

Abstract
The INK4 family members p16INK4a and p15INK4b negatively regulate cell cycle progression by inhibition of cyclin‐dependent kinase (CDK) 4/6. Loss of p16INK4a functional activity is frequently observed in tumor cells, and is thought to be one of the primary causes of carcinogenesis. In contrast, despite the biochemical similarity to p16INK4a, the frequency of defects in p15INK4b was found to be lower than in p16INK4a, suggesting that p15INK4b‐inductive agents may be useful for tumor suppression. Here we report the discovery of a novel pyrido‐pyrimidine derivative, JTP‐70902, which exhibits p15INK4b‐inducing activity in p16INK4a‐inactivated human colon cancer HT‐29 cells. JTP‐70902 also induced another CDK‐inhibitor, p27KIP1, and downregulated the expression of c‐Myc and cyclin D1, resulting in G1 cell cycle arrest. MEK1/2 was identified by compound‐immobilized affinity chromatography as the molecular target of JTP‐70902, and this was further confirmed by the inhibitory activity of JTP‐70902 against MEK1/2 in kinase assays. JTP‐70902 suppressed the growth of most colorectal and some other cancer cell lines in vitro, and showed antitumor activity in an HT‐29 xenograft model. However, JTP‐70902 did not inhibit the growth of COLO320 DM cells; in these, constitutive extracellular signal‐regulated kinase phosphorylation was not detected, and neither p15INK4b nor p27KIP1 induction was observed. Moreover, p15INK4b‐deficient mouse embryonic fibroblasts were found to be more resistant to the growth‐inhibitory effect of JTP‐70902 than wild‐type mouse embryonic fibroblasts. These findings suggest that JTP‐70902 restores CDK inhibitor‐mediated cell cycle control by inhibiting MEK1/2 and exerts a potent antitumor effect. (Cancer Sci 2007; 98: 1809–1916)
Abbreviations:
- BSA
bovine serum albumin
- CDK
cyclin‐dependent kinase
- CKI
CDK‐inhibitor
- DMEM
Dulbecco's modified Eagle's medium
- DTT
dithiothreitol
- EDTA
ethylene diamine tetra‐acetic acid
- ELISA
enzyme‐linked immunosorbent assay
- ERK
extracellular signal‐regulated kinase
- INK4
inhibitor of CDK4
- log GI50
logarithm of the 50% growth inhibition value
- MAPK
mitogen‐activated protein kinase
- MBP
myelin basic protein
- MEF
mouse embryonic fibroblasts
- MEK
MAPK/ERK kinase
- pRb
retinoblastoma protein
- SDS‐PAGE
sodium dodecylsulfate–polyacrylamide gel electrophoresis
- TGF
transforming growth factor
Disruption of cell cycle control, reported in numerous tumor cells, is thought to be one of the primary causes of malignant transformation. CDK are key regulators of the cell cycle, and their activities are tightly controlled in a phase‐specific manner during cell cycle progression.( 1 ) Cyclin D–CDK4/6 and cyclin E–CDK2 play important roles in the G1 to S phase transition of the cell cycle by phosphorylating the tumor suppressor pRb, which forms a complex with E2F.( 1 , 2 ) The phosphorylation of pRb by CDK causes release and activation of E2F, allowing transcription of genes necessary for the progression to S phase.( 3 )
The loss of G1/S transition control in tumor cells often arises from aberrant activation of cell‐proliferative signaling pathways or inactivation of cell cycle‐regulating proteins. Of these pathways, the Ras–MAPK pathway has been investigated extensively. The classical MAPK cascade, comprising c‐Raf or B‐Raf, MEK1/2, and ERK1/2, is constitutively activated in various malignant tumors, often through gain‐of‐function mutations of Ras and Raf family members.( 4 ) These activating mutations cause increased expression of cyclin D, leading to CDK activation and aberrant cell proliferation.( 5 , 6 )
