

Abstract
More than half of human cancers contain mutations in the tumor suppressor protein p53, most of which accumulate in the DNA binding domain of the protein. Here we report the identification of a mutant p53, designated p53–46F, in which Ser‐46 is replaced with phenylalanine. In vitro, adenovirus‐mediated transduction of the p53–46F gene induced apoptosis more efficiently than wild‐type p53 in a number of cancer cell lines, whereas Ser‐15 phosphorylation of p53–46F was enhanced in all cancer cell lines examined. Moreover, the expression level of the cell cycle inhibitor p21/WAF1 was decreased in cell lines infected with adenovirus p53–46F (Ad‐p53–46F). p53–46F caused a more enhanced level of transcriptional activation of several p53‐target genes, including Noxa, p53AIP1 and p53RFP, compared with wild‐type p53. In vivo, adenovirus‐mediated gene transfer of p53–46F enhanced apoptosis, thus suppressing tumor growth of a lung cancer cell line more effectively than wild‐type p53 or p53–121F, another p53 mutant. Collectively, our data suggest that p53–46F is an active version of p53 that demonstrates enhanced induction of p53‐dependent apoptosis. This is probably mediated by upregulated transactivation of genes downstream of p53, increased Ser‐15 phosphorylation and a decrease in p21/WAF1 levels. We propose p53–46F as an alternative candidate to wild‐type p53 for use in developing new therapeutic strategies for the treatment of cancer. (Cancer Sci 2006; 97: 633–641)
The tumor suppressor gene p53 encodes a transcription factor that binds to specific sequences of its target genes and activates their transcription.( 1 , 2 , 3 , 4 ) Although a number of downstream target genes have been identified so far, those involved in cell cycle, DNA repair, antiangiogenesis and apoptosis are thought to mediate the core functions of p53‐regulated tumor suppression.( 5 , 6 , 7 ) p21/WAF1 is an inhibitor of cyclin‐dependent kinases that induce cell cycle arrest at G0/G1 phase,( 8 ) whereas p53R2, a component of ribonucleotide reductase, plays a pivotal role in providing dNTPs for DNA synthesis in the repair of damaged DNA.( 9 ) TSP1 and BAI1 mediate p53‐dependent antiangiogenesis,( 10 , 11 ) and Bax, Noxa, Puma and p53AIP1 are p53 target genes involved in apoptosis.( 12 , 13 , 14 , 15 , 16 ) p53 also activates a number of other target genes with diverse functions, which it is believed to achieve by altering its transcriptional target or through self‐modification such as phosphorylation, acetylation, sumoylation and glycosylation.( 7 ) Recently we demonstrated that p53 phosphorylated at Ser‐46 appears to activate the p53‐dependent apoptotic pathway by altering its transcriptional target from p21/WAF1 to p53AIP1.( 16 ) Thus, specific activation of a target gene or function appears to be important for successful p53‐based therapy against human cancers.( 17 , 18 )
Over 50% of human cancers contain p53 mutations,( 19 , 20 ) more than 80% of which are missense mutations that lead to the stable expression of full‐length p53 protein.( 21 ) Interestingly, most mutations are found in the region of the DNA binding domain, implying the impairment of transcription factor activity.( 19 , 20 , 21 , 22 ) Indeed, p53‐dependent expression of target genes in response to cellular stresses is often downregulated in tumors carrying p53 mutations( 23 , 24 , 25 ) suggesting that the inactivation of transcriptional activity is essential for tumorigenesis. However, recent studies have reported wide variation in transactivation by mutant p53 proteins of target genes. A significant number of target genes demonstrate wild‐type activity; some show increased activity levels whereas decreased activity is seen in a limited number.( 26 , 27 , 28 ) In addition, some mutant p53 proteins demonstrate enhanced induction of apoptosis, implying a gain of tumor suppressive function;( 29 ) p53–121F was the first such p53 mutant to be identified.( 30 ) Although selective transcriptional activation of the cell death inducer Bax by p53–121F has been proposed as the mechanism behind the enhanced apoptotic activity, the details remain to be elucidated.
Here we report a mutant active form of p53, p53–46F, in which Ser‐46 is replaced with phenylalanine. This mutant induces apoptosis more effectively than wild‐type p53 both in vitro and in vivo. Enhanced transactivation of p53 target genes, an increase in Ser‐15 phosphorylation of p53–46F and a decrease in levels of p21/WAF1 protein are likely to play an important role in this phenomenon. The introduction of p53–46F rather than wild‐type p53 to tumor cells is therefore a promising strategy for cancer therapy.
Materials and Methods
Cell lines
U87MG and U373MG (glioblastoma cell lines), A549 and H1299 (lung cancer cell lines), HCT116 and LS174T (colon cancer cell lines) and Hep G2 (hepatoblastoma cell line) were purchased from ATCC (Manassas, VA, USA). LC176 (lung cancer cell line) was kindly provided by Dr Takahashi, Division of Molecular Oncology, Aichi Cancer Center Research Institute, Nagoya, Japan. All cell lines were cultured under the conditions recommended by the suppliers.
Recombinant adenoviruses and infection
Ad‐p53‐WT, Ad‐p53–46F and Ad‐p53–121F viruses were constructed with the Adenovirus Expression Vector Kit (Takara, Otsu, Japan). Adenoviruses were propagated in HEK293 cells and purified by CsCl density centrifugation. Viral titer was measured using the 50% tissue culture infectious dose method in HEK293 cells. Ad‐EGFP viruses encoding enhanced green fluorescence protein (EGFP) were a gift from Dr Tahara, Institute of Medical Science, University of Tokyo, Tokyo, Japan. Prior to infection, 1 × 106 cells were seeded in 10‐cm culture dishes. After 24 h, viral stocks were diluted to their final concentrations with appropriate culture medium, applied to the cell monolayer with brief agitation, and incubated at 37°C until required for assays.
