

Abstract
TRAIL/Apo2L is a pro‐apoptotic cytokine that is capable of inducing apoptosis in a wide variety of cancer cells but not in normal cells. Among various molecular strategies by which cancer cells evade apoptosis, PI3K/Akt signaling represents a dominant survival pathway. In this report, we investigated the role of PI3K/Akt pathway in TRAIL‐induced apoptotic death in human bladder cancer cells. We observed that RT4 cells had very low level of constitutively active Akt and were sensitive to TRAIL, whereas UM‐UC‐3 and T24 cells had higher levels of constitutively active Akt and were resistant to TRAIL. Downregulation of constitutively active Akt by PI3K inhibitors, wortmannin and LY294002, reversed cellular resistance to TRAIL. However, transfecting constitutively active Akt into RT4 cells increased Akt activity and inhibited TRAIL‐induced apoptosis. These results suggest that elevated Akt activity protects UM‐UC‐3 and T24 cells from TRAIL‐induced apoptosis, and the PI3K/Akt signaling might inhibit apoptotic signals. Thus, the modulation of Akt activity by combining pharmacological drugs or genetic alterations of the Akt expression could induce cellular responsiveness to TRAIL and PI3K/Akt signaling pathway could serve as a novel target for therapeutic intervention in bladder cancer. (Cancer Sci 2006; 97: 1093–1098)
Abbreviations:
- FBS
fetal bovine serum
- MTT
3‐(4, 5‐dimethylthiazole‐2‐yl)‐2, 5‐diphenyltetrazolium bromide
- PI3K
phosphatidylinositol‐3 kinase
- SD
standard deviation
- TRAIL
tumor‐necrosis factor‐related apoptosis‐inducing ligand.
TRAIL, also known as Apo‐2 ligand, is a pro‐apoptotic cytokine that constitutes a family of ligands that transduce death signals through death domain‐containing receptors.( 1 , 2 , 3 ) TRAIL is a transmenbrane protein that functions by binding to two closely related receptors, DR4 and DR5. Both receptors are expressed on a high proportion of tumor cell lines, contain intracytoplasmic death domains, and on ligation mediate apoptosis. TRAIL is capable of inducing apoptosis in a wide variety of cancer cells in culture and in tumor implants in mice.( 4 , 5 ) However, its pro‐apoptotic effects are minimal in normal cells.( 6 , 7 ) Despite the ubiquitous expression of TRAIL receptors, some cancer cell lines show either partial or complete resistance to the pro‐apoptotic effects of TRAIL. The reason for the TRAIL resistance is unknown, but it is not regulated solely by the differential expression of the known TRAIL receptors DR4 and DR5. Instead, it appears to be more likely that intracellular inhibitors acting downstream of TRAIL receptors render some cells insensitive to TRAIL, as the resistance of many types of cancer cells to TRAIL can be reversed by treatment with protein synthesis inhibitors( 8 , 9 ) or chemotherapeutic agents.( 5 ) The binding of TRAIL to its receptors DR4 and DR5 leads to the cleavage and activation of caspase‐8, which in turn activates downstream effector caspases such as caspase‐3. Activation of caspase‐8 by TRAIL might also cleave BID, a Bcl‐2 inhibitory protein, which triggers mitochondrial depolarization.( 10 , 11 ) The study of the intracellular mechanisms that control TRAIL resistance might enhance our knowledge of death receptor‐mediated signaling and help to develop TRAIL‐based approaches for cancer treatment. In fact, there are many survival factors operating intracellularly in response to survival signals. Among the cellular signaling pathways that promote cell survival, Akt is one of the important survival factors that contributes resistance to apoptotic signals.( 12 ) Akt is a Ser/Thr protein kinase implicated in mediating a variety of biological responses, which includes the inhibition of apoptosis and the stimulation of cellular growth. Activation of PI3K/Akt pathway generates phosphatidylinositol‐3,4,5‐triphosphate, which in turn binds to the pleckstrin homology domain of Ser/Thr kinase Akt, resulting in recruitment of Akt to the menbrane. Previous studies have shown that Akt plays an important role in survival when cells are exposed to various kinds of apoptotic stimuli. Moreover, it was found to be overexpressed in some gastric adenocarcinomas and in breast, ovarian, prostate, and pancreatic cancers.( 13 , 14 )
The purpose of this study was to investigate the role of the PI3K/Akt pathway in TRAIL‐induced apoptosis in human bladder cancer cells. We showed that TRAIL induces apoptosis with variable responses in RT4 cells, whereas UM‐UC‐3 and T24 cells are highly resistant to TRAIL. Interestingly, UM‐UC‐3 and T24 cells highly express a constitutively active Akt that is inversely correlated with TRAIL sensitivity. Moreover, downregulation of Akt with DN‐Akt, or inhibitors of PI3K, made these cells sensitive to TRAIL. This study is the first to show that the PI3K/Akt pathway could interfere with TRAIL‐induced apoptotic signaling in bladder cancer cells.
