

Abstract
In the present study, irinotecan (CPT‐11) was highly effective not only against the chemosensitive neuroblastoma (NB) xenografts SK‐N‐ASnu and TNB9, but also against the multidrug‐resistant NB xenograft TS‐N‐2nu. SK‐N‐ASnu and TNB9 were significantly more responsive to low‐dose daily CPT‐11 treatment than to intermittent administration of one‐third of the median lethal dose. For TS‐N‐2nu, there was no significant difference in tumor growth inhibition between the two treatment schedules. Treatment with CPT‐11 alone could not completely abolish tumor growth in mice. For TNB9, tumor regrowth seemed to result from an inability to regress host vessels in the stroma during treatment and an inability to suppress host‐derived vascular endothelial growth factor (VEGF) expression throughout therapy. In the multidrug‐resistant TS‐N‐2nu, VEGF was not suppressed by low‐dose therapy with CPT‐11, and neurofilament‐positive tumor cells escaped from apoptosis and were growth arrested at G0/G1 phase. These findings suggest a mechanism for the incomplete responsiveness of TS‐N‐2nu to CPT‐11. Our data demonstrate that diminished VEGF gene and protein expression is closely correlated with tumor growth inhibition and inhibition of angiogenesis by CPT‐11 in NB xenografts. Our results further suggest that a persistent blocker of stroma‐derived VEGF will need to be combined with CPT‐11 to completely inhibit the growth of chemosensitive NB, and that administration of CPT‐11 at higher doses will be required to inhibit the growth of multidrug‐resistant NB. (Cancer Sci 2008; 99: 1209–1217)
- Abbreviations: CPT‐11
irinotecan
- HIF
hypoxia‐inducible factor
- LD50
the median lethal dose
- MIR
maximal growth inhibition rate
- NB
neuroblastoma
- NF
neurofilament protein
- NSE
neuron‐specific enolase
- RTW
relative tumor weight
- SMA
smooth muscle actin
- TUNEL
terminal deoxynucleotidyl transferase‐mediated dUTP‐biotin nick‐end labeling
- VEGF
vascular endothelial growth factor
Neuroblastoma is one of the most common malignant solid tumors in childhood. There have been significant advances in the treatment of NB. However, the treatment results for patients older than 1 year of age with disseminated disease and those with MYCN‐amplified NB are still unsatisfactory. Among the newly developed antitumor drugs with therapeutic activity, CPT‐11 is promising for NB treatment.( 1 , 2 , 3 , 4 ) CPT‐11 is a semisynthetic derivative of camptothecin,( 5 ) and exerts antitumor activity by inhibiting DNA topoisomerase I.( 6 ) Topotecan, a camptothecin analog, has been shown to inhibit transcriptional activity and protein accumulation of HIF‐1α in human glioma cells by a DNA damage‐independent mechanism, and to cause significant tumor growth inhibition in glioblastoma xenografts.( 7 , 8 ) HIF‐1 is a transcription factor that plays a major role in cellular adaptive responses to hypoxia. It is a heterodimer composed of the HIF‐1α and HIF‐1β subunits.( 9 ) HIF‐1β is expressed constitutively in excess, whereas HIF‐1α protein expression is regulated tightly by tissue oxygen concentration. Under normoxia, HIF‐1α is degraded rapidly via the ubiquitin–proteasome pathway through prolyl hydroxylation and binding of the von Hippel‐Lindau protein. This process is suppressed under hypoxia, allowing increased stability of the HIF‐1α protein and transcriptional activation with HIF‐1β.( 10 , 11 ) It has recently been made clear that under hypoxia, HIF‐1α accumulates, establishes a feedback loop, and is kept under control by transcription‐dependent degradation.( 12 ) Under normoxia, HIF‐1α is expressed at low levels. Inhibitors of transcription do not activate the feedback loop. However, under hypoxia, HIF‐1α transcriptionally activates its own degradation independent of the prolyl hydroxylase and von Hippel‐Lindau pathway. Inhibitors of transcription dramatically superinduce HIF‐1α.
More than 60 genes involved in angiogenesis, glycolysis, erythropoiesis, cell survival, and metastasis are activated by HIF‐1.( 13 ) The growth and metastasis of solid tumors depends largely on angiogenesis. One of the most important key molecules in the regulation of neoangiogenesis is VEGF.( 14 ) HIF‐1α mediates angiogenesis by induction of VEGF.( 15 , 16 , 17 )
In NB, high VEGF gene expression correlates with stage IV of the disease.( 18 ) High tumor vascularity is associated with disseminated disease, MYCN amplification, unfavorable histology, and poor outcome.( 19 , 20 ) Therefore, inhibiting VEGF expression and interfering with angiogenesis may be a useful treatment target for aggressive NB. To date, there have been no reports investigating the relationship between tumor growth inhibition, inhibition of HIF‐1α and VEGF gene and protein expression, and inhibition of histopathological angiogenesis in NB. Using three human NB xenografts, we evaluated the antitumor activity of CPT‐11 administered intermittently at one‐third of the LD50 and continually at low doses. We further investigated the relationship between the inhibition of tumor growth, inhibition of HIF‐1α and VEGF gene and protein expression, and inhibition of histological angiogenesis by CPT‐11 in chemosensitive and multidrug‐resistant NB xenografts.
Materials and Methods
Animals and human NB xenografts. Human NB xenografts designated TNB9 and TS‐N‐2nu were derived from stage IV NB with 80 and 13 copies of MYCN amplification in the adrenal gland of a 15‐month‐old boy and the adrenal of a 4‐year‐old girl, respectively. Our preparatory studies confirmed that the original biological characteristics of TNB9 and TS‐N‐2nu, particularly MYCN amplification status and chromosome findings, were retained after serial transplantations. The SK‐N‐AS NB cell line, generously donated by Dr Tohru Sugimoto (Kyoto Prefectural University of Medicine), was maintained in RPMI‐1640 medium supplemented with 10% fetal bovine serum (BioWest, Nuaille, France). SK‐N‐AS cells have no MYCN amplification. The NB xenograft designated SK‐N‐ASnu was generated by injecting ~1 × 106 cultured SK‐N‐AS cells subcutaneously into the flank of Balb/cAJcl‐nu mouse (CLEA Japan, Tokyo, Japan), and the tumor generated was passaged several times before treatment.