Flow cytometry
At the time periods indicated, whole cells were harvested and fixed with 70% ethanol in phosphate‐buffered saline (PBS). Fixed cells were rinsed twice with PBS, and cell nuclear RNA was digested with DNase‐inactivated RNase A (Sigma‐Aldrich, St Louis, MO, USA) at 37°C for 30 min in order to reduce the background. Cell nuclear DNA was stained with 50 µg/mL propidium iodide (Sigma‐Aldrich) in PBS. The percentage of apoptotic cells in the population was calculated from at least 2 × 104 cells using FACScalibur (Becton Dickinson, Franklin Lakes, NJ, USA).
Reverse transcription–polymerase chain reaction analysis
For reverse transcription–polymerase chain reaction (RT‐PCR) analysis, total RNA was isolated from cells using TRIZOL regent (Life Technologies, Rockville, MD, USA). Total RNA was isolated from H1299 tumor tissue using a glass homogenizer and TRIZOL reagent. cDNA was synthesized from 10 µg total RNA with SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). The exponential phase of RT‐PCR was determined on 15–30 cycles to enable semiquantitative comparisons to be carried out between cDNA synthesized in identical reactions. PCR was carried out on a Gene Amp PCR system 9700 (Applied Biosystems, Foster City, CA, USA).
Northern blot analysis
mRNA was purified from total RNA using the OligotexTM‐dT30 Super mRNA Purification kit (Takara). Aliquots of mRNA (1 µg) were electrophoresed on a 1% agarose gel containing 6% formaldehyde and transferred onto a Biodyn A nylon membrane (Pall Corporation, Pensacola, FL, USA). Probes for p53, p21, Puma, Bax, and Noxa transcripts were labeled with [α‐32P] dCTP using a Megaprime Random Primer Labeling Kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Pre‐hybridization and hybridization were carried out using 2% sodium dodecylsulfate (SDS), 50% formamide, 10 × Denhardt's solution, 5 × saline–sodium phosphate–EDTA buffer (SSPE) and 1% salmon sperm DNA. The blots were hybridized with the radiolabeled probes at 42°C for 16 h, washed with 2 × saline–sodium citrate buffer (SSC)/0.05% SDS at 55°C, and northern blot images were detected using the Storm 860 image analysis system (Amersham Pharmacia Biotech).
Antibodies
Mouse monoclonal anti‐p53 Ab‐6 (DO‐1) and anti‐p21 Ab‐1 (EA10) were purchased from Calbiochem (La Jolla, CA, USA). Mouse monoclonal anti‐β‐actin (AC15) was purchased from Sigma‐Aldrich. Mouse monoclonal antiphosphorylated p53 at Ser‐15 (16G8) was purchased from Cell Signaling Technology (Beverly, MA, USA). Rabbit polyclonal anti‐Bax (N‐20) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Immunoblot analysis
Cells were infected with Ad‐p53‐WT or Ad‐p53–46F at the multiplicity of infection (MOI) indicated at various time points. Total cell lysates were prepared with RIPA buffer 1% NP‐40, 50 mM Tris‐HCl [pH 8.0], 150 mM NaCl, 0.5% deoxychorate Na, 0.1% SDS and complete protease‐inhibitor cocktail (Roche, Indianapolis, IN, USA). Soluble proteins (10 µg) were loaded on 10–15% SDS polyacrylamide gels and transferred to Hybond‐P membranes (Amersham Pharmacia Biotech). The membranes were blocked with 3% bovine serum albumin (BSA) in Tris‐buffered saline containing 0.05% Tween 20 (TBS‐T). After a 1‐h incubation with primary antibody at room temperature, the blots were incubated with horse radish peroxidase‐conjugated secondary antibodies for each primary antibody. Immunoblot images were detected using the ECL or ECL plus system (Amersham Pharmacia Biotech).
Gene reporter assays
DNA fragments including the p53 binding site of p21, p53AIP1, p53DINP1, p53R2, p53RDL1, p53RFP, Bax, Puma and semaphorin 3F were amplified by PCR and cloned into the pGL3‐promoter or pGL3‐basic vector (Promega, Madison, WI, USA). The Noxa reporter vector was a gift from Dr Taniguchi, Graduate School of Medicine, University of Tokyo. H1299 cells were plated in six‐well culture plates (1.5 × 105 cells/well) 24 h before cotransfection of 1 µg of reporter plasmid and either 1 µg of wild‐type or mutant (46F and 175H) p53 expression vector in combination with 50 ng of pRL‐CMV vector. At 24 h after infection, cells were rinsed with PBS and lysed in 500 µL of passive lysis buffer (Promega). Cell lysates were used in the Dual Luciferase Reporter Assay System (Promega). Quantification of luciferase activities and calculations of relative ratios were carried out manually with a luminometer (Berthold Technologies, Bad Wildbad, Germany).
Animal experiments
H1299 xenografts were established in 7‐week‐old female nude mice, BALB/cAJcl‐nu (CLEA Japan, Tokyo, Japan). H1299 cells were implanted subcutaneously in four positions (2 × 106 cells/position) on the flank of each mouse. Xenografts were infected with Ad‐p53‐WT, Ad‐p53–46F, Ad‐p53–121F, Ad‐EGFP or PBS (untreated control) when the tumor volume reached approximately 300 mm3. Tumor volume was calculated using the formula a 2 × b/2, where a is the minor axis and b is the major axis in millimeters. Adenoviral injection was carried out once a day on days 0–4, and each tumor was infected with a total dose of 1 × 109 plaque‐forming units (p.f.u.) of virus. Each adenovirus vector was injected in a total volume of 100 µL PBS. To avoid subcutaneous bleeding, the solutions were injected in a single shot, away from tumor vessels. The antitumor effect of Ad‐p53–46F was evaluated by comparing relative tumor size with tumors injected with Ad‐EGFP, Ad‐p53‐WT or Ad‐p53–121F. Data comparing the five treatment groups were analyzed using non‐parametric methods. All mouse procedures were carried out according to the recommendations of the Institutional Animal Care and Use Committee of the National Cancer Center at Tsukiji, Japan.