Materials and Methods
Cells and culture conditions. The human bladder cancer cell lines RT4, UM‐UC‐3 and T24 were obtained from the American Type Culture Collection (Manassas, VA). Human normal urothelial cells SV‐HUC‐1 were also obtained from the same source. RT4, UM‐UC‐3 and T24 cells were grown in RPMI supplemented with 10% heat‐inactivated FBS and antibiotics. SV‐HUC‐1 cells were grown in Ham's F12 medium with 7% heat‐inactivated FBS and antibiotics. All cells were grown as monolayers on plastic tissue culture dishes at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Reagents. Antibodies against phospho‐Akt (Ser‐473) and Akt were from Cell Signaling Technology (Beverly, MA). Wortmannin, LY‐294002, and TRAIL were purchased from Sigma (St Louis, MO). Antibody against β‐actin was purchased from Santa Cruz Biotechology (Santa Cruz, CA). Lipofectamine was purchased from Invitrogen Life Technologies (Carlsbad, CA). JC‐1 dye was purchased from Molecular Probes (Eugene, OR).
Transient transfection. Cells were plated in 60 mm dishes in RPMI‐1640 containing 10% FBS and 1% penicillin‐streptomycin mixture at a density of 1 × 106 cells/dish. Cells were transfected with expression constructs encoding WT‐Akt (pUSE‐WT‐Akt), CA‐Akt (pUSE‐CA‐Akt), DN‐Akt (pUSE‐DN‐Akt), or the empty vector (pUSE) in the presence of an expression vector pCMV‐LacZ (Invitrogen Life Technologies) expressing β‐galactosidase. WT‐Akt, CA‐Akt, and DN‐Akt were purchased from Upstate Biotechnology (Lake Placid, NY). For each transfection, 2 µg of DNA was diluted in 50 µL of medium without serum. After the addition of 3 µL of lipofectamine into 50 µL of Opti‐MEM, the transfection mixture was incubated for 10 min at room temperature. Cells were washed with serum‐free medium, the transfection mixture was added, and cultures were incubated for 24 h in the incubator. The next day, culture medium was replaced with fresh RPMI‐1640 containing 10% FBS and 1% penicillin‐streptomycin mixture, and cells were treated with or without TRAIL (100 ng/mL) for 24 h.
MTT assay. Cells (1 × 104 in 100 µL of culture medium/well) were seeded into 96‐well flat‐bottomed plates, treated with or without drugs, and incubated for various times at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cell viability was assessed by MTT assay. Ten microliters of 5 mg/mL MTT solution (Sigma) was added to each well. After 3 h of incubation, 200 µL of dimethylsulfoxide (Fisher Biotech, Fairlawn, NJ) was added to each well. The absorbance of each well was measured at 495 and 650 nm using a Vmax kinetic microplate reader (Molecular Devices, Sunnyvale, CA).
Western blot analysis. Lysis of cells was done in a buffer containing 10 mM Tris‐HCL (pH 7.6), 150 mM NaCl, 5 mM EDTA, 1% sodium dodecylsulfate, 0.1% Triton X‐100, and 1 mM sodium orthovanadate, supplemented with 2 mM leupeptin, 2 mM aprotinin, 1 mM phenylmethylsulfonyl fluoride, and 2 mM pepstatin. After centrifugation at 12 000 g for 15 min, the supernatant was harvested as the total protein extract and stored at −80 °C. Protein concentrations were measured using a protein assay reagent (Bio‐Rad). Equal amounts of protein were separated by 12.5% sodium dodecylsulfate–polyacrylamide gel electrophoresis and electrophoretically transferred to polyvinylidene difluoride membrane. The membrane was blocked with 5% non‐fat dry milk in phosphate‐buffered saline−0.1% Tween 20 at 4°C overnight. The membrane was incubated with primary antibody for 1 h. Immunoreactivity was detected by sequential incubation with horseradish peroxidase‐conjugated secondary antibody and enhanced chemiluminescence reagents using the enhanced chemiluminescence detection system (Amersham Bioscience, Piscataway, NJ). For densitometric analysis of Western blots, Scion Image (Scion, Frederick, MD) was used.
