

Abstract
The C‐terminal cytoplasmic domain of the metastatic potentiator CXCR4 regulates its function and spatiotemporal expression. However, little is known about the mechanism underlying constitutive internalization of CXCR4 compared to internalization mediated by its ligand, stromal cell‐derived factor‐1 alpha (SDF‐1α)/CXCL12. We established a system to analyze the role of the CXCR4 cytoplasmic tail in steady‐state internalization using the NP2 cell line, which lacks endogenous CXCR4 and SDF‐1α. Deleting more than six amino acids from the C‐terminus dramatically reduced constitutive internalization of CXCR4. Alanine substitution mutations revealed that three of those amino acids Ser344 Glu345 Ser346 are essential for efficient steady‐state internalization of CXCR4. Mutating Glu345 to Asp did not disrupt internalization, suggesting that the steady‐state internalization motif is S(E/D)S. When responses to SDF‐1α were tested, cells expressing CXCR4 mutants lacking the C‐terminal 10, 14, 22, 31 or 44 amino acids did not show downregulation of cell surface CXCR4 or the cell migration induced by SDF‐1α. Interestingly, however, we identified two mutants, one with E344A mutation and the other lacking the C‐terminal 17 amino acids, that were defective in constitutive internalization but competent in ligand‐promoted internalization and cell migration. These data demonstrate that ligand‐dependent and ‐independent internalization is genetically separable and that, between amino acids 336 and 342, there is a negative regulatory element for ligand‐promoted internalization. Potential involvement of this novel motif in cancer metastasis and other CXCR4‐associated disorders such as warts, hypogammaglobulinemia, infections and myelokathexis (WHIM) syndrome is discussed. (Cancer Sci 2007; 98: 373–379)
The chemokine receptor CXCR4 is a class‐A G protein‐coupled receptor (GPCR; reviewed in ( 1 , 2 ) and its natural ligand is stromal cell‐derived factor‐1 alpha (SDF‐1α)/CXCL12. CXCR4 also serves as the receptor for HIV type 1 (HIV‐1). Many cell types express CXCR4, including peripheral blood lymphocytes, monocytes‐macrophages, thymocytes, dendritic cells, endothelial cells, epithelium‐derived tumor cells, microglial cells, neurons and hematopoietic stem cells. CXCR4 plays multiple biological roles from promoting development of neuronal networks to regulating migration of leukocytes, cerebellar granule cells and hematopoietic stem cells.( 3 , 4 , 5 , 6 , 7 , 8 ) Analysis of knockout mice indicates that the CXCR4/SDF‐1α system is essential for maintenance of hematopoiesis and intestinal vascularization.( 9 , 10 )
The CXCR4/SDF‐1α system also functions in pathological processes, including autoimmune diseases, cancer progression and metastasis, and AIDS caused by HIV‐1. Recently, metastasis of breast cancer cells was found to be regulated by the CXCR4/SDF‐1α axis.( 5 ) Similarly, other studies have found that metastasis of other malignancies was controlled by the CXCR4/SDF‐1α system, including colon carcinoma( 11 ) non‐small cell lung cancer( 12 ) and prostate cancer.( 13 ) These observations suggest that the CXCR4/SDF‐1α axis is a potential target for metastatic cancer therapy.
Warts, hypogammaglobulinemia, infections and myelokathexis (WHIM) syndrome is a rare combined immunodeficiency characterized by an unusual form of neutropenia. It is reported that the CXCR4 cytoplasmic tail is mutated and often truncated in WHIM syndrome.( 14 ) Thus, determining the biochemical activity of the CXCR4 cytoplasmic tail should facilitate understanding of the pathogenesis of WHIM syndrome as well as suggest ways to control cancer metastasis.
Following SDF‐1α binding, CXCR4 is activated, triggering multiple signaling cascades via Gα or β‐arrestin 2 (reviewed in( 15 )). To desensitize activated CXCR4, the G protein‐coupled receptor kinase (GRK) is recruited and phosphorylates serine residues on the CXCR4 cytoplasmic tail, thereby inactivating Gα‐mediated signal. Simultaneously, CXCR4 is internalized in a clathrin‐dependent manner. β‐arrestin 2 competes with Gα for CXCR4 binding and can initiate signal transduction independent from Gα. β‐arrestin 2 can also induce clathrin‐dependent CXCR4 endocytosis. Thus, cell surface levels of CXCR4 transiently decrease after agonist binding but, several hours later, surface levels of CXCR4 return to normal. Most internalized CXCR4 is transported to lysosomes and degraded, but some internalized CXCR4 is recycled. It is reported that amino acids within the cytoplasmic tail are required for agonist‐dependent endocytosis of CXCR4.( 16 , 17 , 18 )
By contrast, it is unclear how steady‐state cell surface levels of CXCR4 are maintained in the absence of SDF‐1α. Although cell surface levels of CXCR4 could be regulated at the transcriptional level, it is likely that primary regulation occurs post‐translationally. Given that the cell surface levels of CXCR4 are positively correlated with cancer cells’ ability to metastasize,( 5 , 19 ) understanding the post‐translational behavior of CXCR4 is likely to shed light on metastatic processes. Historically, cells expressing endogenous CXCR4 have been used for analysis of CXCR4 trafficking. However, as is the case with many G protein‐coupled receptors (GPCR), CXCR4 trafficking is influenced by spontaneous oligomerization in the absence of ligand.( 20 , 21 , 22 ) Thus, previous observations might not correctly model phenotypes seen in CXCR4 mutants.
In the present study, we analyzed the contribution of the cytoplasmic tail to the post‐translational trafficking of CXCR4 in a cell line lacking both endogenous CXCR4 and SDF‐1α. Using genetic approaches, we identified two amino acid motifs within the CXCR4 cytoplasmic tail; one that positively regulates spontaneous ligand‐independent internalization and the other that negatively regulates ligand‐dependent CXCR4 internalization.