Some small pieces of minced tumor were implanted subcutaneously with trochars into the unilateral or bilateral flanks of 5–6‐week‐old male nude mice. Treatment began in randomized groups of three to six mice when tumors reached approximately 150 mg. Tumor size and mouse bodyweight were measured every 4 days. The tumor weight was calculated according to the formula:
| tumor weight (mg) = a × b 2 × 1/2, |
where a is the length of the tumor and b is the width. The mean RTW was calculated using the equation:
| RTW = Wx/W0, |
where Wx and W0 are the mean tumor weight on days x and 0 (the initiation of treatment), respectively.
Drug sensitivity of three NB xenografts. Drug sensitivity of SK‐N‐ASnu, TNB9, and TS‐N‐2nu was investigated for eight antitumor drugs: cyclophosphamide, ifosfamide, cisplatin, mitomycin C, vincristine, etoposide, pirarubicin, and CPT‐11. Experiments were carried out according to the protocol of Battelle Columbus laboratories.( 21 , 22 ) Each drug was suspended according to the provider's instructions, and administered intraperitoneally except for pirarubicin, which was administered intravenously. Drugs were administered to six nude mice per group at the total LD50 in three injections at 4‐day intervals (q4d × 3). Control mice were administered an equivalent volume of vehicle (0.1 mL/10 g bodyweight) intraperitoneally. The MIR was obtained by calculating the minimal ratio of RTW in treated mice (RTWt) to that in control mice (RTWc) on the same day:
| MIR = (1‐RTWt/RTWc) × 100(%). |
Tumor response was evaluated as highly effective (++), effective (+), or ineffective (–) when tumor regression (RTW < 1.0) was seen after treatment, when MIR was equal to or greater than 58%, and when MIR was less than 58%, respectively.
Low‐dose prolonged administration of CPT‐11. Besides the q4d × 3 schedule of LD50 × 1/3 per dose (conventional dose) for the drug‐sensitivity experiment, three treatment schedules at a low dose (LD50 × 1/30, 5.9 mg/kg per dose) were evaluated: (1) intraperitoneal administration daily for 14 consecutive days (qd × 14); (2) intraperitoneal administration daily for 20 consecutive days (qd × 20); and (3) intraperitoneal administration daily 5 days per week for 2 consecutive weeks followed by a 7‐day resting period, with one repetition ([(qd × 5)2]2).
Real‐time quantitative reverse transcription–polymerase chain reaction. Total RNA was prepared from frozen tumor tissue according to the acid guanidinium–phenol–chloroform method.( 23 ) One microgram of each RNA sample was incubated with oligo d(T)16 and MuLV reverse transcriptase (Applied Biosystems, Foster City, CA, USA) to yield cDNA. The expression levels of the human HIF‐1α and VEGF genes as well as murine HIF‐1α and VEGF genes were measured at the cDNA level by the ABI Prism 7700 Sequence Detection System (Applied Biosystems). Humanβ‐ACTN or murineβ‐ACTN was used as an internal control gene. The specific primers used were hHIF‐1α‐f 5′‐CCAGTTACGTTCCTTCGATCAGT‐3′ and hHIF‐1α‐r 5′‐TTTGAGGACTTGCGCTTTCA‐3′, hVEGF‐f 5′‐TACCTCCACCATGCCAAGTG‐3′ and hVEGF‐r 5′‐ATGATTCTGCCCTCCTCCTTC‐3′, hβ‐ACTN‐f 5′‐CAACCGCGAGAAGATGACC‐3′ and hβ‐ACTN‐r 5′‐CACAGCCTGGATAGCAACGTAC‐3′, mHIF‐1α‐f 5′‐CCAGTTACGTTCCTTTGATCAGT‐3′ and mHIF‐1α‐r 5′‐GTAACTGTGCTCATACTTGGA‐3′, mVEGF‐f 5′‐AACGATGAAGCCCTGGAGT‐3′ and mVEGF‐r 5′‐CCGCATGATCTGCATGGT‐3′, mβ‐ACTN‐f 5′‐AGACTTCGAGCAGGAGATGG‐3′ and mβ‐ACTN‐r 5′‐TCAGGCAGCTCATAGCTCTTC‐3′. The sequences of the TaqMan probes were hHIF‐1α 5′‐FAM‐CACCATTAGAAAGCAGTTCCGCAAGCC‐TAMRA‐3′, hVEGF 5′‐FAM‐TCCCAGGCTGCACCCATGGC‐TAMRA‐3′, hβ‐ACTN 5′‐FAM‐TTTGAGACCTTCAACACCCCAGCCA‐TAMRA‐3′, mHIF‐1α 5′‐FAM‐CACCATTAGAGAGCAATTCTCCAAGCC‐TAMRA‐3′, mVEGF 5′‐FAM‐TGCCCACGTCAGAGAGCAACAT‐TAMRA‐3′, and mβ‐ACTN 5′‐FAM‐CACTGCCGCATCCTCTTCCTCCCT‐TAMRA‐3′. Twenty microliters of the polymerase chain reaction mixture used for quantification contained template cDNA, 1× qPCR Mastermix (Eurogentec, San Diego, CA, USA), 300 nM of each primer, and 200 nM of TaqMan probe. Each experiment was carried out in triplicate and repeated twice. The thermal cycling conditions were: 50°C for 2 min, 95°C for 10 min, 45 cycles at 95°C for 15 s, and 60°C for 1 min.
Immunoblotting. Frozen tumor samples were crushed in liquid nitrogen and homogenized in a sample buffer consisting of 125 mM Tris‐HCl (pH 6.8), 20 mM dithiothreitol, 4% sodium dodecylsulfate, 10% glycerol, and 0.5% protease inhibitor cocktail (Complete Mini; Roche Applied Science, Mannheim, Germany). The extracts were sonicated for 10 s and centrifuged at 17 000 g for 10 min at 4°C to remove debris. The protein concentrations were determined using the Bio‐Rad DC protein assay (Bio‐Rad Laboratories, Hercules, CA, USA). Samples containing the same amounts of protein (10–70 µg) were separated on 12.5% sodium dodecylsulfate–polyacrylamide gels, electroblotted on polyvinylidene fluoride membranes (Immobilon‐P; Millipore, Bedford, MA, USA), and probed with antibodies. The primary antibodies used were anti‐HIF‐1α clone H1α67 (Novus Biologicals, Littleton, CO, USA) at a dilution of 1:1000, anti‐VEGF clone A‐20 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at a dilution of 1:900, and anti‐actin clone 20–33 (Sigma, St Louis, MO, USA) at a dilution of 1:500. Bound antibodies were detected with horseradish peroxidase‐conjugated secondary antimouse (Amersham Biosciences, Buckinghamshire, UK) or antirabbit (Stressgen, Victoria, BC, Canada) antibodies, followed by an enhanced chemiluminescence system (PerkinElmer Life Sciences, Boston, MA, USA).