Measurement of mitochondrial energization. Retention of JC‐1 staining (Molecular Probes) was used as a measure of mitochondrial energization. This dye, existing as a monomer in solution emitting a green fluorescence, can assume a dimeric configuration emitting red fluorescence in a reaction driven by the mitochondrial transmembrane potential. Cells (1 × 105 in 100 µL of culture medium/well) were seeded and treated with drugs and incubated for various times. JC‐1 (5 µg/mL) was added during the last 30 min of treatment. Cells were washed twice with Hanks’ Balanced Salt Solution (Invitrogen) to remove unbound dye. The concentration of retained JC‐1 dye was measured (490 nm excitation/600 nm emission) by a Spectra Maxi Germini fluorescence plate reader (Molecular Devices, Menlo Park, CA).
Measurement of caspase activity. Caspase activity was measured using fluorogenic caspase substrates. Briefly, cells were harvested after incubation with PI3K inhibitors (wortmannin, LY‐294002) in the presence or absence of TRAIL then lyzed with cell extract buffer (0.03% Nonidet P‐40, 20 mM HEPES (pH 7.5), 1.5 mM MgCl2, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, and 1 mM dithiothreitol). Lysates were centrifuged at 15 000 g for 10 min, and 50 µL of the cytosolic fraction was incubated for 60 min at 37°C in a total volume of 200 µL of caspase buffer (10 mM HEPES [pH 7.5], 50 mM NaCl, 2.5 mM dithiothreitol) containing 25 µM Ac‐DEVD‐AMC, or Ac‐IETD‐AMC, or Ac‐LEHD‐AMC (Bachem, King of Prussia, PA). 7‐amino‐4‐methylcoumarin fluorescence, released by caspase activity, was measured at 405 nm using a Spectra Maxi Germini fluorescence plate reader (Molecular Devices).
Apoptosis assay. Apoptosis was assessed by the accumulation of cells at sub‐G1 fraction.( 15 ) Cells were fixed with 70% ethanol for 30 min at 4 °C, incubated with RNase A (1 mg/mL in phosphate‐buffered saline) for 30 min at 37°C and stained with 50 µg/mL propidium iodide (Sigma) for 15 min. The samples were analyzed by FACScalibur and Cell Quest (Becton Dickinson, San Jose, CA).
Results
Effects of soluble human TRAIL on cell viability of bladder cancer cells. We first tested the cytotoxic effect of TRAIL on three bladder cancer cell lines by MTT assay. Cell viability assays showed that RT4 cells were sensitive to TRAIL in a dose‐ and time‐dependent manner, whereas UM‐UC‐3 and T24 cells were highly resistant to TRAIL (Fig. 1). As controls, we also used human normal urothelial SV‐HUC‐1 cells, which do not express death receptors DR4 and DR5. SV‐HUC‐1 cells were also highly resistant to TRAIL. In order to confirm whether cell death was due to apoptosis, we used flow cytometric analysis. The results further showed that the resistance to TRAIL in UM‐UC‐3 and T24 cells was due to decreased apoptosis (data not shown).
Figure 1.
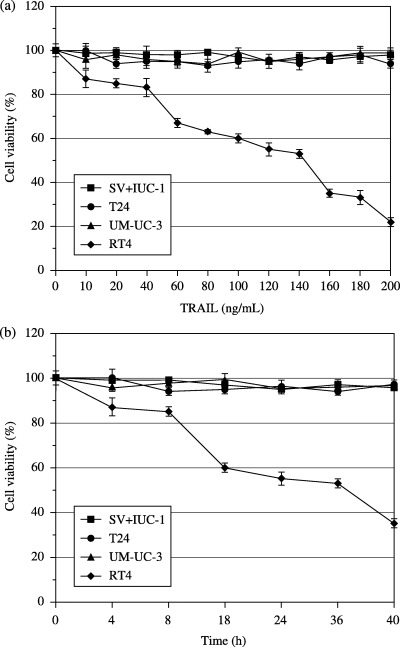
Sensitivity of human bladder normal and cancer cells to soluble human TRAIL. (a) Effects of various doses of TRAIL on the relative viability of bladder normal and cancer cells were assessed. The cells were treated with various doses of TRAIL for 24 h and cell viability was examined by MTT assay. Data are expressed as the mean ± SD. (b) Time‐dependent effect of TRAIL on the viability of bladder normal and cancer cells was assessed. The cells were treated with TRAIL (100 ng/mL) at various time points. Cell viability was examined by MTT assay. Data are expressed as the mean ± SD.
