of hMDR1 and hβ‐ACTIN in the cells, we carried out TaqMan quantitative real‐time RT‐PCR using the ABI PRISM 7900 sequence detection system (Applied Biosystems), using the manufacturer’s standard protocol (hMDR1, Hs00184491_m1; hβ‐ACTIN, 4310881E).
Analysis of intracellular imatinib accumulation. Cells (2 × 106) were incubated in 5 mL incubation buffer (150 mm NaCl, 3 mm KCl, 1 mm CaCl2, 0.5 mm MgCl2, 5 mm d‐glucose, 5 mm HEPES, pH 7.4) containing 1 μm imatinib. The cell pellet was washed once in ice‐cold PBS containing 1% BSA, and twice with ice‐cold PBS. The cellular imatinib was then extracted by incubation with 300 μL of 50% methanol (HPLC mobile phase/methanol = 1/1) for 30 min. Quantification of imatinib was done using the HPLC (model LC‐6A; Shimadzu, Kyoto, Japan) method described previously.( 8 ) For the cellular protein quantitation, the pellet was solubilized with 200 μL of 1 N NaOH and analyzed by the Bradford method, using a Bio Rad Protein Assay kit (Bio Rad, Hercules, CA, USA) with BSA as a standard.
siRNA transfection. An siRNA specific for the b3a2 breakpoint of the BCR–ABL gene (5′‐GCAGAGUUCAAAAGCCCUUdTdT), and a control siRNA composed of the scrambled b3a2 sequence (5′‐GCAGAGUUCUAAAGCGCUUdTdT),( 23 ) were synthesized by Nippon EGT (Toyama, Japan). For electroporation of K562/W and K562/R cells, we used MicroPorator MP‐100 (AR Brown, Tokyo, Japan). Cells were washed twice with PBS, and mixed with 20 μm stock siRNA to a final concentration of 3, 4 or 8 pmol/μL. Subsequently, 5 × 105 cells/10 μL of the cell suspension were electroporated using the following settings: pulse voltage = 1450 V, pulse width = 10 ms, and pulse number = three times. After electroporation, the cells were resuspended in RPMI‐1640 medium and cultured in the incubator for 48 h under the same conditions as other cells.
Statistical analysis. Statistical significance was determined by Welch’s t‐test. P < 0.05 was considered statistically significant.
Results
Characterization of imatinib‐resistant K562/R cells. We established a new K562/R clonal cell line, which indicated a 15‐fold increase in the IC50 of imatinib over the parent K562/W cells (Fig. 1A), had no mutation in the BCR–ABL kinase domain (data not shown), and had neither overexpression nor overactivation of BCR–ABL or LYN kinase (Fig. 1B). Mutation analysis of KRAS showed no missense mutations( 24 , 25 ) in either cell line, although one silent mutation (519T > C) was found in both cells (Supporting information Fig. S1), suggesting that KRAS mutation in K562/R cells is not a factor in imatinib resistance. Both levels of mRNA (300‐fold, P < 0.01; Fig. 1C) and protein in the crude membrane fraction (Supporting information Fig. S2B) of MDR1 were found to be elevated in K562/R cells compared with K562/W cells (almost similar results were obtained in prevK562/R cells(9)), although intracellular accumulation levels of imatinib were similar (Fig. 1D). Moreover, CysA, a P‐gp inhibitor, did not influence intracellular imatinib accumulation levels in either type of cell. These results suggest that the imatinib resistance of K562/R cells is not directly related to the cellular P‐gp expression level or function.
Figure 1.