Materials and methods
Cells. The glioblastoma cell line NP2, human embryonic kidney (HEK) 293T, and HeLa, cells were maintained in RPMI‐1640 (Sigma, Tokyo, Japan) supplemented with 10% FBS (Japan Bioserum, Tokyo, Japan), penicillin and streptomycin (Invitrogen, Tokyo, Japan). All cell lines were incubated at 37°C in the humidified 5% CO2 atmosphere.
Plasmids. Full‐length CXCR4 cDNA was amplified from a plasmid kindly provided by Dr Shioda( 23 ) using the following primers: sense, 5′‐ACCGGTGCCACCATGGAGGGGATCAGTATATACACTTCAG‐3′, and antisense, 5′‐AGATCTCGCTGGAGTGAAAACTTGAAGACTCAGACTC‐3′. CXCR4 lacking the cytoplasmic tail (d‐44) was amplified using the same sense primer and the antisense primer, 5′‐AGATCTTGGCTCCAAGGAAAGCATAGAGGATGGG‐3′. Polymerase chain reaction (PCR) fragments were cloned into the Age I‐Bgl II sites of pEGFP‐C2 (Clontech, Palo Alto, CA, USA) to create pCXCR4 FL and pCXCR4 d‐44, respectively. To construct pCXCR4 FL‐ and d‐44‐GFP, the Sna BI‐Bgl II fragments from pCXCR4 FL and d‐44 were cloned into the Sna BI‐Bgl II sites of pEGFP‐N2, respectively (Clontech). To construct pCXCR4 FL‐ and d‐44‐GFP flag, the Sna BI‐Bgl II fragments from pCXCR4 FL and d‐44 were cloned into the Sna BI‐Bgl II sites of pEGFP‐flag in which the following annealed oligonucleotides had been inserted into the Bsr GI site of pEGFP‐N2: forward, 5′‐GTACGACTACAAAGACGATGACGACTATAAGTAAGC‐3′, and reverse, 5′‐ GGCCGCTTACTTATAGTCGTCATCGTCTTTGTAGTC‐3′. To construct pCMMP CXCR4 FL‐ and d‐44‐GFP, pCXCR4 FL‐ and d‐44‐GFP were digested with Not I, blunted using T4 DNA polymerase, and further digested with Age I. The Age I‐blunted Not I fragments of both constructs were cloned into the pCMMP eGFP plasmid that had been digested with Bam HI, blunted with T4 DNA polymerase, and digested with Age I. pCMMP CXCR4 FL‐ and d‐44‐GFP‐flag were constructed using the same strategy. CXCR4 deletion and point mutants were PCR‐amplified using the sense primer 5′‐ACCGGTGCCACCATGGAGGGGATCAGTGTGAAAACTTGAAGACTCAGACTC‐3′ and the following reverse primers: d‐6, 5′‐AAGCTTGAGCTCGAGATCTCAGACTCAGACTCAGTGGAAAC‐3′; d‐10, 5′‐AAGCTTGAGCTCGAGATCTCAGTGGAAACAGATGAATGTCC‐3′; d‐14, 5′‐AAGCTTGAGCTCGAGATCTCTGAATGTCCACCTCGCTTTCC‐3′; d‐17, 5′‐AAGCTTGAGCTCGAGATCTCACCTCGCTTTCCTTTGG‐3′; d‐22, 5′‐AAGCTTGAGCTCGAGATCTCGGAGAGGATCTTGAGGCTGGACC‐3′; d‐31, 5′‐AAGCTTGAGCTCGAGATCTCGCTCACAGAGGTGAGTGCGTGC‐3′; E343A, 5′‐CGAGATCTCGCTGGAGTGAAAACTTGAAGACTCAGACGCAGTGGAAACAGATGAATGTC‐3′; S344A, 5′‐CGAGATCTCGCTGGAGTGAAAACTTGAAGACTCAGCCTCAGTGGAAACAGATGAATGTC‐3′; E345A, 5′‐CGAGATCTCGCTGGAGTGAAAACTTGAAGACGCAGACTCAGTGGAAACAGATGAATGTC‐3′; S346A, 5′‐CGAGATCTCGCTGGAGTGAAAACTTGAAGCCTCAGACTCAGTGGAAACAGATGAATGTC‐3′; S347E, 5′‐CGAGATCTCGCTGGAGTGAAAACTTTCAGACTCAGACTCAGTGGAAACAGATGAATGTC‐3′; H350E, 5′‐CGAGATCTCGCTGGACTCAAAACTTGAAGACTCAGACTCAGTGGAAACAGATGAATGTC‐3′; S347E/H350E, 5′‐CGAGATCTCGCTGGACTCAAAACTTTCAGACTCAGACTCAGTGGAAACAGATGAATGTC‐3′; and E343/345D, 5′‐CGAGATCTCGCTGGAGTGAAAACTTGAAGAGTCAGAGTCAGTGGAAACAGATGAATGTC‐3′. The PCR fragments were cloned into the Age I‐Bgl II sites of pCMMP CXCR4 FL‐GFP‐flag, replacing wild‐type with mutant CXCR4. Protein expression of each mutant in 293T cells was verified by Western blot analysis.
Immunoblotting. Immunoblotting was performed as described.( 24 , 25 ) The primary antibody was anti‐green fluorescent protein (GFP) polyclonal antibody (Beckton Dickinson, San Jose, CA, USA). The secondary probe was EnVision+ (Dako, Glostrup, Denmark). Signals were visualized with an LAS3000 imager (Fuji Film, Tokyo, Japan) after treating the membranes with the Lumi‐Light Western Blotting Substrate (Roche Diagnostics GmbH, Mannheim, Germany).