PI3K inhibition abrogated TRAIL resistance in UM‐UC‐3 and T24 cells. Pro‐survival function of growth factor signaling occurs through the PI3K/Akt pathway, and Akt is an important regulator of various intracellular events in tumor progression. Therefore, we investigated the involvement of the PI3K/Akt pathway in TRAIL resistance in these cell lines. First, Akt activity was measured by Western blot analysis using an antibody that specifically recognizes the phosphorylated/activated form of Akt (Fig. 2a). High expression of activated Akt was observed in UM‐UC‐3 and T24 cells, whereas low expression of activated Akt was observed in RT4 cells. Total Akt, DR4 and DR5 levels in these cells were almost equal. We next explored whether inhibition of PI3K by wortmannin or LY‐294002 can sensitize UM‐UC‐3 and T24 cells to TRAIL. When incubated with TRAIL (100 ng/mL) alone, no significant difference regarding phosphorylated Akt levels was observed in these cell lines (data not shown). However, as shown in Figure 2b, treatment of UM‐UC‐3 cells with wortmannin (1 µM) or LY‐294002 (25 µM) for 24 h reversed the high constitutive activity of Akt. Similar results were obtained when T24 cells were used (data not shown). The protein expression of DR4 or DR5 was not altered by treatment with these PI3K inhibitors. Next, we sought to examine whether downregulation of constitutively active Akt makes UM‐UC‐3 and T24 cells sensitive to TRAIL, as they highly express constitutively active Akt. RT4, UM‐UC‐3 and T24 cells were pretreated with wortmannin (1 µM) or LY‐294002 (25 µM) for 4 h, followed by treatment with TRAIL (100 ng/mL) for 24 h, and apoptosis was assessed (Fig. 3). TRAIL alone induced apoptosis in RT4 cells, but UM‐UC‐3 and T24 cells were resistant to apoptosis. Pretreatment with wortmannin or LY‐294002 further enhanced the effects of TRAIL on apoptosis in RT4, but not significantly. In contrast, pretreatment of UM‐UC‐3 and T24 cells with wortmannin or LY‐294002 dramatically increased apoptosis when combined with TRAIL. Thus, high levels of constitutively active Akt in UM‐UC‐3 and T24 cells renders them resistant to TRAIL.
Figure 2.
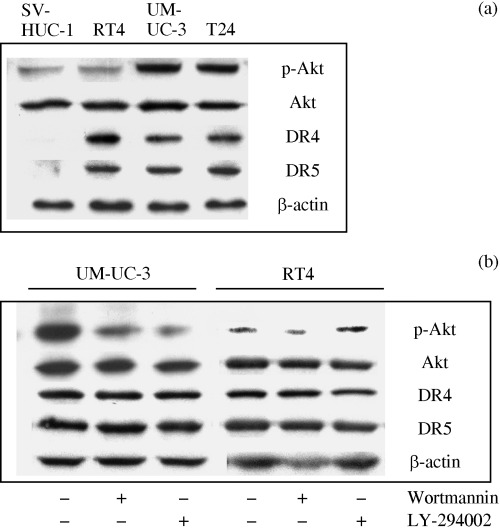
Constitutively active Akt in bladder cancer cells. (a) Cell lysates were subjected to immunoblotting with each antibody. (b) Immunoblot analysis of phospho‐Akt levels in bladder cancer UM‐UC‐3 and RT4 cells incubated with PI3K inhibitors (wortmannin and LY‐294002). The cells were treated with wortmannin (1 µM) or LY‐294002 (25 µM) in the presence of TRAIL (100 ng/mL) for 24 h.
Figure 3.
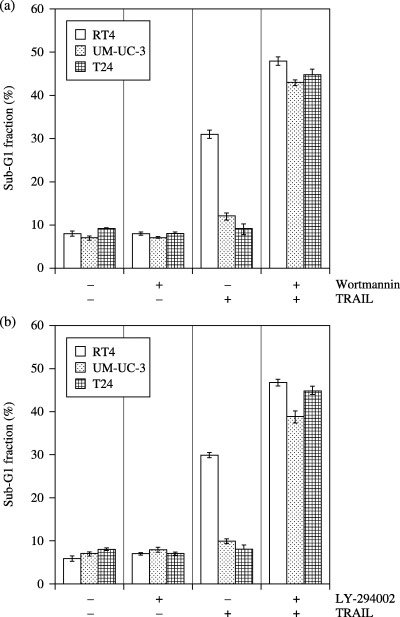
Cell cycle analysis. The bladder cancer cells were treated with wortmannin (1 µM; a) or LY‐294002 (25 µM; b) in the presence or absence of TRAIL (100 ng/mL) for 24 h, and apoptosis was assessed by the accumulation of cells in the sub‐G 1 fraction.Data are expressed as the mean ± SD.