Immunohistochemistry and detection of apoptotic cells. Tumors were fixed in 10% neutral buffered formalin for 24 h prior to paraffin embedding. After deparaffinization, tissue sections were heated at 121°C for 7–30 min either in 50 mM citrate buffer (pH 6.0) for NSE and NF, or in 10 mM Tris buffer with 1 mM ethylenediaminetetraacetic acid (pH 9.0) for αSMA and Ki‐67 antigen. Endogenous peroxidase was blocked with 3% hydrogen peroxide in methanol for 3 min at room temperature. Anti‐NSE (Sigma) and monoclonal anti‐Ki‐67 antigen clone MIB‐1 (DakoCytomation, Carpinteria, CA, USA) were in their ready‐to‐use forms. A monoclonal anti‐NF clone 2F11 (Dako, Glostrup, Denmark) and a monoclonal anti‐αSMA clone 1A4 (Dako) were used at dilutions of 1:100 and 1:50, respectively. The bound antibodies were amplified using peroxidase‐labeled polymer‐conjugated antirabbit or antimouse antibody (EnVision+ System; DakoCytomation). For the coloring reaction, 3,3′‐diaminobenzidine (Sigma) was used as the chromogen and nuclear counterstaining was carried out with hematoxylin.
Apoptotic cells in tumor tissues were detected using the TUNEL assay. After deparaffinization and rehydration, tissue sections were covered with 20 µg proteinase K (Wako Pure Chemical Industries, Osaka, Japan) per mL phosphate‐buffered saline for 15 min at room temperature. After blocking of endogenous peroxidase, slides were washed in 20 µg proteinase K per mL (PBS) and immersed in TdT buffer (Takara Bio, Shiga, Japan). The samples were then incubated with TdT enzyme (Takara) and biotin‐16‐dUTP (Roche Applied Science) in the TdT buffer containing 0.01% bovine serum albumin at 37°C for 1.5 h in a humidity chamber. Biotin‐16‐dUTP nucleotides incorporated onto the free 3′ OH ends of DNA fragments were detected using the standard streptavidin–biotin complex immunoperoxidase technique (Elite ABC reagent; Vector Laboratories, Burlingame, CA, USA) with 3,3′‐diaminobenzidine as the chromogen.
Statistical analysis. The responses of NB xenografts to CPT‐11 were evaluated with respect to tumor growth delay by measuring the mean number of days required to reach two times the original tumor weight for individual tumors. Relative mRNA expression levels for HIF‐1α and VEGF were expressed as the mean ± SD. Student's t‐test or Welch's t‐test was used to assess the significance of differences between the treatment groups.
Results
Antitumor effects of eight drugs on three NB xenografts. Among the eight antitumor drugs examined, CPT‐11 was the most effective when administered at a conventional dose (LD50 × 1/3 per dose) at 4‐day intervals (Fig. 1). However, CPT‐11 has not yet been approved as a therapeutic drug against any pediatric tumors. cyclophosphamide, ifosfamide, cisplatin, and etoposide are standard drugs for NB treatment. SK‐N‐ASnu and TNB9 were sensitive or highly sensitive to all of those drugs except etoposide. TS‐N‐2nu was sensitive to cyclophosphamide, but resistant to the other three drugs. CPT‐11 alone was highly effective against TS‐N‐2nu, a multidrug‐resistant xenograft. No mice died during that experiment.
Figure 1.
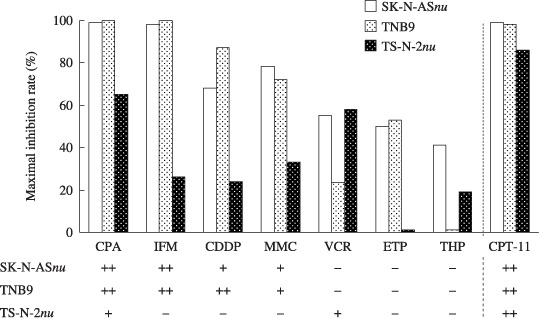
Maximal growth inhibition rates (MIR) and antitumor effects of eight drugs against three neuroblastoma xenografts. The MIR (%) was obtained by calculating the minimal ratio of mean relative tumor weight (RTW) in treated mice (RTWt) over that in control mice (RTWc) on the same day. MIR = (1 – RTWt/RTWc) × 100. The antitumor effect was evaluated as highly effective (++) when tumor regression was seen after treatment, effective (+) when the MIR was equal to or greater than 58%, or ineffective (–) and when the MIR was less than 58%. CDDP, cisplatin; CPA, cyclophosphamide; CPT‐11; irinotecan; ETP, etoposide; IFM, ifosfamide; MMC, mitomycin C; THP, pirarubicin; VCR, vincristine.
Responses of three NB xenografts to intermittent conventional‐dose and prolonged low‐dose administration of CPT‐11. Each treatment schedule for CPT‐11 demonstrated significant tumor growth inhibition against the three NB xenografts (P < 0.01, Student's t‐test) without resulting in substantial weight loss in nude mice (Table 1).
Table 1.