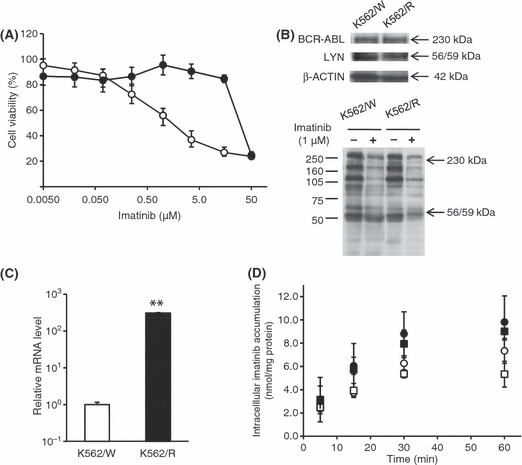
Characteristics of imatinib‐resistant K562/R cells. Imatinib and FBS were depleted from the medium 1 day before all experimental procedures to efflux intracellular imatinib from K562/R cells. (A) K562/W (open circles) and K562/R (closed circles) cells were exposed to various concentrations of imatinib for 48 h. Cell viability was assayed by Alamar Blue. Each point represents the mean ± SD from six cells. (B) The expression levels of BCR–ABL, LYN, and β‐actin and phosphorylation of tyrosine were examined by Western blot analysis. K562/W and K562/R cells were treated with 1 μm imatinib for 15 min. (C) The mRNA levels of hMDR1 and hβ‐ACTIN were examined by real time RT‐PCR analysis. The relative amount of hMDR1 mRNA was normalized to that of hβ‐ACTIN. (D) Intracellular imatinib accumulation was measured by HPLC analysis in K562/W and K562/R cells. The cells (2.0 × 106) were exposed to 1 μm imatinib with (squares) or without (circles) 5 μm cyclosporin A (CysA) at 37°C for varying lengths of time in K562/W (open symbols) and K562/R (closed symbols). Each point represents the mean ± SD from four cells. **P < 0.01 versus K562/W cells.
Imatinib did not inhibit the phosphorylation of ERK1/2 in K562/R cells. To study the specific activation signals in K562/R cells, K562/W and K562/R cells were treated with imatinib for varying lengths of time, as shown in Figure 2(A), after which the phosphorylation of BCR–ABL and its downstream factors, AKT, ERK5, STAT5, and ERK1/2 was examined. The phosphorylation levels of BCR–ABL and STAT5 were similarly high in both cell lines, but those of AKT and ERK1/2 were higher in the K562/R cells. After the treatment with imatinib, the phosphorylation of BCR–ABL, AKT, and STAT5 was effectively inhibited in both cell lines (Fig. 2A). ERK5 expression, which is known to be regulated by BCR–ABL,( 17 ) was not decreased by imatinib in either cell line (Fig. 2A). To analyze the phosphorylation status of ERK5 in both cell lines, we carried out 2D Western blotting using anti‐ERK5 antibodies after a 24‐h treatment with imatinib, and compared the 2D patterns of ERK5‐positive spots (Fig. 2C). Prior to imatinib treatment, both cell lines showed at least two ERK5‐positive spots in 2D Western blotting. After treatment, one of these spots, presumed to be phosphorylated ERK5, underwent a significant shift from left (acid, pI 4.7) to right (basic, pI 4.9), suggesting that the phosphorylation of ERK5 was inhibited by the imatinib treatment in both cell lines. In contrast, ERK1/2 phosphorylation was inhibited in K562/W cells only (Fig. 2A). Interestingly, imatinib inhibitory effects on the phosphorylation of BCR–ABL were significant in a dose‐dependent manner, while phosphorylation of ERK1/2 was never downregulated by imatinib in K562/R cells (Fig. 2B). Results similar to the above were obtained in prevK562/R cells (Supporting information Fig. S2A) and eight other clones also established as imatinib‐resistant K562 cells (Fig. 2D). These results strongly indicate that, unlike in K562/W cells, BCR–ABL does not play a major role in the phosphorylation of ERK1/2 in K562/R cells.
Figure 2.
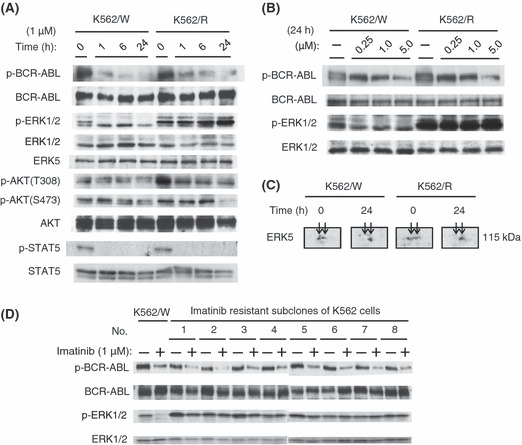
Effects of imatinib on K562/W and K562/R cells were analyzed by immunoblotting. (A) Time course of BCR–ABL, ERK1/2, AKT, and STAT5 phosphorylation levels in K562/W and K562/R cells after 1 μm imatinib treatment. (B) Effect of the indicated concentrations of imatinib treatment for 24 h on the phosphorylation levels of BCR–ABL and ERK1/2 in K562/W and K562/R cells. (C) K562/W and K562/R cells were treated with 1 μm imatinib for 24 h. ERK5 was detected by 2D Western blotting. (D) K562/W and subcloned, imatinib‐resistant K562 cells (from 1 to 8) were treated with 1 μm imatinib for 15 min. Clone cells of no. 2 is K562/R cells.