Previous reports have demonstrated that death‐inducing signaling complex formation is essential not only for TRAIL‐induced signaling, which require Fas‐associated death domain protein,( 10 , 16 ) but for activation of caspase‐8.( 10 ) In order to exclude the possibility of defects in caspase‐8, we examined the capase‐8 activity in UM‐UC‐3, T24, and RT4 cells (Fig. 4). No difference in TRAIL‐induced caspase‐8 activity was seen among these cells. Pretreatment of cells with wortmannin or LY‐294002 did not affect TRAIL‐induced caspase‐8 activity. These results suggest that resistance of UM‐UC‐3 and T24 cells to TRAIL is not due to defects in caspase‐8 activation and that Akt does not affect caspase‐8 activation.
Figure 4.
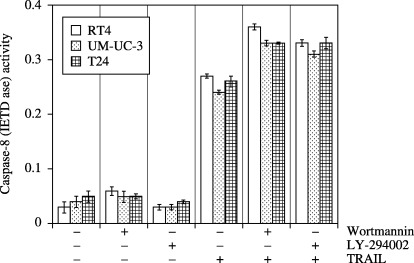
Regulation of caspase‐8 activity by wortmannin, LY294002 and TRAIL. The bladder cancer cells were treated with wortmannin (1 µM) or LY‐294002 (25 µM) in the presence or absence of TRAIL (100 ng/mL) for 12 h, and caspase‐8 activity was measured at 405 nm using a Spectra Maxi Germini fluorescence plate reader. Data are expressed as the mean ± SD.
Akt attenuates TRAIL‐induced drop in mitochondrial membrane potential in UM‐UC‐3 and T24 cells. Mitochondria seem to play an important role in apoptosis.( 10 , 17 ) Both mitochondrial depolarization and the loss of cytochrome c from the mitochondrial intermembrane space have been proposed as the early events during apoptotic cell death.( 17 , 18 ) Therefore, we next investigated mitochondrial dysfunction by measuring ΔΨm (the mitochondrial membrane potential) using the mitochondria‐specific dye, JC‐1 (Fig. 5). In UM‐UC‐3 and T24 cells, no significant effect on ΔΨm was seen when incubated with TRAIL or wortmannin or LY‐294002 alone. However, the combination of TRAIL with wortmannin or LY‐294002 induced a significant decrease (P < 0.05) in ΔΨm. In contrast, the decrease of ΔΨm in RT4 cells when treated with TRAIL alone, or TRAIL plus LY‐294002, or TRAIL plus wortmannin, was not statistically significant.
Figure 5.
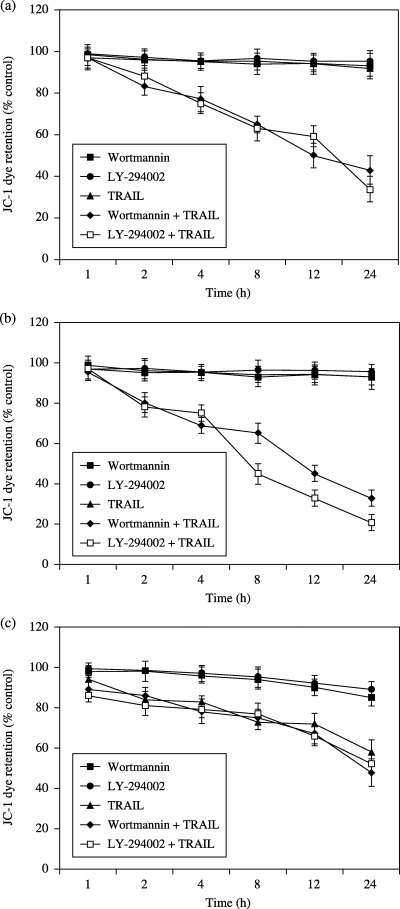
Mitochondrial membrane potential (ΔΨm) in bladder cancer cells treated with wortmannin (1 µM) or LY‐294002 (25 µM) in the presence or absence of TRAIL (100 ng/mL) at the specified time points was assayed. ΔΨm was measured by JC‐1 dye retention using a fluorescence plate reader. (a) UM‐UC‐3 cells. (b) T24 cells. (c) RT4 cells.
TRAIL induced activation of caspase‐9 and caspase‐3 in RT4 cells but not in UM‐UC‐3 or T24 cells. Mitochondrial membrane potential drop is essential for the formation of apoptosomes, which in turn activate caspase‐3 and caspase‐9. We examined the activation of caspase‐3 and caspase‐9 in cells treated with TRAIL in the presence or absence of wortmannin or LY‐294002 (Fig. 6). Treatment of UM‐UC‐3, T24, and RT4 cells with wortmannin or LY‐294002 had no effect on caspase‐3 or caspase‐9 activity. In RT4 cells, caspase‐3 and caspase‐9 were activated by TRAIL, but not in UM‐UC‐3 or T24 cells. When combined with wortmannin or LY‐294002, the increase of TRAIL‐induced activation of caspase‐3 and caspase‐9 was not significant in RT4 cells, but TRAIL significantly enhanced the activation of both caspase‐3 and caspase‐9 in UM‐UC‐3 and T24 cells.