Flow cytometry. Cells were labeled with anti‐CXCR4 antibodies recognizing the N‐terminus conjugated with R‐phycoerythrin (PE; 2B11, BD Pharmingen, San Diego, CA) or recognizing the second extracellular loop (12G5) conjugated with either PE or PE‐Cy5 (Beckton Dickinson) for 30 min at 4°C. Cells were washed once with phosphate‐buffered saline (PBS) supplemented with 1% FBS and analyzed by FACS Aria (Beckton Dickinson). To isolate GFP‐expressing NP2 cells, cells were infected with murine leukemia virus (MLV)‐based retroviral vectors as described.( 25 ) Cells exhibiting similar green fluorescence intensities were gated and sorted by FACS Aria. Efficiency of internalization was measured by comparing mean fluorescence intensities for cell surface CXCR4 detected by a PE‐labeled 2B11 monoclonal antibody before and after SDF‐1α treatment (200 ng/mL, Peprotech EC, London, UK).
Microscopic analysis and imaging of cells. To judge a phenotype of a CXCR4 mutant, three independent scientists investigated the mutant cell phenotype under a fluorescent microscope (Olympus, Tokyo, Japan). Each scientist investigated more than 1000 cells for each mutant. More than 99% of cells of a mutant fell in the indicated phenotypic category. These phenotypes were unchanged for more than a year of continuous cultivation in tissue culture. For imaging, NP2 cells were grown on glass plates for more than 24 h, fixed in 4% formaldehyde in PBS for 5 min, stained with Hoechst 33258, mounted (Vectorshield, Vector Laboratories, Burlingame, CA, USA), and imaged using a confocal microscope META 510 (Carl Zeiss, Tokyo, Japan). A representative cell for each CXCR4 mutant carrying a wide cytoplasm was chosen such that the spatial resolution was high. The focal plane just above the glass surface was scanned with an optical thickness of approximately 1 µm. For the imaging of subcellular compartments, cells were incubated with either BODIPY TR ceramid, ER‐Tracker Blue‐White DPX, or Lysotracker Red DND‐99 (Invitrogen) according to the manufacturer's protocol and imaged without fixation. Image brightness and contrast were processed by META510 software (Carl Zeiss). Unless noted, cells were imaged at ×630 magnification, the GFP signal was displayed in green, and Hoechst 33258‐stained nuclei were blue. To visualize ligand‐induced internalization, cells were treated with 200 ng/mL SDF‐1α before fixation. The live cell imaging was performed using Leica DFC350FX system and the images were processed by FW4000 software (Leica Microsystems, Tokyo, Japan). Cells were plated on the glass‐bottomed dish (Matsunami glass, Kishiwada, Japan) and incubated at 37°C in the humidified 5% CO2 atmosphere during the monitoring.
Cell migration assay. Cell migration was measured using an HTS FluoroBlok Multiwell Insert System (8.0 µm pore size, BD Falcon) according to the manufacturer's protocol. For stimulation assays, cells were incubated without serum overnight before SDF‐1α treatment (200 ng/mL). Cells were allowed to migrate overnight.
Statistical analysis. Significance of differences were determined by a Student's t‐test. P‐values less than 0.05 were considered significant.
RESULTS
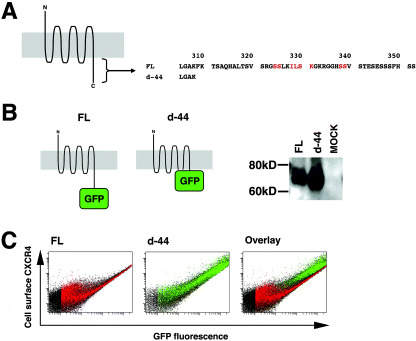
Deleting 10 amino acids from the carboxyl end of CXCR4 alters the efficiency of constitutive internalization. Previous studies indicated that the cytoplasmic tail of CXCR4 amino acids 308–352 plays a critical role in ligand‐dependent internalization (Fig. 1a). Also, it has been shown in transfected cells that cell surface levels of CXCR4 lacking the cytoplasmic tail (equivalent to the d‐44 mutant here) are higher than those of the full length, wild‐type protein (hereafter designated FL), suggesting that the cytoplasmic tail of CXCR4 regulates steady‐state internalization.( 26 , 27 ) To confirm this, we constructed expression plasmids of CXCR4 FL and d‐44 fused to GFP or GFP‐FLAG at the C‐terminus. Previous studies and data reported here indicated that CXCR4 function is not affected by this modification.( 28 ) The expression of each construct was verified by Western blot analysis (Fig. 1b). Single cell‐based quantitative analyzes revealed that the ratio of cell surface levels to the total amount of CXCR4 FL (Fig. 1c, left) was consistently lower than that of d‐44 (Fig. 1c, middle) at any expression levels (Fig. 1c, right for the comparison). These data supported previous findings and demonstrate that constitutive internalization occurs at any level of CXCR4 expression.
Figure 1.