Responses of three neuroblastoma xenografts to different irinotecan (CPT‐11) treatment schedules
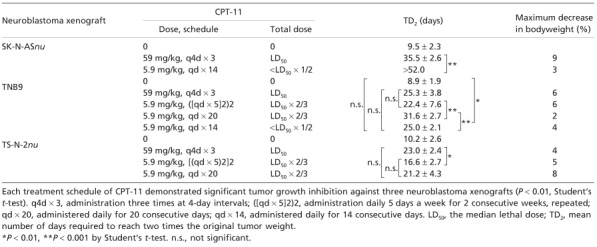
Initially, the responses to CPT‐11 for a conventional‐dose intermittent schedule (q4d × 3) and a low‐dose prolonged schedule (qd × 14) were compared in SK‐N‐ASnu. The results are presented in Figure 2a and Table 1. The low‐dose qd × 14 schedule was excellent for growth inhibition. Two out of the six tumors treated with low‐dose CPT‐11 showed complete regression at the end of therapy on day 52. Although the individual total dose for CPT‐11 was less than half of LD50, the low‐dose qd × 14 schedule was much more efficacious than the conventional‐dose q4d × 3 in which the LD50 for CPT‐11 was administered (P < 0.001, Student's t‐test).
Figure 2.
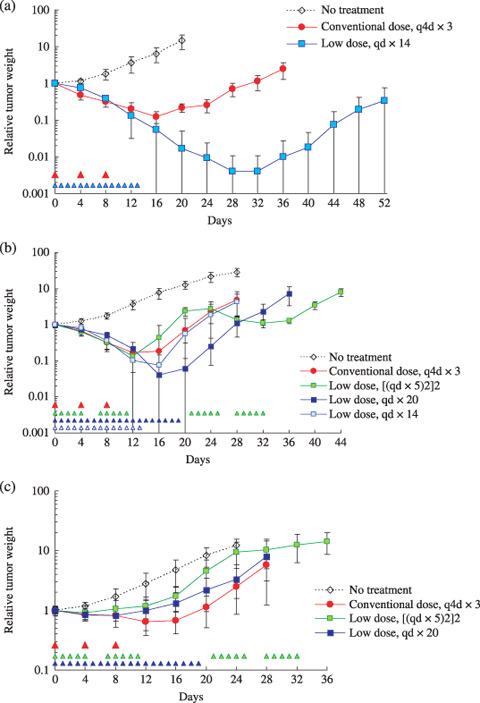
Responses of (a) SK‐N‐ASnu, (b) TNB9, and (c) TS‐N‐2nu xenografts to irinotecan (CPT‐11). Mice were administered CPT‐11 intraperitoneally for a conventional‐dose (59 mg/kg per dose) intermittent schedule, and for a low‐dose (5.9 mg/kg per dose) prolonged schedule. The arrowhead indicates the day CPT‐11 was administered. Tumor weight was calculated according to the formula: tumor weight (mg) = a × b 2 × 1/2, where a is the length of the tumor and b is the width. Tumors were measured every 4 days. The results are presented as mean relative tumor weight ± SD to that on day 0 when treatment was started.
The relationship between administration schedules and tumor responses to CPT‐11 was further examined using chemosensitive TNB9 and multidrug‐resistant TS‐N‐2nu xenografts. TNB9 responded well to all four administration schedules for CPT‐11 in the first 2 weeks of treatment (Fig. 2b). Low‐dose daily CPT‐11 administration without a resting period resulted in a duration‐dependent suppression of tumor growth. The low‐dose qd × 20 schedule was not only more effective than low‐dose qd × 14 (P < 0.001, Student's t‐test), but also the most effective of all four schedules. It showed significant tumor growth inhibition when compared to conventional‐dose q4d × 3 (P = 0.0077, Student's t‐test), which was as effective as low‐dose qd × 14 (P = 0.890, Student's t‐test). Low‐dose ([qd × 5]2)2, in spite of having the longest treatment period at 5 weeks, resulted in rapid tumor regrowth during a 7‐day resting period and showed no significant growth delay when compared to conventional‐dose q4d × 3 (P = 0.431, Student's t‐test) or to low‐dose qd × 14 (P = 0.448, Welch's t‐test) (Table 1). These results suggest that CPT‐11 administration on a low‐dose prolonged schedule without long resting periods is more effective against chemosensitive NB compared with conventional‐dose intermittent administration.
In the multidrug‐resistant TS‐N‐2nu, no low‐dose prolonged schedule showed greater tumor growth inhibition than the conventional q4d × 3 schedule (Table 1; Fig. 2c). There was no significant difference in growth delay between the conventional q4d × 3 and low‐dose qd × 20 schedules (P = 0.412, Student's t‐test). Most tumors did not show increased response to low‐dose daily CPT‐11 administration throughout treatment. The conventional q4d × 3 schedule was significantly more effective than the low‐dose ([qd × 5]2)2, which hardly achieved tumor regression after a 7‐day resting period (P = 0.0029, Student's t‐test) (Table 1). These findings suggest that CPT‐11 administered at a higher dose without a long resting period has successful antitumor activity against multidrug‐resistant NB.
Human and murine HIF‐1α and VEGF gene expression. We quantified human (tumor‐derived) and murine (host‐derived) HIF‐1α and VEGF gene expression levels in TNB9 and TS‐N‐2nu xenografts (Fig. 3a–d). In the untreated TNB9, the tumor‐derived human HIF‐1α gene expression level was decreased significantly according to the degree of tumor growth (day 10 vs day 14, P = 0.0331, Student's t‐test; day 14 vs day 19, P = 0.0182, Student's t‐test), whereas tumor‐derived human VEGF gene expression was increased significantly (day 10 vs day 14, P = 0.0061, Student's t‐test; day 14 vs day 19, P = 0.0020, Student's t‐test) (Fig. 3a). Both tumor‐derived HIF‐1α and VEGF genes in TNB9 treated with CPT‐11 were significantly downregulated regardless of the administration schedule of CPT‐11. Gene expression of tumor‐derived HIF‐1α in tumors treated with conventional‐dose CPT‐11 was reduced significantly compared to tumors treated with low‐dose CPT‐11 (P < 0.001, Student's t‐test). Suppression of tumor‐derived VEGF expression was maintained even after the discontinuation of CPT‐11 (Fig. 3a). Interestingly, host‐derived murine HIF‐1α and VEGF genes in TNB9 treated with conventional doses of CPT‐11 were upregulated in a compensatory manner. Host‐derived VEGF gene expression was not suppressed throughout the course of treatment (Fig. 3b).
Figure 3.