Figure 6.
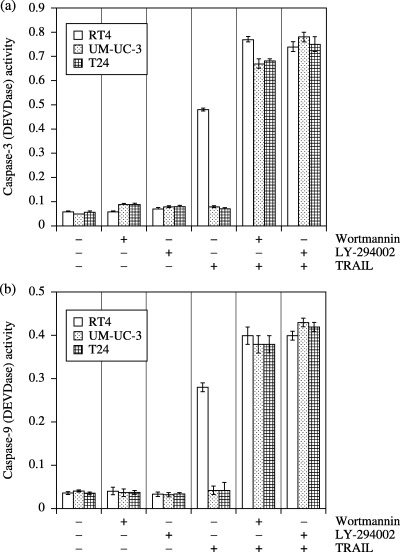
Effect of wortmannin, LY‐294002 and TRAIL on caspase‐3 and caspase‐9 activity. (a) The bladder cancer cells were treated with wortmannin (1 µM) or LY‐294002 (25 µM) in the presence or absence of TRAIL (100 ng/mL) for 12 h, and caspase‐3 activity was measured at 405 nm using a Spectra Maxi Germini fluorescence plate reader. Data are expressed as the mean ± SD. (b) The bladder cancer cells were treated with wortmannin (1 µM) or LY‐294002 (25 µM) in the presence or absence of TRAIL (100 ng/mL) for 12 h, and caspase‐9 activity was measured at 405 nm using a Spectra Maxi Germini fluorescence plate reader.
Constitutively active Akt in RT4 cells confers TRAIL resistance. If Akt is the only signaling factor for TRAIL sensitivity in RT4 cells, then upregulation of Akt by constitutively active Akt will inhibit TRAIL‐induced apoptosis. As RT4 cells express very low levels of constitutively active Akt, we examined the effects of the overexpression of constitutively active Akt on TRAIL‐induced apoptosis (Fig. 7). RT4 cells were transfected with empty vector, WT‐Akt, CA‐Akt and DN‐Akt, and incubated with or without TRAIL for 24 h. Apoptosis induced by TRAIL was observed in cells transfected with empty vector, WT‐Akt and DN‐Akt, whereas CA‐Akt‐transfected cells inhibited TRAIL‐induced apoptosis (Fig. 7a). We also examined the effects of CA‐Akt and DN‐Akt on TRAIL‐induced drop in ΔΨm (Fig. 7b). TRAIL‐induced drop in ΔΨm was observed in RT4 cells transfected with empty vector, WT‐Akt and DN‐Akt. In contrast, transfection of CA‐Akt in RT4 cells inhibited TRAIL‐induced drop in ΔΨm. These results showed that increased Akt activity in RT4 cells makes them resistant to TRAIL by improving mitochondrial integrity.
Figure 7.
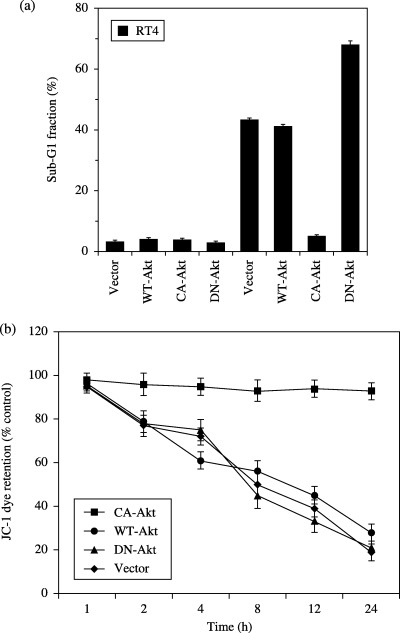
Effect of CA‐Akt and DN‐Akt on TRAIL‐induced apoptosis. (a) RT4 cells were transfected with empty vector, WT‐Akt, CA‐Akt, DN‐Akt. Cells were treated with (first four columns) or without TRAIL (last four columns) (100 ng/mL). Apoptosis was measured after 24 h. Data are expressed as the mean ± SD. (b) The effects of CA‐Akt and DN‐Akt on TRAIL‐induced drop in mitochondrial membrane potential (ΔΨm) at various time points. RT4 cells were transfected with empty vector, WT‐Akt, CA‐Akt, DN‐Akt. Cells were treated with or without TRAIL (100 ng/mL). ΔΨm was measured by JC‐1 dye retention using a fluorescence plate reader. Data are expressed as the mean ± SD.