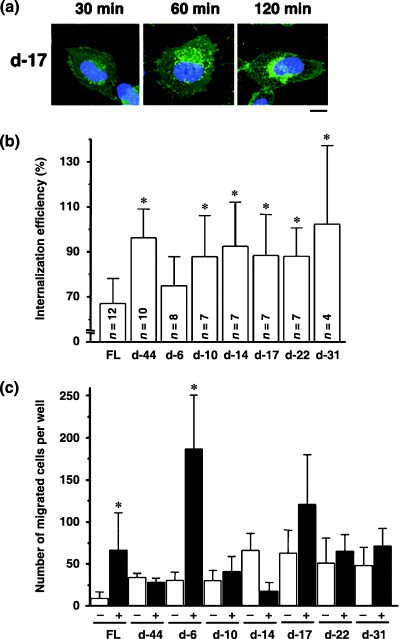
The effect of stromal cell‐derived factor‐1 alpha (SDF‐1α) treatment on NP2 cells expressing CXCR4 mutants. (a) Cells expressing d‐17 were treated with SDF‐1α, incubated at 37°C for the indicated times, fixed and imaged. The blue signal represents the Hoechst‐stained nucleus. (Original magnification, ×630; bar, 10 µm). (b) FACS analysis to measure internalization efficiency of cell surface CXCR4 and mutant forms 2 h after SDF‐1α exposure. The average and standard deviation from the indicated number of independent experiments are shown. Asterisks represent statistically significant difference from the FL levels (P < 0.01). (c) Cell migration assay to assess response of cells expressing CXCR4 and mutants to SDF‐1α. The number of migrated cells in three to six randomly selected fields was counted and the average and standard deviation were calculated. (□) number of migrated cells in the absence of ligand; (▪) migration in the presence of ligand. (*) statistically significant differences in the number of migrated cells between SDF‐1α‐untreated and ‐treated cells (P < 0.01).
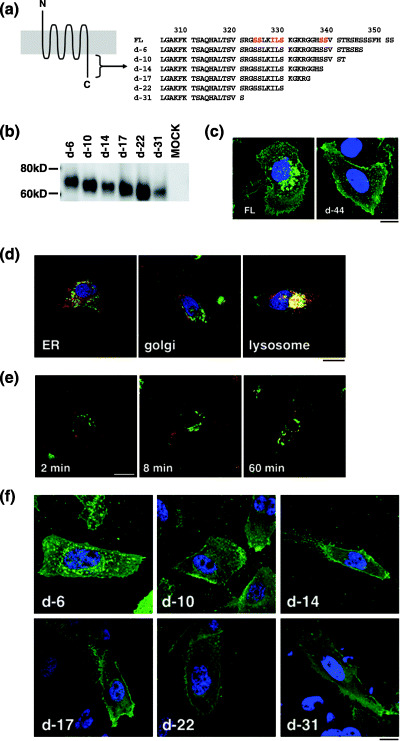
To further examine the contribution of the cytoplasmic tail to post‐translational trafficking of CXCR4, we devised a system utilizing the human NP2 glioma line: NP2 cells are flat and exhibit a large cytoplasmic space such that intracellular compartments can be well resolved under the microscope. NP2 cells also lack endogenous CXCR4( 29 ) and SDF‐1α (data not shown), both of which could potentially affect distribution of transduced CXCR4. However, NP2 cells are capable of appropriate signaling in response to CXCR4/SDF‐1α interaction. We generated a series of CXCR4 deletion mutants lacking the cytoplasmic tail (Fig. 2a) and transduced them into NP2 cells using MLV vectors. Cells bearing similar green fluorescence intensities were collected by FACS sorter. The expression of each mutant was verified by Western blot analysis (Fig. 2b). Microscopic observations revealed that cells expressing FL were bordered by green fluorescence, and significant green fluorescence was detected in vesicular compartments of varying diameters lying close to the nucleus surrounding the nucleus (hereafter designated the FL phenotype, Fig. 2c, left). Vesicles around the nucleus were mostly lysosomes, as demonstrated by fluorescent organella marker analyses in which cells expressing CXCR4 FL‐GFP stained with the lysosomal marker yielded a substantial amount of co‐localization signal. On the other hand, only a mall amount of co‐localization signal was detected when the ER or Golgi markers were used (Fig. 2d), consistent with our biochemical fractionation (unpublished data) and previous publications.( 16 , 27 , 28 , 30 ) The active constitutive internalization was visualized by labeling cell surface CXCR4 by PE‐Cy5‐conjugated monoclonal antibody followed by fluorescence imaging after cells were incubated at 37°C (Fig. 2e). The live cell imaging revealed that internalizing GFP‐positive vesicles trafficked at an average velocity of 4.7 mm/h (n = 15), which is within the range of clathrin‐dependent vesicular transport (2–20 mm/h), not that of caveolin‐dependent vesicular transport (25–170 mm/h).( 31 , 32 , 33 , 34 , 35 ) These data suggest that the FL is constitutively internalized from the cell surface to the cytoplasmic compartment. In sharp contrast, most green fluorescent signals from d‐44 mutant‐expressing cells were detected at the cell surface, and only a few small GFP‐positive vesicles were seen in the cytoplasm near the nucleus (hereafter designated the d‐44 phenotype, Fig. 2c, right). Similar observations were made in d‐10, d‐14, d‐17, d‐22 and d‐31 mutant‐expressing cells (Fig. 2f). The d‐6 construct displayed a phenotype similar to FL, although the intracellular GFP signal was less prominent (Fig. 2c). Similar results were obtained in HeLa and 293 cells (data not shown). These data suggest that wild‐type CXCR4 was trafficked to the plasma membrane but was internalized spontaneously. Thus, steady‐state internalization appeared to be regulated by amino acids located between d‐6 and d‐10 (e.g. amino acids 343–346).
Figure 2.