BCR–ABL‐targeting siRNA decreased the phosphorylation of ERK1/2 in K562/W cells, but not in K562/R cells. To confirm that ERK1/2 was activated independently of BCR–ABL in K562/R cells, BCR–ABL‐targeting siRNA was transfected into cells from both cell lines. Since K562 cells express the b3a2 form of the BCR–ABL fusion mRNA, the specific sequence for b3a2 can effectively silence cellular BCR–ABL expression.( 23 ) Transient transfection with varying amounts of b3a2 BCR–ABL siRNA significantly reduced the expression of BCR–ABL protein but not that of c‐ABL in K562/W cells, compared with cells transfected with a control scrambled siRNA (Fig. 3A). The same treatment also induced the downregulation of phosphorylated‐ERK1/2 in K562/W cells; however, it did not have this effect on K562/R cells (Fig. 3B).
Figure 3.
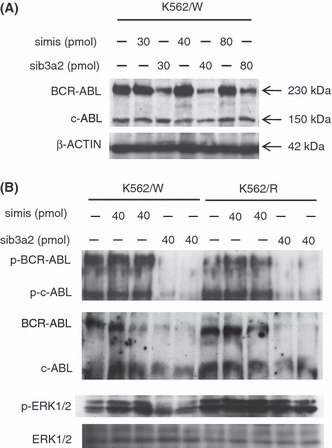
Effects of BCR–ABL targeting siRNA on K562/W and K562/R cells. (A) K562/W cells were electroporated with different amounts of siRNA ranging from 30 to 80 pmol per 5 × 105 cells. (B) Effect of 40 pmol siRNA on the phosphorylation levels of BCR–ABL and ERK1/2 in K562/W and K562/R cells. sib3a2 and simis indicate BCR‐ABL‐specific siRNA and mismatch control siRNA, respectively.
Inhibition of ERK1/2 overcame imatinib resistance in K562/R cells. Because the results obtained from the above experiments strongly suggested that the BCR–ABL‐independent activation of ERK1/2 is directly related to the mechanism of imatinib resistance in K562/R cells, we examined the effect of ERK1/2 inhibition on these cells. Treatment with an ERK1/2 inhibitor, U0126,( 26 ) alone for 48 h showed similar dose‐dependent toxicity for K562/W and K562/R cells (Fig. 4A). Treatment with U0126 (1 μm) alone had little cytotoxic effect on either cells; however, in combination with imatinib, cell death increased dramatically, with both cells showing a similar sensitivity to imatinib (Fig. 4B). These results suggest that co‐administration of the ERK1/2 inhibitor with imatinib could overcome imatinib resistance in K562/R cells.
Figure 4.
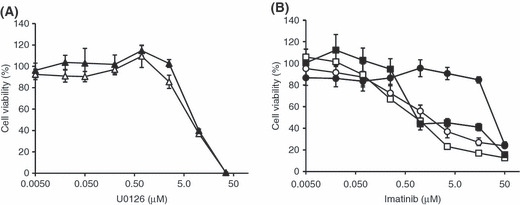
Cytotoxicity of U0126 alone and cotreatment with imatinib in K562/W and K562/R cells. (A) K562/W (open triangles) and K562/R (closed triangles) cells were exposed to 0.0050 μM U0126 alone for 48 h. (B) K562/W (open symbols) and K562/R (closed symbols) cells were exposed to 0.0050–50 μm imatinib with (squares) or without (circles) 1 μm U0126 for 48 h. Cell viability was assayed by Alamar Blue. Each point represents the mean ± SD from six cells.
Co‐treatment of imatinib and U0126 inhibited phosphorylation of ERK1/2 in both K562/W and K562/R cells. Because co‐administration of imatinib with U0126 restored imatinib sensitivity in K562/R cells, we next analyzed the inhibitory effect of imatinib on ERK1/2 phosphorylation by Western blotting in both cell lines exposed to U0126 alone or combined with imatinib. After U0126 treatment for 24 h, a dose‐dependent downregulation of ERK1/2 phosphorylation in both cell lines was observed (Fig. 5). Interestingly, co‐administration of 1 μm imatinib with 1 μm U0126 remarkably inhibited the phosphorylation of ERK1/2 in K562/R cells. These results confirm that the BCR–ABL‐independent ERK1/2 activation is essential for imatinib resistance in K562/R cells.