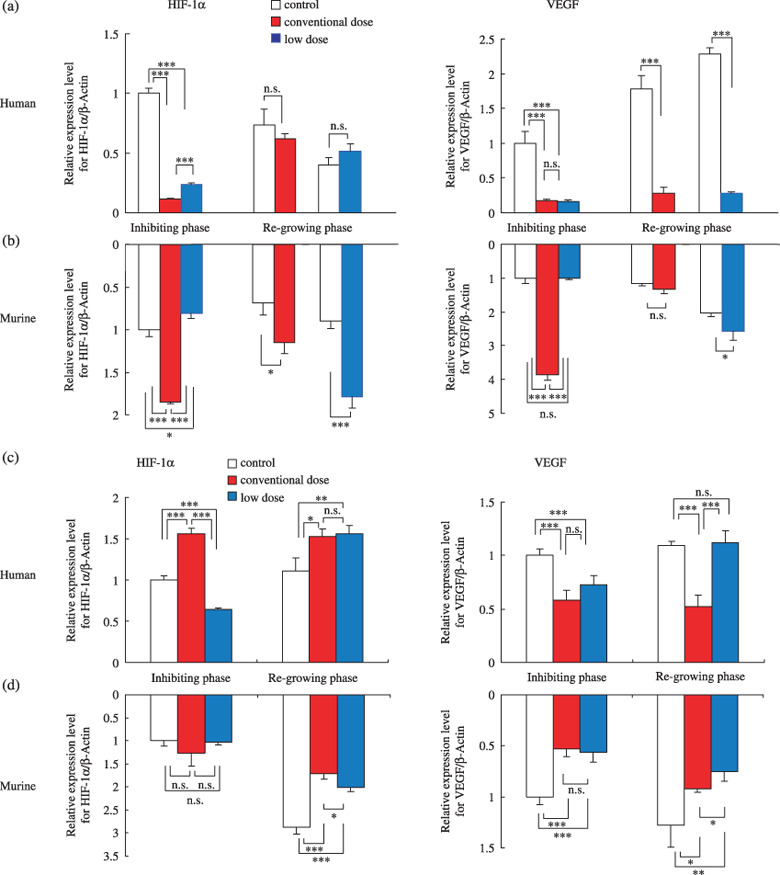
Quantitative reverse transcription–polymerase chain reaction gene expression analysis of (a,b) TNB9 and (c,d) TS‐N‐2nu xenografts. Relative mRNA expression levels for (a,c) human (tumor‐derived) and (b,d) murine (host‐derived) hypoxia‐inducible factor (HIF)‐1α and vascular endothelial growth factor (VEGF) in total RNA. Inhibiting phase, tumors on day 10 in TNB9, and on day 7 in TS‐N‐2nu. Regrowing phase, tumors on day 14 with conventional‐dose treatment and on day 19 with low‐dose treatment in TNB9, and on day 15 in TS‐N‐2nu. Results are presented as mean relative expression level ± SD to that in untreated tumors in inhibiting phase. *P < 0.05, **P < 0.01, ***P < 0.001; by Student's t‐test. n.s., not significant.
In the untreated TS‐N‐2nu, the gene expression levels of tumor‐derived human HIF‐1α as well as VEGF remained nearly unchanged regardless of the degree of tumor growth (Fig. 3c). Gene expression of tumor‐derived HIF‐1α in TS‐N‐2nu treated with conventional doses of CPT‐11 was upregulated to compensate for inhibition of VEGF expression. VEGF gene expression was not inhibited by the low‐dose daily treatment with CPT‐11. Treatment with CPT‐11 did not induce host‐derived murine HIF‐1α or VEGF gene expression in TS‐N‐2nu (Fig. 3d).
These results demonstrate that in NB xenografts treated with CPT‐11, there is a close relationship between the gene expression levels of tumor‐derived VEGF and the degree of tumor growth inhibition, regardless of the administration schedule. In TNB9, host‐derived HIF‐1α and VEGF produced by the host stroma throughout the course of CPT‐11 therapy appear to be responsible for rapid tumor regrowth. In contrast, in TS‐N‐2nu, host‐derived HIF‐1α and VEGF gene expression seem to contribute less to tumor regrowth.
Hypoxia‐inducible factor‐1α and VEGF protein expression in NB xenografts. In TNB9, HIF‐1α protein was induced dramatically during conventional‐dose treatment with CPT‐11, whereas VEGF protein expression was inhibited (Fig. 4a). HIF‐1α protein expression was reduced to a level almost as low as that observed in untreated tumors after 1 week of conventional‐dose therapy. At that time, VEGF protein expression was still inhibited. Low‐dose daily CPT‐11 treatment did not cause strong induction of HIF‐1α protein, although expression was not inhibited (Fig. 4a). VEGF protein in tumor treated with daily low doses of CPT‐11 was induced soon after the discontinuation of CPT‐11.
Figure 4.
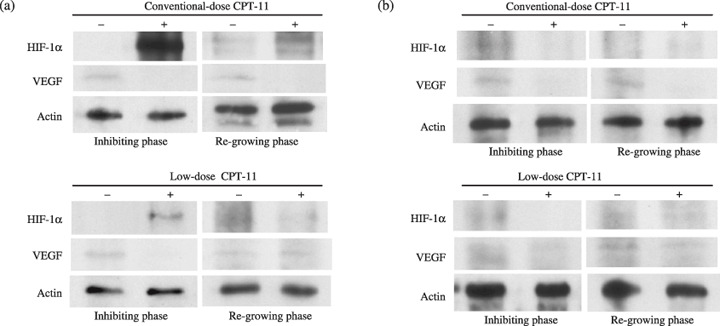
Immunoblot analysis of hypoxia‐inducible factor (HIF)‐1α and vascular endothelial growth factor (VEGF) proteins in (a) TNB9 and (b) TS‐N‐2nu xenografts. Inhibiting phase, tumors on day 10 in TNB9, and on day 7 in TS‐N‐2nu. Regrowing phase, tumors on day 14 with conventional‐dose treatment and on day 19 with low‐dose treatment in TNB9, and on day 15 in TS‐N‐2nu. For protein loading controls, actin levels were checked.
In TS‐N‐2nu, HIF‐1α protein expression was not induced strongly throughout CPT‐11 treatment. Intermittent conventional, but not low‐dose daily, treatment with CPT‐11 strongly inhibited VEGF protein expression (Fig. 4b).