Expression profiles of CXCR4 and a mutant with cytoplasmic tail deletion. (a) Schematic representation of CXCR4. The N‐terminus CXCR4 is exposed in the extracellular space and the C‐terminus is intracellular. Gray represents the lipid bilayer. The amino acid sequence of the cytoplasmic tail is shown. Residues in red are required for ligand‐induced endocytosis. The CXCR4 d‐44 mutant lacks amino acid 309–351. (b) Schematic representation and Western blot of FL and d‐44 constructs. (c) Flow cytometry profiles of FL and d‐44 expressed in 293T cells. The horizontal axis represents green fluorescence intensity indicative of green fluorescent protein (GFP)‐tagged CXCR4 protein levels, and the vertical axis is PE‐Cy5 fluorescence intensity, reflecting cell surface CXCR4 detected by the anti‐CXCR4 antibody. GFP‐positive cells expressing FL are colored in red (left) and those expressing d‐44 in green (middle). The expressional differences between FL and d‐44 is highlighted on the overlay plot (right).
Steady‐state and SDF‐1α‐induced CXCR4 internalization is genetically separable. Next, we investigated distribution of CXCR4 protein and cell migration after SDF‐1α treatment. Confocal analysis showed that after SDF‐1α exposure, cells expressing FL, d‐6 and d‐17 mutants showed GFP signals in intracellular compartments, which were enhanced 60 min after SDF‐1α treatment, an effect most clearly shown in d‐17‐expressing cells (Fig. 3a). GFP signals from intracellular vesicles gradually disappeared 1–2 h after exposure to ligand. Such redistribution of GFP signals was not observed in cells expressing d‐10, d‐14, d‐22, d‐31 and d‐44 (data not shown). Cell surface levels of CXCR4 before and after SDF‐1α treatment were measured by FACS analysis undertaken with an antibody directed against the CXCR4 N‐terminus, because that antibody did not interfere with ligand–receptor interaction (Fig. 3b). The downregulation of cell surface levels of FL 2 h after ligand exposure was 67.1 ± 11.1%, whereas that of d‐44 was 96.3 ± 12.3% (average and standard deviation from 12 and 10 independent experiments, respectively), consistent with previous reports.( 17 , 27 , 28 ) Ligand‐induced downregulation of d‐6 was 74.9 ± 12.9% (n = 8), similar to FL levels. Ligand‐induced internalization was significantly less efficient in cells expressing d‐10, d‐14, d‐17, d‐22, d‐31 and d‐44 mutants when compared with FL (P < 0.001). Although the d‐17 mutant supported ligand‐facilitated internalization, as evidenced by microscopic observation, cell surface levels remained unchanged (Fig. 3a,b). This may be due in part to rapid recruitment of newly synthesized d‐17 to the cell surface.
Figure 3.
Identification of the amino acids required for steady‐state CXCR4 internalization. (a) Amino acid sequences of the cytoplasmic tail of FL and deletion mutants. Residues in red are required for ligand‐induced endocytosis. (b) The protein expression of each mutant in 293T cells was verified by Western blot analysis. (c) Confocal micrographs of NP2 cells expressing FL and d‐44 mutant proteins. The blue signal represents the Hoechst‐stained nucleus. (Original magnification, ×630; bar, 10 µm.) (d) Confocal micrographs showing NP2 cells expressing CXCR4 FL stained with ER, Golgi, or lysosome organella markers. The organella marker signal is shown in red, the GFP signal is in green. The pixels that both red and green signals co‐localized are shown in yellow. (Original magnification, ×630; bar, 10 µm.) (e) CXCR4 FL trafficking in the absence of SDF‐1α in NP2 cells. Cell surface CXCR4 FL was labeled with an antibody conjugated with PE‐Cy5 (red), incubated at 37°C for the indicated times, fixed and imaged. (Original magnification, ×630; bar, 10 µm.) (f) Confocal micrographs of NP2 cells expressing FL and mutant proteins. The intracellular vesicular green fluorescence reflecting steady‐state internalization can be seen in the d‐6 mutant. The blue signal represents the Hoechst‐stained nucleus. (Original magnification, ×630; bar, 10 µm.)
Next, we examined cells expressing CXCR4 mutants in response to SDF‐1α. Migration results from intracellular signaling initiated by SDF‐1α/CXCR4 interaction. Induction of cell migration by SDF‐1α in cells expressing FL was 7.2‐fold that of untreated cells (P < 0.05). In contrast, migration of cells expressing d‐44 in response to SDF‐1α was undetectable. These data are in agreement with a previous report.( 26 ) The d‐6 mutant, which is internalized upon SDF‐1α treatment, supported ligand‐promoted cell migration by 6.1‐fold (P < 0.01) relative to untreated cells, similar to FL. Other deletion mutants tested did not display enhanced cell migration following ligand treatment, except for d‐17, which showed modestly enhanced (1.9‐fold) migration relative to untreated cells, which was not statistically significant. When basal migratory activities were compared, removal of six or more amino acids from the cytoplasmic tail appeared to potentiate migration in the absence of ligand (open bars, Fig. 3c). These data suggest that constitutive internalization is regulated independently of ligand‐facilitated internalization.