Figure 5.
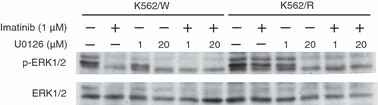
The effects of imatinib and U0126, alone or in combination, on ERK1/2 phosphorylation. K562/W and K562/R cells were treated with or without 1 μm imatinib and U0126 (1 and 20 μm) for 24 h, as indicated.
Discussion
Despite the significant efficacy of imatinib in treating CML, the development of primary and acquired imatinib resistance is a problem in patients with CML. We previously reported that, although P‐gp partly contributes to imatinib resistance in prevK562/R cells, P‐gp function alone cannot fully explain the resistance to imatinib. We thus postulated the involvement of a separate mechanism, and established a new imatinib‐resistant K562 clonal cell line (K562/R) from prevK562/R cells for use in studying this mechanism. In the new clonal cells, BCR–ABL was inhibited by both imatinib and BCR–ABL siRNA, while neither treatment suppressed ERK1/2 phosphorylation. Co‐treatment with imatinib and the ERK1/2 inhibitor U0126 restored imatinib sensitivity in K562/R cells. These data suggested for the first time that ERK1/2, activated through a BCR–ABL‐independent pathway, contributes to imatinib resistance in K562 cells.
Imatinib resistance is most often caused by BCR–ABL‐dependent mechanisms, including point mutations in the functional kinase domain of BCR–ABL( 5 , 6 ) and amplification of the BCR–ABL fusion gene.( 7 ) Point mutations in BCR–ABL reduce the binding of imatinib to the protein by either a direct or an indirect mechanism. However, in our study, the K562/R cells showed neither BCR–ABL mutations nor overexpression (Fig. 1B), indicating that their imatinib resistance mechanism could involve a BCR–ABL‐independent pathway. Similar mechanisms have been reported, namely, the overexpression of P‐gp and LYN kinase.( 10 , 11 , 12 , 13 ) Constitutively active mutants of KRAS are also known to be related to drug resistance in several cancers, including CML.( 24 , 25 , 27 , 28 , 29 ) For example, mutations in KRAS codon 12 involving a substitution of valine for glycine or aspartic acid are known; however, these were not observed in our K562/W or K562/R cells. Although both cell lines had the 519T > C silent mutation at the translated region (Supporting information Fig. S1), this particular mutation would not seem to influence the sensitivity to imatinib. In addition, LYN, which is a member of the Src tyrosine kinase family and is reported to be involved in imatinib resistance through overexpression of the anti‐apoptotic protein BCL‐2,( 12 , 13 ) is neither overexpressed nor overactivated in K562/R cells (Fig. 1B).
It is well known that imatinib interacts with P‐gp as a substrate, and that overexpressed P‐gp inhibits intracellular accumulation of imatinib.( 8 , 9 , 10 ) In our study, although MDR1 mRNA and protein expression were higher in K562/R than K562/W cells, there was no difference in intracellular imatinib accumulation, which was not decreased by the P‐gp inhibitor CysA (Fig. 1C,D). From these results, we hypothesized that P‐gp functional activity was modulated by unknown factors in K562/R cells. Intracellular imatinib levels are possibly controlled by many factors, such as drug transporters and plasma carrier proteins, in vivo. Even though upregulation of MDR1 mRNA or protein is observed in imatinib‐resistant CML patients, it will be necessary to measure intracellular imatinib levels to understand individual cases of imatinib resistance. Therefore, we ruled out a contribution from known mechanisms including P‐gp, and postulated the involvement of a separate mechanism.