These findings demonstrate that strong induction of HIF‐1α protein with inhibition of VEGF protein expression in TNB9 is caused by conventional‐dose CPT‐11 treatment, and that HIF‐1α protein expression is not always inhibited by low‐dose daily CPT‐11 treatment. These results suggest that VEGF but not HIF‐1α protein expression is related to inhibition of tumor growth.
Histology and immunohistochemistry. The anti‐αSMA antibody reacts with the smooth muscle of blood vessels. Tumor endothelial microvessels are completely covered by a layer of αSMA‐positive pericytes.( 24 ) Therefore, we identified tumor vessels in NB xenografts via αSMA immunostaining.
Untreated TNB9 contained poorly differentiated, NSE‐positive tumor cells with fibrovascular stroma (Fig. 5a, left panels). The density of the vessels increased according to the degree of tumor growth (data not shown). TNB9 treated daily with low doses of CPT‐11 showed obvious necrosis, calcification, and fibrosis. Nests of NSE‐positive tumor cells were scattered among the peripheral fibrous capsules (Fig. 5a, middle panels). Immunostaining for αSMA showed high vascularity in the stromal areas. Soon after low‐dose CPT‐11 therapy, nests of viable tumor cells were dominant with sparse necrotic areas, and tumor vascularity was reduced (Fig. 5a, right panels).
Figure 5.
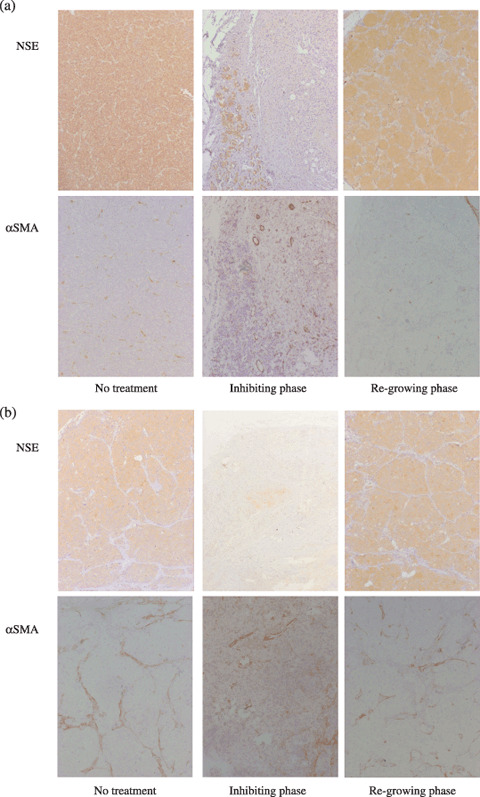
Immunohistochemistry of (a) TNB9 and (b) TS‐N‐2nµ xenografts during CPT‐11 treatment. Tumor sections from mice without CPT‐11 treatment, in inhibiting phase, and in regrowing phase were stained using the immunoperoxidase technique for neuron‐specific enolase (NSE) and α‐smooth muscle actin (SMA). (Original magnification, ×60.) (a) Untreated TNB9 contained poorly differentiated NSE‐positive tumor cells with fibrovascular stroma (left panels). In inhibiting phase, nests of NSE‐positive tumor cells were scattered among the peripheral fibrous capsules. High vascularity was observed in the stromal areas (middle panels). In regrowing phase, nests of viable tumor cells were dominant with sparse necrotic areas, and tumor vascularity was reduced (right panels). (b) Untreated TS‐N‐2nu contained uniform NSE‐positive tumor cell masses intermingled with microvessels that had developed in the fibroconnective stroma (left panels). In tumors in inhibiting phase, the number of NSE‐positive cells was markedly diminished, and a small number of large vessels was scattered in the stromal area (middle panels). Tumors in regrowing phase showed limited areas of necrosis, calcification, and connective tissue stroma. Large vessels were seen in the stroma (right panels).
Untreated TS‐N‐2nu contained uniform NSE‐positive tumor cell masses intermingled with microvessels that had developed in the fibroconnective stroma (Fig. 5b, left panels). TUNEL‐positive apoptotic cells were seen sparsely in the tumor (data not shown). In TS‐N‐2nu treated with conventional doses of CPT‐11, wide areas of necrosis, calcification, and stromal ingrowth were observed. The number of NSE‐positive cells was diminished markedly, and a small number of large vessels was scattered in the stromal area (Fig. 5b, middle panels). TS‐N‐2nu treated daily with low doses of CPT‐11 showed limited areas of necrosis, calcification, and connective tissue stroma. Large vessels were seen in the stroma (Fig. 5b, right panels). TS‐N‐2nu treated daily with low doses of CPT‐11 strongly expressed NF and contained a large number of apoptotic cells. Immunohistochemical findings for NF, TUNEL, and Ki‐67 antigens in mirror sections of TS‐N‐2nu treated daily with low doses of CPT‐11 revealed that NF‐positive cells had escaped from apoptosis (Fig. 6a,b). Furthermore, nearly all NF‐positive cells were negative for Ki‐67 (Fig. 6c,d), which indicated that they were arrested at the G0/G1 phase.( 25 )
Figure 6.
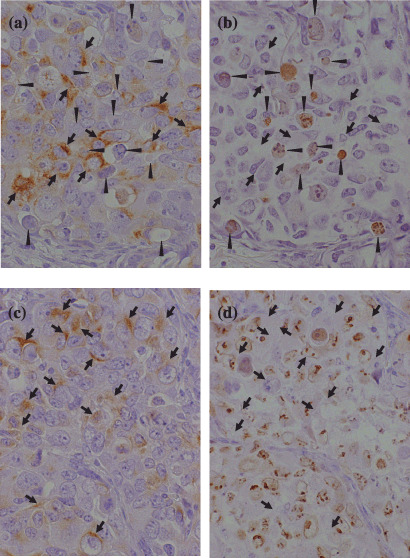
Immunohistochemistry for (a,c) neurofilament protein (NF), (b) terminal deoxynucleotidyl transferase‐mediated dUTP‐biotin nick‐end labeling and (d) Ki‐67 antigen in mirror sections of TS‐N‐2nu treated with low‐dose qd×15. (Original magnification, ×300.) (a,b) NF‐positive cells (arrows) escaped from apoptosis (arrowheads). (c,d) Nearly all NF‐positive cells (arrows) were negative for Ki‐67 and arrested at G0/G1 phase.