Identification of CXCR4 S(E/D)S as a ligand‐independent internalization motif. The above data indicated that the carboxy‐terminal four amino acids (ESES; residues 343–346) likely function in ligand‐independent CXCR4 internalization. To further characterize which amino acids are required for ligand‐independent internalization, we generated alanine substitution mutants for each of the four amino acids in the context of FL and examined their phenotypes (Fig. 4a). Protein expression of mutants was verified in Western blot analysis (Fig. 4a). Among the four mutants, the E343A mutant showed the FL phenotype, while the others displayed the d‐44 phenotype in the absence of ligand (Fig. 4b). These data demonstrate that Ser344‐Glu345‐Ser346 constitute the core motif for SDF‐1α‐independent CXCR4 internalization. Both E345A and S346A mutants exhibited the Thr342‐Glu343‐Ser344 sequence adjacent to the original SES sequence. However, this ‘SES‐like’ motif did not support constitutive internalization, suggesting that Thr cannot substitute for Ser to maintain functionality as a constitutive internalization motif. We reasoned that if such a motif requires an acidic amino acid between two serine residues, changing Glu to Asp should maintain the motif's function. Thus, we constructed a mutant in which Glu was replaced with Asp (E343/345D; Fig. 4a). Also, to determine whether two adjacent SES sequences could augment the FL phenotype, we substituted Ser347 with Glu (S347E), creating an additional SES motif next to the original SES one (Fig. 4a). As controls, we created H350E and S347E/H350E mutants (Fig. 4a). Expression of these mutants was verified by Western blot analysis (Fig. 4a). Interestingly, the E343/345D mutant retained the FL phenotype (Fig. 4b), indicating that an acidic residue is required to maintain function of the constitutive internalization motif. S347E showed an intermediate phenotype in which numerous fine GFP‐positive vesicles were seen close to the nucleus (Fig. 4b). These data indicate that the two adjacent SES sequences do not augment the FL phenotype but actually interfere with steady‐state internalization. Both H350E and S347E/H350E mutants also showed an intermediate phenotype (Fig. 4b), suggesting that more than three acidic amino acids close to the SES motif may inhibit its function, potentially by generating a negative charge cluster. Overall, we conclude that the SDF‐1α‐independent internalization motif is located at amino acids 344–346 of the CXCR4 cytoplasmic tail.
Figure 4.
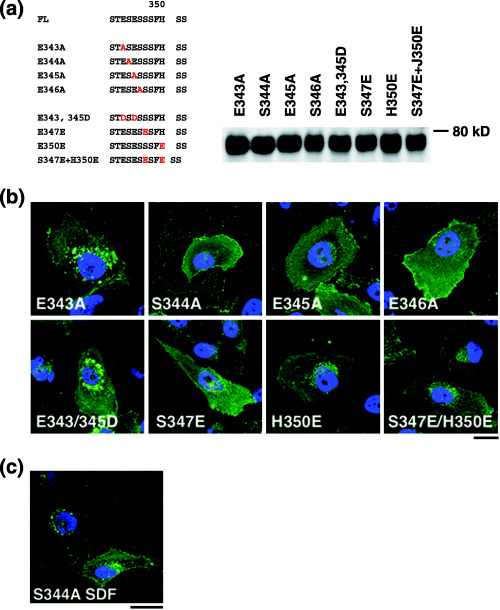
Characterization of the SDF‐1α‐independent internalization motif of CXCR4. (a) Left, amino acid sequences of CXCR4 FL and substitution mutants. Letters in red indicate introduced mutations. Right, protein expression of each mutant in 293T cells was verified by Western blot analysis. (b) Confocal micrographs of NP2 cells expressing each mutant. The blue signal represents the Hoechst‐stained nucleus. (Original magnification, ×630; bar, 10 µm.) (c) NP2 cells expressing the S344A mutant treated with SDF‐1α for 2 h are shown. The blue signal represents the Hoechst‐stained nucleus. (Original magnification, ×630; bar, 10 µm.)
Finally, we analyzed phenotypes of the S344A mutant in greater detail. Two hours after SDF‐1α treatment, cells expressing this mutant showed accumulation of GFP signals at perinuclear regions, similar to the d‐17 mutant (1, 4). FACS analysis revealed that cell surface levels of S344A decreased to 70.8 ± 11.7% (n = 7) following SDF‐1α treatment relative to untreated cells, almost as efficient as FL (Fig. 3b). Migratory activity of cells expressing the S344A mutant was stimulated 3.0‐fold by SDF‐1α, while that of cells expressing FL assayed in parallel showed a 5.8‐fold increase relative to untreated cells. These data demonstrate that the S344A mutant, which is defective in constitutive internalization, can undergo ligand‐dependent internalization and stimulate migration. Along with the d‐17 data, our observations strongly suggest that genetic elements required for the ligand‐dependent and ‐independent internalization are separable.
Discussion
We demonstrated here that CXCR4 is constitutively internalized in the absence of SDF‐1α and that steady‐state trafficking of CXCR4 is regulated by its cytoplasmic tail. We show that the three amino acid motif, Ser344‐Glu345‐Ser346, within the cytoplasmic tail is essential for efficient steady‐state internalization of CXCR4. Our work indicates that ligand‐independent internalization of CXCR4 is genetically separable from ligand‐dependent internalization: mutants defective in steady‐state internalization (d‐17 and S344A) were competent to respond to SDF‐1α‐promoted internalization signals. That residues required for ligand‐dependent endocytosis (Ser324, 325, 330, 338, 339, Ile328 Leu329 and Lys331; summarized in Fig. 1a)( 16 , 17 , 18 ) do not overlap with those required for ligand‐independent internalization, further supports the idea that these activities are separable.
Interestingly, the d‐17 mutant displayed SDF‐1α‐promoted internalization, whereas the d‐14 and d‐22 mutants did not. These data suggest that an element between amino acids 336 and 342 negatively regulates ligand‐initiated CXCR4 internalization. We are currently determining what amino acids are required for that motif. What is unique about the constitutive internalization motif is its position effect in terms of the distance of the motif from the “body” of the receptor. SES‐like motifs can be found in the cytoplasmic tails of both CXC‐chemokine receptors (for example, CXCR3) and CC‐chemokine receptors including CCR2, CCR5 and CCR7. Indeed, these receptors share similar amino acid sequences in which two acidic amino acids (mostly Asp) positioned between the 36th and 45th amino acids of the cytoplasmic tail, where Ser and Thr residues are often in the close proximity to the acidic amino acids but positively charged amino acids, are infrequent. We hypothesize that for the ligand‐independent internalization motif to function, the SES motif or its equivalent must be positioned at approximately the 40th residue of the cytoplasmic tail.