Discussion
In the present study, we have shown that the PI3K/Akt pathway plays an essential role in regulating cells to escape from TRAIL‐induced apoptosis. TRAIL is expressed in a number of tissues and shows potent apoptotic activity against selected targets, including a variety of cancers.( 2 , 19 ) In addition to its well‐described effects on cell death, TRAIL can inhibit cell cycle progression, whereas blockade of TRAIL results in hyperproliferation in autoreactive lymphocytes, resistant to TRAIL‐induced apoptosis. Analysis of TRAIL‐resistant apoptosis in vitro has shown that there are some TRAIL‐resistant human melanoma and colon carcinoma cell lines.( 8 , 20 ) It appears that intracellular inhibitors acting downstream of the TRAIL receptors render specific transformed cell lines resistant to TRAIL.
A variety of reports has suggested the role of Akt in chemotherapeutic resistance to apoptosis and indicated its prosurvival function.( 21 ) It has been demonstrated that Akt might inhibit a variety of apoptotic stimuli in multiple ways.( 22 ) These include direct phosphorylation and modulation of proapoptotic proteins BAD,( 23 ) and caspase‐9,( 24 ) and activation of anti‐apoptotic NF‐κB‐mediated transcriptional pathways,( 25 , 26 ) preventing them from inducing the transcription of proapoptotic genes.( 27 ) However, once expressed, active Akt is under tight regulation by PI3K and other kinases of the signaling pathway that promote cell survival. Signaling of growth factors translocates Akt to the inner surface of the plasma membrane in proximity to regulatory kinases that phosphorylate and activate Akt.( 23 )
As PI3K targets Akt for survival, we have modulated the activation of Akt by two approaches. First, we used the PI3K inhibitors wortmannin and LY‐294002 to sensitize UM‐UC‐3 and T24 cells to TRAIL. Although RT4 cells undergo cell death on treatment with TRAIL alone, increased cell death of UM‐UC‐3 and T24 cells was observed only when combined with wortmannin or LY‐294002. This suggests that a high level of CA‐Akt in UM‐UC‐3 and T24 cells is responsible for TRAIL resistance.
Active Akt inhibits the drop in ΔΨm in UM‐UC‐3 and T24 cells. Although ineffective alone, TRAIL in combination with wortmannin or LY‐294002 induced a drop in ΔΨm and subsequently activated caspase‐9 in UM‐UC‐3 and T24 cells.
In addition to the use of PI3K inhibitors we used a genetic approach to downregulate active Akt by transfecting DN‐Akt. Downregulation of Akt by DN‐Akt transfection rendered UM‐UC‐3 and T24 cells susceptible to TRAIL‐induced apoptosis. In contrast, upregulation of Akt activity in RT4 cells, which express very low CA‐Akt, restored TRAIL resistance. In fact, Akt has been demonstrated to inhibit apoptosis and cytochrome c release induced by several pro‐apoptotic Bcl‐2 family members.( 24 , 28 ) Taken together, Akt promotes cell survival by intervening in the apoptosis cascade upstream of cytochrome c release and downstream of caspase‐8 activation through a mechanism involving mitochondria in apoptotic cells.
In conclusion, these findings suggest that the cytotoxic effects of TRAIL on bladder cancer cells vary significantly and inversely correlate with the levels of constitutively active Akt. Cells having higher constitutively active Akt were more resistant to undergo apoptosis by TRAIL and downregulation of constitutively active Akt by pharmacological or genetic approaches altered the cellular responsiveness to TRAIL.
TRAIL in combination with agents that downregulate Akt activity can have clinical applicability in treating TRAIL‐resistant bladder cancer cells.