BCR–ABL has multiple downstream survival pathways, such as ERK1/2, ERK5, AKT, and JAK/STAT.( 14 , 15 , 16 , 17 ) We examined whether imatinib treatment inhibited these downstream factors and found that the phosphorylation of BCR–ABL, ERK5, AKT, and STAT5 was indeed downregulated in both K562/W and K562/R cells, whereas ERK1/2 was not inhibited by imatinib in K562/R cells (Fig. 2A,C). A similar result was obtained by treatment using BCR–ABL siRNA (Fig. 3B), which further indicated that ERK1/2 is phosphorylated by a BCR–ABL‐independent mechanism in the K562/R cells. Next, the contribution of ERK1/2 to imatinib resistance in the K562/R cells was examined (4, 5). Although the cells were not sensitive to treatment with 1 μm imatinib alone, co‐administration with 1 μm U0126 inhibited ERK1/2 phosphorylation and dramatically induced K562/R cell death. U0126 is a known inhibitor of ERK5 as well,( 30 ) but because imatinib inhibited ERK5 phosphorylation (Fig. 2C), most of the synergistic effect of U0126 was presumably mediated by its downregulation of ERK1/2, rather than ERK5. These results demonstrate that inhibition of not only BCR–ABL, but also ERK1/2, due to its activation being independent of the former, is necessary to overcome imatinib resistance in the K562/R cells.
Concerning ERK1/2 activation in K562/R cells, the factors responsible have not been determined. It is known, however, that U0126 inhibits ERK1/2 through MEK1/2, which may be directly upstream of ERK1/2 in this BCR–ABL‐independent pathway. We have also shown that the aberrant activation of LYN or KRAS is not involved (Fig. 1B; Supporting Information Fig. S1). However, the possibility remains that unknown factors could increase ERK1/2 sensitivity and activate this signal via weak activation of BCR–ABL.
Recently, new drugs have been developed to target the BCR–ABL‐dependent and ‐independent imatinib‐resistance mechanisms. Nilotinib and dasatinib have a greater affinity for BCR–ABL than imatinib, and inhibit BCR–ABL and Src kinase, respectively.( 31 , 32 ) U0126 co‐treatment with dasatinib reverses LYN‐dependent imatinib resistance,( 33 ) suggesting the efficacy of its combination with molecular target drugs. Sorafenib, which inhibits multiple kinases, induces apoptosis in both BCR–ABL‐expressing imatinib‐sensitive and ‐resistant cells.( 34 , 35 , 36 ) It is possible that the combination of sorafenib with a BCR–ABL inhibitor works by inhibiting ERK1/2 activation in K562/R cells.
In conclusion, we demonstrate that ERK1/2, activated through a BCR–ABL‐independent mechanism, contributes to imatinib resistance in certain CML cells (Supporting Information Fig. S3). Although further work is required to confirm whether or not this resistance mechanism occurs in patients with CML, our study shows that this mechanism can be overcome by inhibiting the ERK1/2 signaling pathway with specific drugs that could be candidates for targeting the CML cells resistant to imatinib.
Disclosure Statement
None.
Abbreviations
- ABL
Abelson
- AKT
v‐akt murine thymoma viral oncogene homolog
- BCL
B‐cell CLL/lymphoma 2
- BCR
breakpoint cluster region
- CML
chronic myeloid leukemia
- CysA
cyclosporin A
- LYN
v‐yes‐1 Yamaguchi sarcoma viral related oncogene homolog
- P‐gp
P‐glycoprotein
- PI3K
phosphoinositide 3‐kinase
- RAF
v‐raf murine sarcoma viral oncogene homolog
- RAS
rat sarcoma oncogene
- STAT
signal transducer and activator of transcription
Supporting information
Fig. S1. Mutation analysis of KRAS in (A) K562/W and (B) K562/R cells. The T519C silent mutation was observed in both cell lines.
Fig. S2. (A) Expression of P‐glycoprotein in crude membrane fractions of K562/W, K562/R, and prevK562/R cells. (B) Effects of imatinib treatment on the phosphorylation levels of ERK1/2 in K562/W, K562/R, and prevK562/R cells. Cells were treated with the indicated concentrations of imatinib for 24 h, and phosphorylation levels were analyzed by western blotting.
Fig. S3. Scheme of a BCR–ABL‐independent imatinib‐resistant mechanism in K562/R cells. In K562/R cells, ERK1/2 is activated by a BCR–ABL‐independent pathway. Imatinib inhibits BCR–ABL and decreases phosphorylated AKT, but not phosphorylated ERK1/2, which is activated by an unknown protein independent of BCR–ABL.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) for Hideyuki Saito (KAKENHI 21390048) and Akinobu Hamada (KAKENHI 19590149) and from Cancer Research, Kiban Research, Houga Research from the Ministry of Education, Science, and Culture of Japan for Norie Araki (KAKENHI 17015034, 19390482, and 20659223, respectively). We thank Anthony Wilson, of the Tumor Genetics and Biology Department at Kumamoto University, for assistance with the English language presentation of this manuscript.