Many GPCR, including α1a‐adrenoceptor, and the µ‐opioid receptor, are spontaneously internalized.( 36 , 37 ) Therefore, we conclude that various GPCR actively and continuously undergo endocytosis in the absence of ligand in a manner similar to CXCR4 and hypothesize that the function of constitutive receptor internalization is to fine‐tune the threshold at which cells sense ligand. Cells should be able to rapidly post up‐ and downregulate cell surface levels of CXCR4 using post‐translational mechanisms. Such regulation should enable cells to migrate toward SDF‐1α‐rich tissues as needed and should also prevent inappropriate cells from migrating.
Our work is relevant to cancer cell metastasis and the pathogenesis of WHIM syndrome. Cell surface levels of CXCR4 positively correlate with cancer cells’ ability to metastasize.( 5 , 19 ) We hypothesize that enhanced metastatic capabilities of cancer cells could be due in part to mutations that disrupt the function of SES motif, which would result in upregulation of cell surface levels of signal‐competent CXCR4 (as exemplified by the E344A mutant). As for WHIM syndrome, it was recently reported that it is due to mutations within CXCR4's cytoplasmic domain.( 14 , 38 ) Interestingly, these mutations result in loss of SES motif. We predict that loss of the SES motif should increase cell surface CXCR4 levels. Although CXCR4 mutations generated here are not identical to reported WHIM mutations, the d‐10 mutant resembles mutations seen in WHIM syndrome, and it exhibits enhanced basal cell migratory activity. Increased cell surface CXCR4 or increased migratory potential may contribute to WHIM pathogenesis. The response of d‐10‐expressing cells to SDF‐1α, however, was not as robust as that of cells derived from WHIM.( 39 , 40 ) This discordance may be partly due to the cell type differences, as we have employed a glioblastoma cell line for our studies.
Thus, CXCR4 is a potentially important therapeutic target not only for cancers but for other conditions such as HIV‐1 infection, chronic autoimmune disease, and genetic disorders including WHIM syndrome. CXCR4 also plays critical roles in embryogenesis, homeostasis and inflammation. Although there are potential caveats for treating cancer with CXCR4 antagonists, our data furthers the understanding of mechanisms regulating CXCR4 and could be useful in devising therapeutic strategies.
Acknowledgments
We thank Drs Toshitada Takemori and Tsutomu Murakami for critical reading of the manuscript. This work was supported in part by the Japan Human Science Foundation, the Japanese Ministry of Health, Labor and Welfare, and the Japanese Ministry of Education, Culture, Sports, Science and Technology.
References
- 1. Gether U. Uncovering molecular mechanisms involved in activation of G protein‐coupled receptors. Endocr Rev 2000; 21: 90–113. [DOI] [PubMed] [Google Scholar]
- 2. Ferguson SS. Evolving concepts in G protein‐coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev 2001; 53: 1–24. [PubMed] [Google Scholar]
- 3. Sapede D, Rossel M, Dambly‐Chaudiere C, Ghysen A. Role of SDF1 chemokine in the development of lateral line efferent and facial motor neurons. Proc Natl Acad Sci USA 2005; 102: 1714–8. Epub 2005 January 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coughlan CM, McManus CM et al. Expression of multiple functional chemokine receptors and monocyte chemoattractant protein‐1 in human neurons. Neuroscience 2000; 97: 591–600. [DOI] [PubMed] [Google Scholar]
- 5. Muller A, Homey B, Soto H et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001; 410: 50–6. [DOI] [PubMed] [Google Scholar]
- 6. Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998; 393: 595–9. [DOI] [PubMed] [Google Scholar]
- 7. Ma Q, Jones D, Borghesani PR et al. Impaired B‐lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4‐ and SDF‐1‐deficient mice. Proc Natl Acad Sci USA 1998; 95: 9448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang JF, Park IW, Groopman JE. Stromal cell‐derived factor‐1alpha stimulates tyrosine phosphorylation of multiple focal adhesion proteins and induces migration of hematopoietic progenitor cells: roles of phosphoinositide‐3 kinase and protein kinase C. Blood 2000; 95: 2505–13. [PubMed] [Google Scholar]
- 9. Kawabata K, Ujikawa M, Egawa T et al. A cell‐autonomous requirement for CXCR4 in long‐term lymphoid and myeloid reconstitution. Proc Natl Acad Sci USA 1999; 96: 5663–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peled A, Petit I, Kollet O et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 1999; 283: 845–8. [DOI] [PubMed] [Google Scholar]
- 11. Zeelenberg IS, Ruuls‐Van Stalle L, Roos E. The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res 2003; 63: 3833–9. [PubMed] [Google Scholar]
- 12. Phillips RJ, Burdick MD, Lutz M, Belperio JA, Keane MP, Strieter RM. The stromal derived factor‐1/CXCL12‐CXC chemokine receptor 4 biological axis in non‐small cell lung cancer metastases. Am J Respir Crit Care Med 2003; 167: 1676–86. [DOI] [PubMed] [Google Scholar]
- 13. Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell‐derived factor‐1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res 2002; 62: 1832–7. [PubMed] [Google Scholar]
- 14. Hernandez PA, Gorlin RJ, Lukens JN et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet 2003; 34: 70–4. [DOI] [PubMed] [Google Scholar]