TUNEL assays and immunofluorescence
The in situ terminal transferase‐mediated dUTP nick end‐labeling (TUNEL) assay was carried out with the ApopTag Fluorescein Direct In Situ Apoptosis Detection Kit (Intergen, Purchase, NY, USA) according to the manufacturer's instructions. Frozen tumor sections were obtained from mice killed on days 5 and 32, after receiving five viral injections on days 0–4. Air‐dried frozen sections were fixed in 4% paraformaldehyde in PBS for 15 min and washed for 5 min with PBS three times. Fixed sections were permeabilized with 0.5% Triton‐X 100 for 2 min, and then washed for 5 min with PBS four times. Premeasured sections were immersed for 90 min in 5% BSA in PBS, then incubated overnight at 4°C in PBS containing 5% BSA and mouse anti‐p53 antibody (Ab‐6) diluted 1 : 200. For immunofluorescence staining, fluorescein‐isothiocyanate‐conjugated horse anti‐mouse IgG (Vector Laboratories, Burlingame, CA, USA) was used as a secondary antibody.
Results
p53–46F is a more potent inducer of apoptosis than wild‐type p53
While carrying out functional analysis of Ser‐46‐phosphorylated p53,( 16 ) a p53 mutant (mt‐p53) was discovered with a phenylalanine substitution at Ser‐46, which demonstrated enhanced p53‐dependent apoptotic activity.
To characterize this mutant, its apoptotic activity was compared with that of wild‐type p53 (wt‐p53) in six cancer cell lines: the lung cancer cell lines A549 (wt‐p53), LC‐176 (wt‐p53) and H1299 (p53‐null), the hepatoblastoma cell line HepG2 (wt‐p53), and the colorectal cancer cell lines LS‐174T (wt‐p53) and HCT116 (wt‐p53). These cell lines were infected with either adenovirus wild‐type p53 (Ad‐p53‐WT) or adenovirus p53–46F (Ad‐p53–46F) at 30 MOI, and apoptotic cells were measured by fluorescence activated cell sorter (FACS) scan analysis. Ad‐p53–46F induced apoptosis more effectively than Ad‐p53‐WT in all cell lines (Fig. 1). An additional 24 cancer cell lines were examined, including those derived from five lung cancers, four brain tumors, two colorectal cancers, four breast cancers, one melanoma, one embryoma, one gastric cancer and five esophageal cancers. In 17 of the 24 cell lines (70%, four wt‐p53 and 13 mt‐p53 cancer cell lines), apoptosis was induced following infection with Ad‐p53–46F (data not shown). Moreover, in 15 of the 17 cell lines (90%, four wt‐p53 and 11 mt‐p53 cancer cell lines), the number of apoptotic cells induced by Ad‐p53–46F exceeded those induced by Ad‐p53‐WT infection (data not shown). Thus p53–46F is more effective in promoting p53‐dependent apoptosis irrespective of the p53 status in cancer cells.
Figure 1.
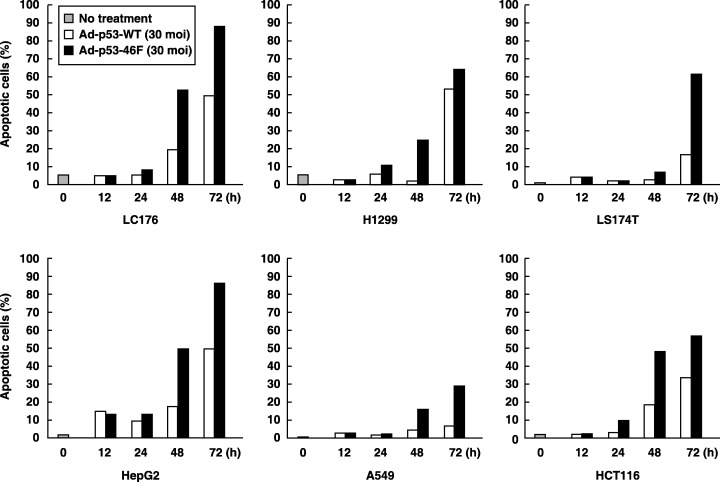
p53–46F activity predominates over wild‐type p53 in p53‐dependent apoptosis. The six cancer cell lines H1299 (p53‐null), A549 (wt p53), HepG2 (wt p53), LC176 (wt p53), LS174T (wt p53) and HCT116 (wt p53) were infected with Ad‐p53‐WT (30 multiplicity of infection [MOI]) or Ad‐p53–46F (30 MOI), and apoptotic cells were evaluated by fluorescence activated cell sorter analysis at the times indicated.
Enhanced Ser‐15 phosphorylation contributes to the stability of p53–46F
Western blot analysis was carried out to determine the expression levels of p53 and p53 target genes. In the six cancer cell lines studied, p53 expression increased in a time‐dependent manner after infection with Ad‐p53‐WT or Ad‐p53–46F (Fig. 2). Although all cells were transduced with the same adenoviral titer, p53 expression levels in some cancer cell lines (A549, LC176 and LS174T) was higher following infection with Ad‐p53–46F than with Ad‐p53‐WT, indicating that the p53–46F protein is more stable than the wild‐type protein.
Figure 2.