References
- 1. Kalidas M, Kantarjian H, Talpaz M. Chronic myelogenous leukemia. JAMA 2001; 286: 895–8. [DOI] [PubMed] [Google Scholar]
- 2. Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov 2002; 1: 493–502. [DOI] [PubMed] [Google Scholar]
- 3. Druker BJ, Guilhot F, O’Brien SG et al. Five‐year follow‐up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355: 2408–17. [DOI] [PubMed] [Google Scholar]
- 4. Daley GQ. Dodging the magic bullet: understanding imatinib resistance. Cancer Biol Ther 2003; 2: 109–10. [DOI] [PubMed] [Google Scholar]
- 5. Branford S, Rudzki Z, Walsh S et al. High frequency of point mutations clustered within the adenosine triphosphate‐binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph‐positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood 2002; 99: 3472–5. [DOI] [PubMed] [Google Scholar]
- 6. Roche‐Lestienne C, Soenen‐Cornu V, Grardel‐Duflos N et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre‐exist to the onset of treatment. Blood 2002; 100: 1014–8. [DOI] [PubMed] [Google Scholar]
- 7. Gorre ME, Mohammed M, Ellwood K et al. Clinical resistance to STI‐571 cancer therapy caused by BCR–ABL gene mutation or amplification. Science 2001; 293: 876–80. [DOI] [PubMed] [Google Scholar]
- 8. Hamada A, Miyano H, Watanabe H, Saito H. Interaction of imatinib mesilate with human P‐glycoprotein. J Pharmacol Exp Ther 2003; 307: 824–8. [DOI] [PubMed] [Google Scholar]
- 9. Hirayama C, Watanabe H, Nakashima R et al. Constitutive overexpression of P‐glycoprotein, rather than breast cancer resistance protein or organic cation transporter 1, contributes to acquisition of imatinib‐resistance in K562 cells. Pharm Res 2008; 25: 827–35. [DOI] [PubMed] [Google Scholar]
- 10. Mahon FX, Belloc F, Lagarde V et al. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 2003; 101: 2368–73. [DOI] [PubMed] [Google Scholar]
- 11. Gambacorti‐Passerini C, Barni R, Le Coutre P et al. Role of alpha1 acid glycoprotein in the in vivo resistance of human BCR–ABL(+) leukemic cells to the abl inhibitor STI571. J Natl Cancer Inst 2000; 92: 1641–50. [DOI] [PubMed] [Google Scholar]
- 12. Donato NJ, Wu JY, Stapley J et al. BCR–ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood 2003; 101: 690–8. [DOI] [PubMed] [Google Scholar]
- 13. Dai Y, Rahmani M, Corey SJ, Dent P, Grant S. A Bcr/Abl‐independent, Lyn‐dependent form of imatinib mesylate (STI‐571) resistance is associated with altered expression of Bcl‐2. J Biol Chem 2004; 279: 34227–39. [DOI] [PubMed] [Google Scholar]
- 14. Jin A, Kurosu T, Tsuji K et al. BCR/ABL and IL‐3 activate Rap1 to stimulate the B‐Raf/MEK/Erk and Akt signaling pathways and to regulate proliferation, apoptosis, and adhesion. Oncogene 2006; 25: 4332–40. [DOI] [PubMed] [Google Scholar]
- 15. Sonoyama J, Matsumura I, Ezoe S et al. Functional cooperation among Ras, STAT5, and phosphatidylinositol 3‐kinase is required for full oncogenic activities of BCR/ABL in K562 cells. J Biol Chem 2002; 277: 8076–82. [DOI] [PubMed] [Google Scholar]
- 16. Kirchner D, Duyster J, Ottmann O, Schmid RM, Bergmann L, Munzert G. Mechanisms of BCR–ABL‐mediated NF‐kappaB/Rel activation. Exp Hematol 2003; 31: 504–11. [DOI] [PubMed] [Google Scholar]
- 17. Buschbeck M, Hofbauer S, Di Croce L, Keri G, Ullrich A. Abl‐kinase‐sensitive levels of ERK5 and its intrinsic basal activity contribute to leukaemia cell survival. EMBO Rep 2005; 6: 63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Quintas‐Cardama A, Cortes J. Molecular biology of BCR–ABL1‐positive chronic myeloid leukemia. Blood 2009; 113: 1619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pendergast AM, Quilliam LA, Cripe LD et al. BCR–ABL‐induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB‐2 adaptor protein. Cell 1993; 75: 175–85. [PubMed] [Google Scholar]
- 20. McCubrey JA, Steelman LS, Abrams SL et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul 2006; 46: 249–79. [DOI] [PubMed] [Google Scholar]