- 15. Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta‐arrestins. Science 2005; 308: 512–7. [DOI] [PubMed] [Google Scholar]
- 16. Marchese A, Benovic JL. Agonist‐promoted ubiquitination of the G protein‐coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem 2001; 276: 45 509–12. [DOI] [PubMed] [Google Scholar]
- 17. Orsini MJ, Parent JL, Mundell SJ, Benovic JL, Marchese A. Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c‐terminal tail that mediate receptor internalization. J Biol Chem 1999; 274: 31 076–86. [DOI] [PubMed] [Google Scholar]
- 18. Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein‐coupled receptor CXCR4. Dev Cell 2003; 5: 709–22. [DOI] [PubMed] [Google Scholar]
- 19. Darash‐Yahana M, Pikarsky E, Abramovitch R et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB 2004; J 18: 1240–2. [DOI] [PubMed] [Google Scholar]
- 20. Vila‐Coro AJ, Rodriguez‐Frade JM, Martin De Ana A, Moreno‐Ortiz MC, Martinez AC, Mellado M. The chemokine SDF‐1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J 1999; 13: 1699–710. [PubMed] [Google Scholar]
- 21. Babcock GJ, Farzan M, Sodroski J. Ligand‐independent dimerization of CXCR4, a principal HIV‐1 coreceptor. J Biol Chem 2003; 278: 3378–85. [DOI] [PubMed] [Google Scholar]
- 22. Terrillon S, Bouvier M. Roles of G‐protein‐coupled receptor dimerization. EMBO Rep 2004; 5: 30–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu H, Shioda T, Hori T et al. Dissociation of ligand‐induced internalization of CXCR‐4 from its co‐receptor activity for HIV‐1 Env‐mediated membrane fusion. Arch Virol 1998; 143: 851–61. [DOI] [PubMed] [Google Scholar]
- 24. Yanagida M, Hayano T, Yamauchi Y et al. Human fibrillarin forms a sub‐complex with splicing factor 2‐associated p32, protein arginine methyltransferases, and tubulins alpha 3 and beta 1 that is independent of its association with preribosomal ribonucleoprotein complexes. J Biol Chem 2004; 279: 1607–14. [DOI] [PubMed] [Google Scholar]
- 25. Komano J, Miyauchi K, Matsuda Z, Yamamoto N. Inhibiting the Arp2/3 complex limits infection of both intracellular mature vaccinia virus and primate lentiviruses. Mol Biol Cell 2004; 15: 5197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roland J, Murphy BJ, Ahr B et al. Role of the intracellular domains of CXCR4 in SDF‐1‐mediated signaling. Blood 2003; 101: 399–406. [DOI] [PubMed] [Google Scholar]
- 27. Haribabu B, Richardson RM, Fisher I et al. Regulation of human chemokine receptors CXCR4. Role of phosphorylation in desensitization and internalization. J Biol Chem 1997; 272: 28 726–31. [DOI] [PubMed] [Google Scholar]
- 28. Tarasova NI, Stauber RH, Michejda CJ. Spontaneous and ligand‐induced trafficking of CXC‐chemokine receptor 4. J Biol Chem 1998; 273: 15 883–6. [DOI] [PubMed] [Google Scholar]
- 29. Soda Y, Shimizu N, Jinno A et al. Establishment of a new system for determination of coreceptor usages of HIV based on the human glioma NP‐2 cell line. Biochem Biophys Res Commun 1999; 258: 313–21. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Y, Foudi A, Geay JF et al. Intracellular localization and constitutive endocytosis of CXCR4 in human CD34+ hematopoietic progenitor cells. Stem Cells 2004; 22: 1015–29. [DOI] [PubMed] [Google Scholar]
- 31. Mundy DI, Machleidt T, Ying YS, Anderson RG, Bloom GS. Dual control of caveolar membrane traffic by microtubules and the actin cytoskeleton. J Cell Sci 2002; 115: 4327–39. [DOI] [PubMed] [Google Scholar]
- 32. Rappoport JZ, Taha BW, Lemeer S, Benmerah A, Simon SM. The AP‐2 complex is excluded from the dynamic population of plasma membrane‐associated clathrin. J Biol Chem 2003; 278: 47 357–60. [DOI] [PubMed] [Google Scholar]
- 33. Rappoport JZ, Simon SM. Real‐time analysis of clathrin‐mediated endocytosis during cell migration. J Cell Sci 2003; 116: 847–55. [DOI] [PubMed] [Google Scholar]
- 34. Keyel PA, Watkins SC, Traub LM. Endocytic adaptor molecules reveal an endosomal population of clathrin by total internal reflection fluorescence microscopy. J Biol Chem 2004; 279: 13 190–204. [DOI] [PubMed] [Google Scholar]
- 35. Yarar D, Waterman‐Storer CM, Schmid SL. A dynamic actin cytoskeleton functions at multiple stages of clathrin‐mediated endocytosis. Mol Biol Cell 2005; 16: 964–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pediani JD, Colston JF, Caldwell D, Milligan G, Daly CJ, McGrath JC. Beta‐arrestin‐dependent spontaneous alpha1a‐adrenoceptor endocytosis causes intracellular transportation of alpha‐blockers via recycling compartments. Mol Pharmacol 2005; 67: 992–1004. [DOI] [PubMed] [Google Scholar]
- 37. Segredo V, Burford NT, Lameh J, Sadee W. A constitutively internalizing and recycling mutant of the mu‐opioid receptor. J Neurochem 1997; 68: 2395–404. [DOI] [PubMed] [Google Scholar]
- 38. Gulino AV, Moratto D, Sozzani S et al. Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood 2004; 104: 444–52. [DOI] [PubMed] [Google Scholar]
- 39. Kawai T, Choi U, Whiting‐Theobald NL et al. Enhanced function with decreased internalization of carboxy‐terminus truncated CXCR4 responsible for WHIM syndrome. Exp Hematol 2005; 33: 460–8. [DOI] [PubMed] [Google Scholar]
- 40. Balabanian K, Lagane B, Pablos JL et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood 2005; 105: 2449–57. [DOI] [PubMed] [Google Scholar]