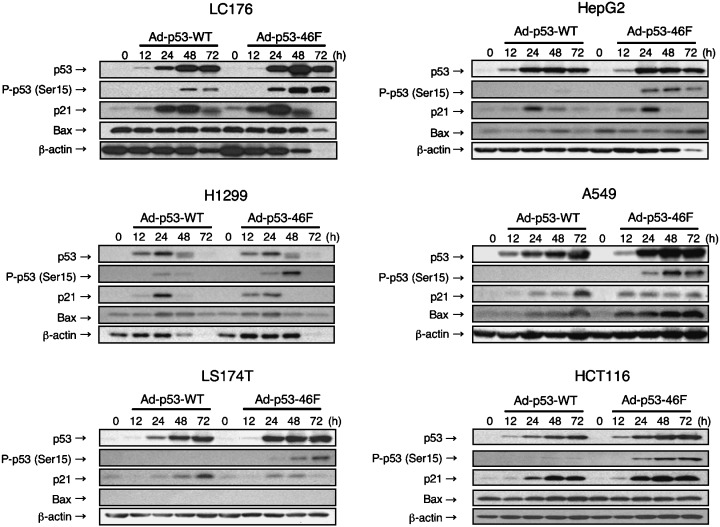
Infection of Ad‐p53–46F increases expression of p53 and some of its target genes as well as increasing p53 Ser‐15 phosphorylation. Western blot analysis of p53, p53 phosphorylated at Ser‐15, p21/WAF1 and Bax was carried out in six cancer cell lines infected with Ad‐p53‐WT or Ad‐p53–46F. β‐Actin was used as a loading control.
Therefore, we examined the level of p53 phosphorylation at Ser‐15, a factor that has previously been associated with the extent of stability of p53.( 31 ) As expected, the enhancement of Ser‐15 phosphorylation was greater following infection with Ad‐p53–46F than with Ad‐p53‐WT (Fig. 2). Consistent with enhanced Ser‐15‐phosphorylation, expression of the apoptotic protein Bax was higher after infection with Ad‐p53–46F than with Ad‐p53‐WT in H1299, A549 and HepG2 cells. Curiously, expression levels of the p21/WAF1 protein in cells infected with Ad‐p53–46F were lower than in Ad‐p53‐WT‐infected cells, and decreased after 48 h in all cells except HCT116.
p53–46F upregulates transcription of p53 target genes more effectively than wild‐type p53
To further explore the mechanism of p53–46F‐regulated apoptosis, an investigation was carried out into the potential correlation between apoptosis and the expression levels of p53 target genes in H1299, one of the Ad‐p53–46F‐sensitive cancer cell lines. To exclude the effects of increased p53–46F stability due to enhanced Ser‐15‐phosphorylation, Ad‐p53–46F was transduced at a 30% lower MOI than Ad‐p53‐WT. As shown in Fig. 3A, more efficient apoptosis was induced by infection of Ad‐p53–46F than by Ad‐p53‐WT both 12 h and 24 h post infection.
Figure 3.
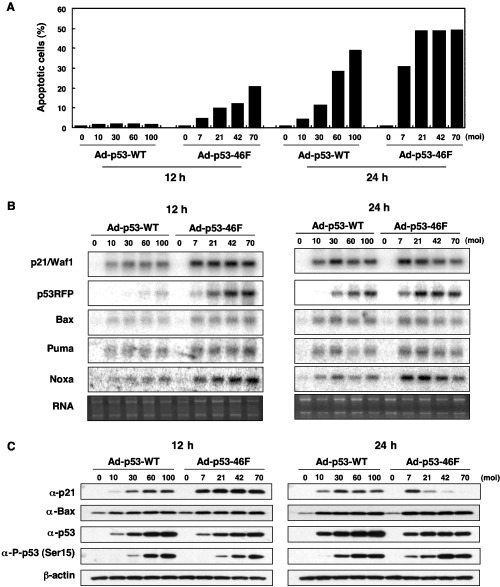
p53–46F‐regulated apoptosis increases Ser‐15 phosphorylation of p53 and decreases p21/WAF1 expression. (A) Comparison of wild‐type p53 and p53–46F induction of apoptosis in H1299. H1299 cells were infected with either Ad‐p53‐WT or Ad‐p53–46F at the multiplicity of infection indicated, and apoptotic cells were evaluated by fluorescence activated cell sorter analysis 12 h and 24 h post infection. (B) Northern blot analysis of p21/WAF1, p53RFP, Bax, Puma and Noxa expression in H1299 cells. The ethidium gel of RNA samples is shown as a loading control. (C) Western blot analysis of p53, p53 phosphorylated at Ser‐15, p21/WAF1 and Bax proteins in H1299. β‐Actin was used as a loading control.
mRNA expression levels of five p53 target genes were analyzed by northern blot analysis following transduction of Ad‐p53‐WT or Ad‐p53–46F. All p53 target genes demonstrated a higher level of transcriptional activation by p53–46F than by p53‐wt at both 12 h and 24 h post infection (Fig. 3B), suggesting a possible mechanism for p53–46F‐regulated apoptosis. It is of interest to note that p53RFP expression was more enhanced by Ad‐p53–46F than other p53 target genes. The transcription of Noxa, PUMA, p21/WAF1 and Bax was enhanced to a similar extent.
p21/WAF1 protein expression is regulated post‐transcriptionally or post‐translationally by p53–46F
To validate the phenomena of Ser‐15 phosphorylation and decreased p21/WAF1 protein levels (Fig. 2), the amount of Ser‐15 phosphorylation and the expression levels of p53, p21/WAF1 and Bax were further examined in these cells. As indicated in Fig. 3C, expression levels of the p21/WAF1 protein decreased 24 h post infection with Ad‐p53–46F in a dose‐dependent manner, despite the maintenance of high levels of p21/WAF1 mRNA (Fig. 3B). This implies that p53–46F regulates the p21/WAF1 protein at the post‐transcriptional or post‐translational level. In contrast, and consistent with the level of mRNA (Fig. 3B), Bax protein expression was induced more efficiently by Ad‐p53–46F than by Ad‐p53‐WT (Fig. 3C). Although, as expected, p53–46F protein levels were relatively lower than those of wild‐type p53, and Ser‐15 phosphorylation of p53–46F was equivalent to (at 12 h) or higher than (at 24 h) that of wild‐type p53. This suggests that an increase in Ser15 phosphorylation of p53–46F, but not the stability of p53–46F protein, may have a specific role in an enhanced ability for p53–46F to induce apoptosis. Therefore, we conclude that a decrease in p21/WAF1 protein levels and enhanced Ser‐15 phosphorylation of p53–46F might play an important role in this phenomenon.