Discussion
Most anticancer drugs cause DNA damage and inhibit tumor cell proliferation. They are usually administered at high doses to kill as many tumor cells as possible. However, standard chemotherapeutic drugs such as vincristine, bleomycin, adriamycin, etoposide, 5‐fluorouracil, carboplatin, paclitaxel, and cyclophosphamide can target angiogenesis when the dose and frequency of administration are optimized.( 26 , 27 , 28 , 29 ) Continuous low‐dose chemotherapy with antitumor drugs was found to be more active against endothelial cells in comparison to tumor cells.( 26 ) Daily administration of topotecan, a camptothecin analog, caused significant tumor growth inhibition associated with a marked decrease in angiogenesis concomitant with HIF‐1α inhibition in glioblastoma xenografts.( 8 ) SN‐38, an active metabolite of CPT‐11, selectively inhibited endothelial cell proliferation and significantly decreased both HIF‐1α and VEGF expression of glioma cells in a dose‐ and time‐dependent manner.( 30 )
In our study, chemosensitive SK‐N‐ASnu and TNB9 were significantly more responsive to daily low‐dose CPT‐11 treatment than intermittent conventional dosing. Tumor‐derived HIF‐1α and VEGF gene expression in TNB9 treated daily with a low dose of CPT‐11 were downregulated significantly. However, host‐derived VEGF gene expression was not suppressed by the treatment. High vascularity was observed in the stromal areas during daily administration of CPT‐11. Administration of CPT‐11 alone resulted in the incomplete inhibition of tumor growth.
We carried out immunohistochemical experiments for HIF‐1α and VEGF in TNB9 and TS‐N‐2nu xenografts. HIF‐1α was hardly detected, whereas VEGF was positive in stromal cells in both untreated xenografts. NSE‐positive tumor cells were negative for VEGF. In TNB9 treated with CPT‐11, both HIF‐1α and VEGF were sparsely positive in stromal cells surrounding necrotic areas, whereas VEGF alone was detected in TS‐N‐2nu. In TNB9, VEGF protein expression was induced soon after discontinuation of CPT‐11 treatment. VEGF but not HIF‐1α expression was closely related to tumor growth inhibition and inhibition of angiogenesis by CPT‐11 in NB xenografts. The rapid tumor regrowth of TNB9 xenografts seemed to be the result of an inability to regress host vessels in the stroma during treatment as well as an inability to suppress host‐derived VEGF expression. Combined therapy with a persistent blocker of stroma‐derived VEGF will be needed to completely block the growth of chemosensitive NB.
Gerber and colleagues reported that complete inhibition of tumor growth and neovascularization in xenografts requires the blockade of both tumor‐ and host‐derived VEGF.( 31 ) They showed that systemic administration of an antihuman VEGF monoclonal antibody with a chimeric murine soluble VEGF receptor protein, mFlt(1–3)‐IgG, resulted in complete suppression of tumor growth in a human rhabdomyosarcoma xenograft, indicating that host‐derived VEGF significantly contributes to the overall process of tumor angiogenesis and growth.( 31 )
In TS‐N‐2nu, there was no significant difference in tumor growth inhibition between intermittent conventional‐dose CPT‐11 treatment and low‐dose daily CPT‐11 treatment. Both VEGF protein and tumor‐derived VEGF gene expression levels were not suppressed by low‐dose daily CPT‐11. Vessels in tumors treated with low‐dose CPT‐11 were larger and fewer in number than those in untreated tumors; this pattern of results seemed to correspond to the hypoxia‐induced apoptosis observed in a large number of TS‐N‐2nu tumor cells. However, NF‐positive cells escaped apoptosis and survived. Furthermore, nearly all NF‐positive cells were arrested at G0/G1 phase. The TS‐N‐2nu tumor cells probably became more aggressive as an adaptation to the hypoxic conditions, resulting in decreased responsiveness to the treatment and increased proliferation. These results show a lack of responsiveness of TS‐N‐2nu xenografts to low doses of CPT‐11. Therefore, administration of CPT‐11 at a higher dose will be required to inhibit the growth of multidrug‐resistant NB.
Acknowledgments
CPT‐II was provided by Yakult Honsha. This work was supported by a Grant‐in‐Aid for Exploratory Research from the Ministry of Education, Culture, Sports, Science and Technology (KAKENHI 19659456). Animal experiments were carried out in a humane manner after receiving approval from the Institutional Animal Experiment Committee of the University of Tsukuba, and in accordance with the university's Regulation for Animal Experiments as well as the Fundamental Guideline for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions set forth by the Ministry of Education, Culture, Sports, Science and Technology.