- 21. Nishimoto S, Nishida E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep 2006; 7: 782–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O’Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem 2000; 267: 5421–6. [DOI] [PubMed] [Google Scholar]
- 23. Brauer KM, Werth D, Von Schwarzenberg K et al. BCR–ABL activity is critical for the immunogenicity of chronic myelogenous leukemia cells. Cancer Res 2007; 67: 5489–97. [DOI] [PubMed] [Google Scholar]
- 24. Pao W, Wang TY, Riely GJ et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005; 2: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Agarwal A, Eide CA, Harlow A et al. An activating KRAS mutation in imatinib‐resistant chronic myeloid leukemia. Leukemia 2008; 22: 2269–72. [DOI] [PubMed] [Google Scholar]
- 26. Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J 2003; 371: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garnett MJ, Marais R. Guilty as charged: B‐RAF is a human oncogene. Cancer Cell 2004; 6: 313–9. [DOI] [PubMed] [Google Scholar]
- 28. Weinstein‐Oppenheimer CR, Henriquez‐Roldan CF, Davis JM et al. Role of the Raf signal transduction cascade in the in vitro resistance to the anticancer drug doxorubicin. Clin Cancer Res 2001; 7: 2898–907. [PubMed] [Google Scholar]
- 29. Davis JM, Navolanic PM, Weinstein‐Oppenheimer CR et al. Raf‐1 and Bcl‐2 induce distinct and common pathways that contribute to breast cancer drug resistance. Clin Cancer Res 2003; 9: 1161–70. [PubMed] [Google Scholar]
- 30. Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem 1999; 274: 26563–71. [DOI] [PubMed] [Google Scholar]
- 31. Kantarjian H, Giles F, Wunderle L et al. Nilotinib in imatinib‐resistant CML and Philadelphia chromosome‐positive ALL. N Engl J Med 2006; 354: 2542–51. [DOI] [PubMed] [Google Scholar]
- 32. Talpaz M, Shah NP, Kantarjian H et al. Dasatinib in imatinib‐resistant Philadelphia chromosome‐positive leukemias. N Engl J Med 2006; 354: 2531–41. [DOI] [PubMed] [Google Scholar]
- 33. Nguyen TK, Rahmani M, Harada H, Dent P, Grant S. MEK1/2 inhibitors sensitize Bcr/Abl+ human leukemia cells to the dual Abl/Src inhibitor BMS‐354/825. Blood 2007; 109: 4006–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilhelm SM, Carter C, Tang L et al. BAY 43‐9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64: 7099–109. [DOI] [PubMed] [Google Scholar]
- 35. Rahmani M, Nguyen TK, Dent P, Grant S. The multikinase inhibitor sorafenib induces apoptosis in highly imatinib mesylate‐resistant bcr/abl+ human leukemia cells in association with signal transducer and activator of transcription 5 inhibition and myeloid cell leukemia‐1 down‐regulation. Mol Pharmacol 2007; 72: 788–95. [DOI] [PubMed] [Google Scholar]
- 36. Kurosu T, Ohki M, Wu N, Kagechika H, Miura O. Sorafenib induces apoptosis specifically in cells expressing BCR/ABL by inhibiting its kinase activity to activate the intrinsic mitochondrial pathway. Cancer Res 2009; 69: 3927–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Mutation analysis of KRAS in (A) K562/W and (B) K562/R cells. The T519C silent mutation was observed in both cell lines.
Fig. S2. (A) Expression of P‐glycoprotein in crude membrane fractions of K562/W, K562/R, and prevK562/R cells. (B) Effects of imatinib treatment on the phosphorylation levels of ERK1/2 in K562/W, K562/R, and prevK562/R cells. Cells were treated with the indicated concentrations of imatinib for 24 h, and phosphorylation levels were analyzed by western blotting.
Fig. S3. Scheme of a BCR–ABL‐independent imatinib‐resistant mechanism in K562/R cells. In K562/R cells, ERK1/2 is activated by a BCR–ABL‐independent pathway. Imatinib inhibits BCR–ABL and decreases phosphorylated AKT, but not phosphorylated ERK1/2, which is activated by an unknown protein independent of BCR–ABL.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item