p53RFP is a potential mediator of p53–46F‐induced apoptosis
Reporter gene assays were carried out to validate the observed enhancement of p53 target gene transcription by p53–46F. Consistent with the results of endogenous gene expression (Fig. 3B), transcription of the luciferase gene fused to the p53‐binding sequence of p53RFP was strikingly enhanced by p53–46F. p53–46F‐induced luciferase activity was more than two‐fold higher than wild‐type p53‐induced activity, suggesting that p53RFP may play a pivotal role in p53–46F‐induced apoptosis (Fig. 4). The p53‐binding sequences of other p53 target genes fused to luciferase, including p21/WAF1, p53AIP1, Bax, Puma, Noxa, p53DINP1, p53R2, p53RDL1 and Sema 3F (semaphorin 3F), also demonstrated enhanced activities when cotransfected with the p53–46F expression plasmid. With the exception of p53AIP1, p53DINP1 and p53RDL1, the elevated activities were less than two‐fold higher than those induced by wild‐type p53 (Fig. 4). Overall, p53–46F was shown to activate transcription of all p53‐target genes examined more efficiently than wild‐type p53.
Figure 4.
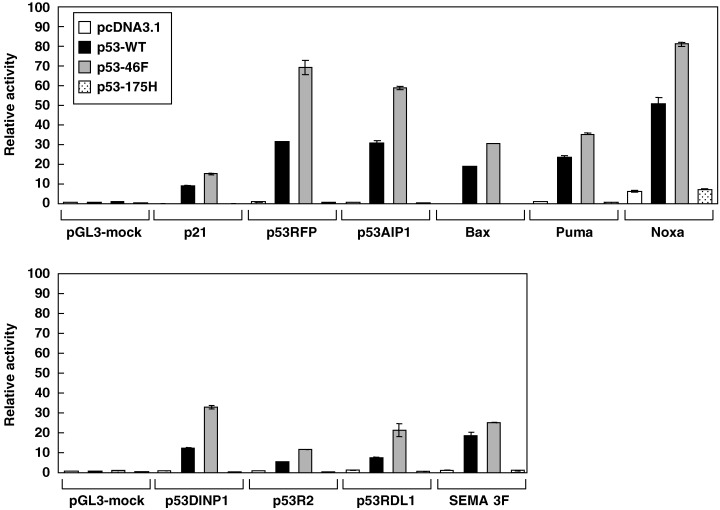
p53–46F demonstrates enhanced transcriptional activation of p53 target genes. A reporter vector containing the p53‐binding sequence of p21/WAF1, p53RFP, p53AIP1, Bax, Puma, Noxa, p53DINP1, p53R2, p53RDL1 or Semaphorin 3F was cotransfected with a mock plasmid, p53‐WT plasmid, p53–46F plasmid or p53–175H plasmid into H1299 cells. Luciferase activity was evaluated 24 h post transfection. The bars indicate the relative activity of luciferase. Error bars indicate the standard deviation of the mean.
p53–46F suppresses tumor growth in vivo
The effect of p53–46F on tumor growth in vivo was evaluated by implanting H1299 lung cancer cells subcutaneously into nude mice, allowing the tumors to grow to 300 mm3 and injecting each tumor with 109 p.f.u./50 µL of Ad‐p53–46F, Ad‐p53‐WT, Ad‐p53–121F or Ad‐EGFP once daily over the course of 5 days. Tumors injected with PBS or EGFP (Ad‐EGFP) were isolated at day 12 post injection, at which time they had attained a volume of 800–1800 mm3 (Fig. 5A), thus preventing further observation. Until this time, the growth of tumors injected with Ad‐p53‐WT, Ad‐p53–46F or Ad‐p53–121F was suppressed; however, after day 12, tumors injected with Ad‐p53‐WT increased in size, reaching a volume of 3000 mm3 by day 32. Ad‐p53–46F‐injected tumors did not grow until day 19, after which time they gradually increased in size until day 32, but remained below 300 mm3. Surprisingly, several tumors injected with Ad‐p53–46F disappeared. Tumors injected with Ad‐p53–121F grew at a moderate rate, approximately halfway between that of the tumors injected with Ad‐EGFP and those injected with Ad‐p53–46F. These results were confirmed by measuring the weight of each tumor isolated at day 32 (Fig. 5B). The experiment was repeated twice and similar results were obtained each time.
Figure 5.