References
- 1. Komuro H, Li P, Tsuchida Y et al . Effects of CPT‐11 (a unique DNA topoisomerase I inhibitor) on a highly malignant xeno‐transplanted neuroblastoma. Med Pediatr Oncol 1994; 23: 487–92. [DOI] [PubMed] [Google Scholar]
- 2. Shitara T, Shimada A, Tsuchida Y et al . Successful clinical response to irinotecan in relapsed neuroblasatoma. Med Pediatr Oncol 2003; 40: 126–8. [DOI] [PubMed] [Google Scholar]
- 3. Tsuchida Y, Shitara T, Kuroiwa M et al . Current treatment and future directions in meuroblastoma. Indian J Pediatr 2003; 70: 809–12. [DOI] [PubMed] [Google Scholar]
- 4. Inagaki J, Yasui M, Sakata N et al . Successful treatment of chemoresistant stage 3 neuroblastoma using irinotecan as a single agent. J Pediatr Hematol Oncol 2005; 27: 604–6. [DOI] [PubMed] [Google Scholar]
- 5. Kunimoto T, Nitta K, Tanaka T et al . Antitumor activity of 7‐ethyl‐10‐[4‐(1‐piperidino)‐1‐piperidino] carbonyloxy‐camptothecin, a novel water‐soluble derivative of camptothecin, against murine tumors. Cancer Res 1987; 47: 5944–7. [PubMed] [Google Scholar]
- 6. Hsiang YH, Hertzberg R, Hecht S et al . Camptothecin induces protein‐linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 1985; 260: 14 873–8. [PubMed] [Google Scholar]
- 7. Rapisarda A, Uranchimeg B, Sordet O et al . Topoisomerase I‐mediated inhibition of hypoxia‐inducible factor 1: mechanism and therapeutic implications. Cancer Res 2004; 64: 1475–82. [DOI] [PubMed] [Google Scholar]
- 8. Rapisarda A, Zalek J, Hollingshead M et al . Schedule‐dependent inhibition of hypoxia‐inducible factor‐1α protein accumulation, angiogenesis, and tumor growth by topotecan in U251‐HRE glioblastoma xenografts. Cancer Res 2004; 64: 6845–8. [DOI] [PubMed] [Google Scholar]
- 9. Wang GL, Jiang BH, Rue EA et al . Hypoxia‐inducible factor 1 is a basic‐helix‐loop‐helix‐PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92: 5510–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang LE, Gu J, Schau M et al . Regulation of hypoxia‐inducible factor 1 alpha is mediated by an O2‐dependent degradation domain via the ubiquitin–proteasome pathway. Proc Natl Acad Sci USA 1998; 95: 7987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jaakkola P, Mole D, Tian YM et al . Targeting of HIF‐α to the von Hippel‐Lindau ubiquitylation complex by O2‐regulated prolyl hydroxylation. Science 2001; 292: 468–72. [DOI] [PubMed] [Google Scholar]
- 12. Demidenko ZN, Rapisarda A, Garayoa M et al . Accumulation of hypoxia‐inducible factor‐1α is limited by transcription‐dependent depletion. Oncogene 2005; 24: 4829–38. [DOI] [PubMed] [Google Scholar]
- 13. Semenza GL. HIF‐1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol 2000; 88: 1474–80. [DOI] [PubMed] [Google Scholar]
- 14. Toi M, Matsumoto T, Bando H. Vascular endothelial growth factor: its prognostic, predictive, and therapeutic implications. Lancet Oncol 2001; 2: 667–73. [DOI] [PubMed] [Google Scholar]
- 15. Iyer NV, Kotch LE, Agani F et al . Cellular and developmental control of O2 homeostasis by hypoxia‐inducible factor 1 αλπηα. Genes Dev 1998; 12: 149–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ryan HE, Lo J, Johnson RS. HIF‐1α is required for solid tumor formation and embryonic vascularization. EMBO J 1998; 17: 3005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carmeliet P, Dor Y, Herbert JM et al . Role of HIF‐1α in hypoxia‐mediated apoptosis, cell proliferation and tumor angiogenesis. Nature 1998; 394: 485–90. [DOI] [PubMed] [Google Scholar]
- 18. Komuro H, Kaneko S, Kaneko M et al . Expression of angiogenic factors and tumor progression in human neuroblastoma. J Cancer Res Clin Oncol 2001; 127: 739–43. [DOI] [PubMed] [Google Scholar]
- 19. Meitar D, Crawford SE, Rademaker AW et al . Tumor angiogenesis correlates with metastatic disease, N‐myc amplification, and poor outcome in human neuroblastoma. J Clin Oncol 1996; 14: 405–14. [DOI] [PubMed] [Google Scholar]
- 20. Ribatti D, Marimpietri D, Pastorino F et al . Angiogenesis in neuroblastoma. Ann NY Acad Sci 2004; 1028: 133–42. [DOI] [PubMed] [Google Scholar]
- 21. Geran RI, Greenberg NH, MacDonald MM et al . Protocols for screening chemical agents and natural products against tumors and biological systems. Cancer Chemother Rep 1972; 3: 51–61. [Google Scholar]
- 22. Ovejera AA, Houchens DP, Barker AD. Chemotherapy of human tumor xenografts in genetically athymic mice. Ann Clin Lab Sci 1978; 8: 50–6. [PubMed] [Google Scholar]
- 23. Chomczynski P, Sacchi N. Single‐step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem 1987; 162: 156–9. [DOI] [PubMed] [Google Scholar]
- 24. Pezzolo A, Parodi F, Valeria M et al . Tumor origin of endothelial cells in human neuroblastoma. J Clin Oncol 2007; 25: 376–83. [DOI] [PubMed] [Google Scholar]
- 25. Gerdes J, Lemke H, Baisch H et al . Cell cycle analysis of a cell proliferation‐associated human nuclear antigen defined by the monoclonal antibody Ki‐67. J Immunol 1984; 133: 1710–15. [PubMed] [Google Scholar]
- 26. Schirner M, Hoffmann J, Menrad A et al . Antiangiogenic chemotherapeutic agents: characterization in comparison to their tumor growth inhibition in human renal cell carcinoma models. Clin Cancer Res 1998; 4: 1331–6. [PubMed] [Google Scholar]
- 27. Bello L, Carrabba G, Giussani C et al . Low‐dose chemotherapy combined with an antiangiogenic drug reduces human glioma growth in vivo . Cancer Res 2001; 61: 7501–6. [PubMed] [Google Scholar]
- 28. Drevs J, Fakler J, Eisele S et al . Antiangiogenic potency of various chemotherapeutic drugs for metronomic chemotherapy. Anticancer Res 2004; 24: 1759–63. [PubMed] [Google Scholar]
- 29. Kieran MW, Turner CD, Rubin JB et al . A feasibility trial of antiangiogenic (metronomic) chemotherapy in pediatric patients with recurrent or progressive cancer. J Pediatr Hematol Oncol 2005; 27: 573–81. [DOI] [PubMed] [Google Scholar]
- 30. Kamiyama H, Takano S, Tsuboi K et al . Anti‐angiogenic effects of SN38 (active metabolite of irinotecan): inhibition of hypoxia‐inducible factor 1 alpha (HIF‐1α/vascular endothelial growth factor (VEGF) expression of glioma and growth of endothelial cells. J Cancer Res Clin Oncol 2005; 131: 205–13. [DOI] [PubMed] [Google Scholar]
- 31. Gerber HP, Kowalski J, Sherman D et al . Complete inhibition of rhabdomyosarcoma xenograft growth and neovascularization requires blockade of both tumor and host vascular endothelial growth factor. Cancer Res 2000; 60: 6253–8. [PubMed] [Google Scholar]