References
- 1. Schulze‐Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem 1998; 254: 439–59. [DOI] [PubMed] [Google Scholar]
- 2. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998; 281: 1305–8. [DOI] [PubMed] [Google Scholar]
- 3. Walczak H, Krammer PH. The CD95 (APO‐1/Fas) and the TRAIL (APO‐2L) apoptosis systems. Exp Cell Res 2000; 256: 58–66. [DOI] [PubMed] [Google Scholar]
- 4. Griffith TS, Lynch DH. TRAIL: a molecule with multiple receptors and control mechanisms. Curr Opin Immunol 1998; 10: 559–63. [DOI] [PubMed] [Google Scholar]
- 5. Keane MM, Ettenberg SA, Nau MM, Russell EK, Lipkowitz S. Chemotherapy augments TRAIL‐induced apoptosis in breast cell lines. Cancer Res 1999; 59: 734–41. [PubMed] [Google Scholar]
- 6. French LE, Tschopp J. The TRAIL to selective tumor death. Nat Med 1999; 5: 146–7. [DOI] [PubMed] [Google Scholar]
- 7. Gura T. How TRAIL kills cancer cells, but not normal cells. Science 1997; 277: 768. [DOI] [PubMed] [Google Scholar]
- 8. Griffith TS, Chin WA, Jackson GC, Lynch DH, Kubin MZ. Intracellular regulation of TRAIL‐induced apoptosis in human melanoma cells. J Immunol 1998; 161: 2833–40. [PubMed] [Google Scholar]
- 9. Muhlenbeck F, Haas E, Schwenzer R et al. TRAIL/Apo2L activates c‐Jun NH2‐terminal kinase (JNK) via caspase‐dependent and caspase‐independent pathways. J Biol Chem 1998; 273: 33091–8. [DOI] [PubMed] [Google Scholar]
- 10. Suliman A, Lam A, Datta R, Srivastava RK. Intracellular mechanisms of TRAIL: apoptosis through mitochondrial‐dependent and ‐independent pathways. Oncogene 2001; 20: 2122–33. [DOI] [PubMed] [Google Scholar]
- 11. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–90. [DOI] [PubMed] [Google Scholar]
- 12. Kennedy SG, Wagner AJ, Conzen SD et al. The PI 3‐kinase/Akt signaling pathway delivers an anti‐apoptotic signal. Genes Dev 1997; 11: 701–13. [DOI] [PubMed] [Google Scholar]
- 13. Cheng JQ, Ruggeri B, Klein WM et al. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci USA 1996; 93: 3636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng JQ, Godwin AK, Bellacosa A et al. AKT2, a putative oncogene encoding a member of a subfamily of protein‐serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci USA 1992; 89: 9267–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takenaka Y, Fukumori T, Yoshii T et al. Nuclear export of phosphorylated galectin‐3 regulates its antiapoptotic activity in response to chemotherapeutic drugs. Mol Cell Biol 2004; 24: 4395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim EJ, Suliman A, Lam A, Srivastava RK. Failure of Bcl‐2 to block mitochondrial dysfunction during TRAIL‐induced apoptosis. Tumor necrosis‐related apoptosis‐inducing ligand. Int J Oncol 2001; 18: 187–94. [PubMed] [Google Scholar]
- 17. Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med 2000; 6: 513–9. [DOI] [PubMed] [Google Scholar]
- 18. Green DR, Reed JC. Mitochondria and apoptosis. Science 1998; 281: 1309–12. [DOI] [PubMed] [Google Scholar]
- 19. Nagata S. Apoptosis by death factor. Cell 1997; 88: 355–65. [DOI] [PubMed] [Google Scholar]
- 20. Wajant H, Haas E, Schwenzer R et al. Inhibition of death receptor‐mediated gene induction by a cycloheximide‐sensitive factor occurs at the level of or upstream of Fas‐associated death domain protein (FADD). J Biol Chem 2000; 275: 24357–66. [DOI] [PubMed] [Google Scholar]
- 21. Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non‐small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res 2001; 61: 3986–97. [PubMed] [Google Scholar]
- 22. Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev 1999; 13: 2905–27. [DOI] [PubMed] [Google Scholar]
- 23. Datta SR, Dudek H, Tao X et al. Akt phosphorylation of BAD couples survival signals to the cell‐intrinsic death machinery. Cell 1997; 91: 231–41. [DOI] [PubMed] [Google Scholar]
- 24. Cardone MH, Roy N, Stennicke HR et al. Regulation of cell death protease caspase‐9 by phosphorylation. Science 1998; 282: 1318–21. [DOI] [PubMed] [Google Scholar]
- 25. Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF‐kappaB activation by tumour necrosis factor requires the Akt serine‐threonine kinase. Nature 1999; 401: 82–5. [DOI] [PubMed] [Google Scholar]
- 26. Romashkova JA, Makarov SS. NF‐kappaB is a target of AKT in anti‐apoptotic PDGF signalling. Nature 1999; 401: 86–90. [DOI] [PubMed] [Google Scholar]
- 27. Kops GJ, Burgering BM. Forkhead transcription factors: new insights into protein kinase B (c‐akt) signaling. J Mol Med 1999; 77: 656–65. [DOI] [PubMed] [Google Scholar]
- 28. Uslu R, Borsellino N, Frost P et al. Chemosensitization of human prostate carcinoma cell lines to anti‐fas‐mediated cytotoxicity and apoptosis. Clin Cancer Res 1997; 3: 963–72. [PubMed] [Google Scholar]