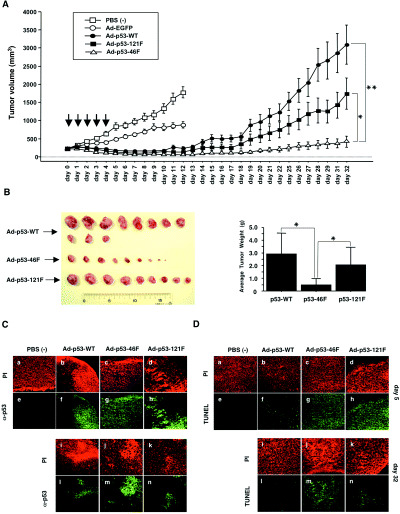
In vivo tumor growth suppression by p53–46F. (A) Tumor volumes were measured every day following the establishment of xenografts in BALB/cAJcl‐nu nude mice. H1299 cells were injected daily for 5 days (indicated by arrows) with phosphate‐buffered saline (PBS; □), Ad‐EGFP (○), Ad‐p53‐WT (•), Ad‐p53–121F (▪) and Ad‐p53–46F (▵). P‐values less than 0.05 (*) and less than 0.001 (**) were deemed statistically significant. Error bars indicate the standard error of the mean. (B) Tumors isolated at day 32. (Left) Macroscopic appearance of tumors infected with Ad‐p53‐WT, Ad‐p53–46F and Ad‐p53–121F. (Right) The average tumor weight of each group is shown on the bar graph. A P‐value less than 0.05 (*) was deemed statistically significant. Error bars indicate the standard deviation of the mean. (C) Expression of p53, p53–46F and p53–121F. Tumors infected with Ad‐p53‐WT, Ad‐p53–46F and Ad‐p53–121F at day 5 (a–h) and day 32 (i–n) post infection were subjected to immunohistochemical analysis. p53 was visualized using an anti‐p53 antibody (Ab‐6) (green) in the tumors injected with PBS (e), Ad‐p53‐WT (f,l), Ad‐p53–46F (g,m) and Ad‐p53–121F (h,n). DNA was stained by propidium iodide (PI) (red) in the tumors injected with PBS (a), Ad‐p53‐WT (b,i), Ad‐p53–46F (c,j) and Ad‐p53–121F (d,k). (D) Apoptotic cells in the tumors. Tumors infected with Ad‐p53‐WT, Ad‐p53–46F and Ad‐p53–121F at day 5 (a–h) and day 32 (i–n) post infection were subjected to transferase‐mediated dUTP nick end‐labeling (TUNEL) analysis. Apoptotic cells were stained using TUNEL (green) in the tumors injected with PBS (e), Ad‐p53‐WT (f,l), Ad‐p53–46F (g,m) and Ad‐p53–121F (h,n). DNA was stained using PI.
An investigation into p53 expression revealed that similar levels of p53 protein were present in the tumors 5 days after injection with Ad‐p53‐WT, Ad‐p53–46F and Ad‐p53–121F, but only Ad‐p53–46F‐injected tumors contained high levels at day 32 (Fig. 5C). Moreover, the number of apoptotic cells was higher in tumors injected with Ad‐p53–46F and Ad‐p53–121F than Ad‐p53‐WT at day 5. Consistent with the profile of p53 expression, a high number of apoptotic cells was observed only in the tumor injected with Ad‐p53–46F at day 32 (Fig. 5D).
Discussion
In the present study, we report on a p53 mutant, p53–46F, with an enhanced ability to induce p53‐dependent apoptosis. An increased level of p53 Ser‐15 phosphorylation was observed in all cell lines infected with Ad‐p53–46F. This has previously been suggested to induce stabilization of the p53 protein( 31 ) and to be mediated by the ATM protein, encoded by the gene mutated in the human autosomal recessive disorder ataxia telangiectasia.( 32 ) In our experimental system, expression levels of the p53–46F protein were increased compared to wild‐type p53, in concert with an increased level of p53 Ser‐15 phosphorylation. In addition, northern blot analysis (Fig. 3B) and a luciferase reporter gene assay (Fig. 4) revealed enhanced transcriptional activation by p53–46F of all of the p53 target genes examined. Although it might be speculated that increased Ser‐15 phosphorylation is responsible for enhanced transactivation of genes downstream of p53, p53–46F is capable of inducing apoptosis more efficiently than p53‐WT, even when expressed at lower levels than the wild‐type protein, and with a similar extent of Ser‐15 phosphorylation. It is possible that the substitution of phenylalanine with serine in p53–46F causes a conformational change in the p53 molecule, resulting both in the acceleration of the interaction between p53–46F, a phosphorylating kinase and Ser‐15, and the enhanced affinity of p53–46F to p53‐binding sequences of target genes. Further structural analysis and investigation of proteins interacting with p53–46F are required to clarify this mechanism.
Although p53–46F activated transcription of all of the p53 target genes examined, it decreased expression levels of the p21/WAF1 protein (2, 3). Moreover, this decrease appears to be related to the enhancement of p53‐induced apoptosis (1, 3). Consistent with this notion, a number of studies have reported that p21/WAF1 inhibits apoptosis and that, conversely, suppression of p21/WAF1 accelerates apoptosis.( 33 , 34 , 35 , 36 , 37 , 38 ) Thus, we postulate that the decrease in p21/WAF1 protein expression by p53–46F might play a role in p53–46F‐induced apoptosis. If this is the case, it is possible that p53–46F preferentially activates transcription of a p53 target gene involved in regulation of p21/WAF1 stability. In support of this hypothesis, we recently observed that p53 induces the expression of p53RFP mRNA, a ubiquitin ligase that regulates the stability of p21/WAF1.( 39 ) As described above, all p53 target genes examined were induced more efficiently by p53–46F than by p53‐WT. However, endogenous p53RFP mRNA levels were most strongly enhanced by p53–46F (Fig. 3C). Moreover, the luciferase activity of the heterologous reporter gene fused to the p53‐binding sequence of p53RFP was elevated more than two‐fold higher by p53–46F than by p53‐WT. p53RFP therefore appears to be a good candidate mediator for this phenomenon. However, further investigations are required to determine whether there is an alternative regulator for p21/WAF1 or another explanatory mechanism.
Several mutant forms of p53 have been shown to demonstrate a more potent ability to induce apoptosis than wild‐type p53 in vitro, including S121F (p53–121F in the present paper), S121C, S121A, S121Y, R290G, K291E, K291Q, K291T, K292T and K292I.( 29 ) Interestingly, there are no significant correlations between the sequence‐specific transcriptional activities of these mutants on six p53 target genes and their ability to induce apoptosis.( 29 ) Therefore, the study suggests that transactivation‐dependent apoptosis does not always play a major role in p53‐dependent apoptosis, indirectly supporting the claim that the transactivation‐independent mechanism plays an important role. In contrast to these p53 mutants, p53–46F demonstrated enhanced transactivational activity for all p53 target genes examined in the present study. Although the transactivation‐independent mechanism is not completely excluded, this result implies that transactivation‐dependent apoptosis is likely to play a critical role in p53–46F‐induced apoptosis. Indeed, microarray screening of p53–46F‐inducible transcripts clearly indicates that a number of genes are induced in a similar way, but at a higher level than those induced by wild‐type p53 (M. Futamura and H. Arakawa, unpublished data). This is an important finding for the application of p53–46F in cancer gene therapy. There are understandable concerns regarding the introduction of p53 mutants into the human body and the potential for undesirable side effects. As the transcriptional activity of p53–46F is very similar to wild‐type, in contrast to other active p53 mutants such as 121F, p53–46F might provide a new strategy for cancer therapy.
Although adenovirus‐mediated p53 gene transfer was expected to be a promising method of delivery for cancer therapy, a number of clinical investigations have indicated that it is ineffective.( 40 , 41 , 42 , 43 ) Recently, some studies have provided evidence that modifications of p53, such as phosphorylation and acetylation, are critical for induction of p53‐regulated apoptosis.( 16 , 44 , 45 , 46 , 47 ) Based on these facts, it seems likely that introduction of the p53 gene itself into cancer cells does not necessarily lead to induction of apoptosis. For example, if the p53 protein is insufficiently modified in cancer cells due to alterations of phosphorylation kinases, a mere increase in p53 protein level may fail to activate transcription of the downstream genes and induce apoptosis. p53–46F offers the advantage of enhancing transcription of p53 target genes irrespective of its level of phosphorylation or protein stability. Indeed, p53–46F increased the extent of Ser‐15 phosphorylation without gamma‐irradiation or treatment with anticancer drugs in many wild‐type p53 cell lines that are resistant to Ad‐p53‐WT.( 18 )
In the present study, apoptosis was accelerated by a p53–46F‐mediated decrease in the level of apoptotic inhibitor p21/WAF1, probably through induction of p53RFP. Adenoviral gene transfer of p53–46F induced suppression of H1299 tumor growth in nude mice (Fig. 5A,B), and was more effective in its suppression of tumor growth than wild‐type p53 and p53–121F (Fig. 5A,B). Interestingly, p53–46F expression in the tumors was observed until day 32 after injection with Ad‐p53–46F (Fig. 5C), which might explain its remarkable ability to reduce tumor growth.
These results provide strong support for the use of p53–46F as a new treatment against p53‐resistant tumors. Furthermore, the finding that p21/WAF1 expression is decreased by p53–46F confirms previous observations of the anti‐apoptotic role of p21/WAF1 and tumor resistance to anticancer drugs.( 33 , 34 , 35 , 36 , 37 , 38 ) We conclude that the specific activation of apoptosis‐related p53 target genes and inhibition of p21/WAF1 could provide a more effective strategy for cancer therapy.
Acknowledgments
We thank Sakiko Usuda, Ayumi Ishizaka and Izumi Hyo for their excellent technical assistance. This work was supported in part by grants from the Ministry of Health, Labor and Welfare, Japan, and the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
- 1. Fields S, Jang SK. Presence of a potent transcription activating sequence in the p53 protein. Science 1990; 249: 1046–9. [DOI] [PubMed] [Google Scholar]
- 2. Raycroft L, Wu HY, Lozano G. Transcriptional activation by wild‐type but not transforming mutants of the p53 anti‐oncogene. Science 1990; 249: 1049–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kern SE, Kinzler KW, Bruskin A et al. Identification of p53 as a sequence‐specific DNA‐binding protein. Science 1991; 252: 1708–11. [DOI] [PubMed] [Google Scholar]
- 4. El‐Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet 1992; 1: 45–9. [DOI] [PubMed] [Google Scholar]
- 5. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408: 307–10. [DOI] [PubMed] [Google Scholar]
- 6. Nakamura Y. Isolation of p53‐target genes and their functional analysis. Cancer Sci 2004; 95: 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arakawa H. p53, apoptosis and axon‐guidance molecules. Cell Death Differ 2005; 12: 1057–65. [DOI] [PubMed] [Google Scholar]
- 8. El‐Deiry WS, Tokino T, Velculescu VE et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–25. [DOI] [PubMed] [Google Scholar]
- 9. Tanaka H, Arakawa H, Yamaguchi T et al. A ribonucleotide reductase gene involved in a p53‐dependent cell‐cycle checkpoint for DNA damage. Nature 2000; 404: 42–9. [DOI] [PubMed] [Google Scholar]
- 10. Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin‐1. Science 1994; 265: 1582–4. [DOI] [PubMed] [Google Scholar]
- 11. Nishimori H, Shiratsuchi T, Urano T et al. A novel brain‐specific p53‐target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene 1997; 15: 2145–50. [DOI] [PubMed] [Google Scholar]
- 12. Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995; 80: 293–9. [DOI] [PubMed] [Google Scholar]
- 13. Oda E, Ohki R, Murasawa H et al. Noxa, a BH3‐only member of the Bcl‐2 family and candidate mediator of p53‐induced apoptosis. Science 2000; 288: 1053–8. [DOI] [PubMed] [Google Scholar]
- 14. Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7: 673–82. [DOI] [PubMed] [Google Scholar]
- 15. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7: 683–94. [DOI] [PubMed] [Google Scholar]
- 16. Oda K, Arakawa H, Tanaka T et al. p53AIP1, a potential mediator of p53‐dependent apoptosis, and its regulation by Ser‐46‐phosphorylated p53. Cell 2000; 102: 849–62. [DOI] [PubMed] [Google Scholar]
- 17. Matsuda K, Yoshida K, Taya Y, Nakamura K, Nakamura Y, Arakawa H. p53AIP1 regulates the mitochondrial apoptotic pathway. Cancer Res 2002; 62: 2883–9. [PubMed] [Google Scholar]
- 18. Yoshida K, Monden M, Nakamura Y, Arakawa H. Adenovirus‐mediated p53AIP1 gene transfer as a new strategy for treatment of p53‐resistant tumors. Cancer Sci 2004; 95: 91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991; 253: 49–53. [DOI] [PubMed] [Google Scholar]
- 20. Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1994; 54: 4855–78. [PubMed] [Google Scholar]
- 21. Soussi T, Beroud C. Assessing TP53 status in human tumors to evaluate clinical outcome. Nat Rev Cancer 2001; 1: 233–40. [DOI] [PubMed] [Google Scholar]
- 22. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor–DNA complex: understanding tumorigenic mutations. Science 1994; 265: 346–55. [DOI] [PubMed] [Google Scholar]
- 23. Mori T, Anazawa Y, Iiizumi M, Fukuda S, Nakamura Y, Arakawa H. Identification of the interferon regulatory factor 5 gene (IRF‐5) as a direct target for p53. Oncogene 2002; 21: 2914–18. [DOI] [PubMed] [Google Scholar]
- 24. Mori T, Anazawa Y, Matsui K, Fukuda S, Nakamura Y, Arakawa H. Cyclin K as a direct transcriptional target of the p53 tumor suppressor. Neoplasia 2002; 4: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kimura T, Gotoh M, Nakamura Y, Arakawa H. hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer Sci 2003; 94: 431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kato S, Han SY, Liu W et al. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high‐resolution missense mutation analysis. Proc Natl Acad Sci USA 2003; 100: 8424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Resnick MA, Inga A. Functional mutants of the sequence‐specific transcription factor p53 and implications for master genes of diversity. Proc Natl Acad Sci USA 2003; 100: 9934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soussi T, Kato S, Levy PP, Ishioka C. Reassessment of the TP53 mutation database in human disease by data mining with a library of TP53 missense mutations. Hum Mutat 2005; 25: 6–17. [DOI] [PubMed] [Google Scholar]
- 29. Kakudo Y, Shibata H, Otsuka K, Kato S, Ishioka C. Lack of correlation between p53‐dependent transcriptional activity and the ability to induce apoptosis among 179 mutant p53s. Cancer Res 2005; 65: 2108–14. [DOI] [PubMed] [Google Scholar]
- 30. Saller E, Tom E, Brunori M et al. Increased apoptosis induction by 121F mutant p53. EMBO J 1999; 18: 4424–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage‐induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997; 91: 325–34. [DOI] [PubMed] [Google Scholar]
- 32. Banin S, Moyal L, Shieh S et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998; 281: 1674–7. [DOI] [PubMed] [Google Scholar]
- 33. Tian H, Wittmack EK, Jorgensen TJ. p21WAF1/CIP1 antisense therapy radiosensitizes human colon cancer by converting growth arrest to apoptosis. Cancer Res 2000; 60: 679–84. [PubMed] [Google Scholar]
- 34. Chattopadhyay D, Ghosh MK, Mal A, Harter ML. Inactivation of p21 by E1A leads to the induction of apoptosis in DNA‐damaged cells. J Virol 2001; 75: 9844–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mahyar‐Roemer M, Roemer K. p21 Waf1/Cip1 can protect human colon carcinoma cells against p53‐dependent and p53‐independent apoptosis induced by natural chemopreventive and therapeutic agents. Oncogene 2001; 20: 3387–98. [DOI] [PubMed] [Google Scholar]
- 36. Gartel AL, Tyner AL. The role of the cyclin‐dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther 2002; 1: 639–49. [PubMed] [Google Scholar]
- 37. Javelaud D, Besancon F. Inactivation of p21WAF1 sensitizes cells to apoptosis via an increase of both p14ARF and p53 levels and an alteration of the Bax/Bcl‐2 ratio. J Biol Chem 2002; 277: 37 949–54. [DOI] [PubMed] [Google Scholar]
- 38. Fan X, Liu Y, Chen JJ. Down‐regulation of p21 contributes to apoptosis induced by HPV E6 in human mammary epithelial cells. Apoptosis 2005; 10: 63–73. [DOI] [PubMed] [Google Scholar]
- 39. Ng CC, Arakawa H, Fukuda S, Kondoh H, Nakamura Y. p53RFP, a p53‐inducible RING‐finger protein, regulates the stability of p21WAF1 . Oncogene 2003; 22: 4449–58. [DOI] [PubMed] [Google Scholar]
- 40. Connell PP, Weichselbaum RR. Gene therapy: the challenges of translating laboratory research into clinical practice. J Clin Oncol 2003; 21: 2230–1. [DOI] [PubMed] [Google Scholar]
- 41. Pagliaro LC, Keyhani A, Williams D et al. Repeated intravesical instillations of an adenoviral vector in patients with locally advanced bladder cancer: a phase I study of p53 gene therapy. J Clin Oncol 2003; 21: 2247–53. [DOI] [PubMed] [Google Scholar]
- 42. Lang FF, Bruner JM, Fuller GN et al. Phase I trial of adenovirus‐mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol 2003; 21: 2508–18. [DOI] [PubMed] [Google Scholar]
- 43. Zeimet AG, Marth C. Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol 2003; 4: 415–22. [DOI] [PubMed] [Google Scholar]
- 44. Shono T, Tofilon PJ, Schaefer TS et al. Apoptosis induced by adenovirus‐mediated p53 gene transfer in human glioma correlates with site‐specific phosphorylation. Cancer Res 2002; 62: 1069–76. [PubMed] [Google Scholar]
- 45. Terui T, Murakami K, Takimoto R et al. Induction of PIG3 and NOXA through acetylation of p53 at 320 and 373 lysine residues as a mechanism for apoptotic cell death by histone deacetylase inhibitors. Cancer Res 2003; 63: 8948–54. [PubMed] [Google Scholar]
- 46. Luo J, Li M, Tang Y et al. Acetylation of p53 augments its site‐specific DNA binding both in vitro and in vivo . Proc Natl Acad Sci USA 2004; 101: 2259–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lyer NG, Chin SF, Ozdag H et al. p300 regulates p53‐dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc Natl Acad Sci USA 2004; 101: 7386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]