The cell cycle progression through cyclin–CDK activation is negatively regulated by CKI. Two families of CKI have been identified in mammalian cells: one includes p21WAF1, p27KIP1 and p57KIP2, each of which inhibit cyclin E/A–CDK2 complexes, and the other family, referred to as the INK4 family, is composed of p16INK4a, p15INK4b, p18INK4c and p19INK4d, which inhibit cyclin D–CDK4/6. p16INK4a is known to function as a tumor suppressor. Mutation or inactivation of p16INK4a has been detected in many human malignant tumors,( 7 ) and mice deficient in p16INK4a are highly susceptible to malignant tumors.( 8 , 9 ) In contrast, it has been reported that the frequency of mutation or defects in p15INK4b of tumor cells is lower than in p16INK4b.( 10 ) TGFβ inhibits cell proliferation at G1 phase and induces the expression of p15INK4b approximately 30‐fold in human keratinocyte HaCaT cells, suggesting that p15INK4b may act as an effector of TGFβ‐mediated cell cycle arrest.( 11 ) It has also been demonstrated that overexpression of p15INK4b in cancer cells induces cell cycle arrest at G1 phase.( 10 , 12 ) These observations suggest that p15INK4b may function as a replacement for p16INK4a, and that agents with the ability to induce p15INK4b may provide new strategies for the prevention or therapy of malignancies, especially against tumors that lack p16INK4a function.( 13 , 14 , 15 , 16 )
Here we report that the pyrido‐pyrimidine derivative JTP‐70902, identified by screening for p15INK4b‐inducing activity, induces cell cycle arrest at G1 phase in the p16INK4a‐inactivated human colon cancer cell line HT‐29.( 17 , 18 ) Analysis of the p15INK4b‐inducing mechanism revealed that JTP‐70902 binds to MEK1/2 and inhibits its kinase activity. We will discuss the importance of CKI induction in the suppression of tumor growth by MEK1/2 inhibition.
Materials and Methods
Chemicals and materials. JTP‐70902 (N‐[3‐{5‐(4‐bromo‐2‐fluorophenylamino)‐3‐cyclopropyl‐8‐methyl‐2,4,7‐trioxo‐3,4,7,8‐tetrahydro‐2H‐pyrido(2,3‐d)pyrimidin‐1‐yl}‐phenyl]‐methanesulfonamide) was synthesized at Japan Tobacco (Osaka, Japan). Screening of our compound library for p15INK4b‐inducing activity was conducted by a branched‐DNA method, using a QuantiGene High Volume Kit for the Direct Quantitation of Cellular mRNA (Bayer Corporation, Tarrytown, NY, USA) with p15INK4b‐specific probes. U0126 was purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against p15INK4b, p18INK4c, p27KIP1, cyclin A, cyclin E, CDK2, CDK4, CDK6, c‐Myc and α‐tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies for MEK1/2, ERK1/2, phospho‐ERK1/2, p38MAPK, phospho‐p38MAPK, Akt, phospho‐Akt, pRb, phospho‐pRb (Ser780) and cyclin D1 were from Cell Signaling Technology. Horseradish peroxidase‐labeled secondary antibodies and ECL Western Blot Detection Reagent were purchased from GE Healthcare Bio‐Science (Piscataway, NJ, USA). Recombinant kinases of MEK1 (inactive), MEK2 (inactive), ERK2 (inactive), c‐Raf (truncated form, active) and B‐Raf (V599E, active) were obtained from Upstate (Lake Placid, NY, USA).
Cell culture and growth inhibition assay. Human colorectal cancer cell line HT‐29 (American Type Culture Collections, Manassas, VA, USA) and COLO320 DM (Health Science Research Resources Bank, Osaka, Japan) were maintained in McCoy's 5 A medium and DMEM‐low glucose, respectively, both of which were supplemented with 10% fetal bovine serum and antibiotics. Cells were precultured for 24 h and then exposed to JTP‐70902. Cell growth was determined by Trypan‐blue dye exclusion test or evaluated using an In Vitro Toxicology Assay Kit, Sulforhodamine B Based (Sigma‐Aldrich, St Louis, MO, USA). Evaluation of growth inhibition against the panel of 39 human cell lines and COMPARE analysis were carried out in accordance with the methods of Yamori et al.( 19 ) Wild‐type and p15INK4b‐deficient (p15INK4b–/–) MEF,( 20 ) a kind gift from Dr Paul Krimpenfort (Netherlands Cancer Institute, Amsterdam, the Netherlands), were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mM glutamine, 0.1 mM minimum essential medium (MEM) non‐essential amino acids, 55 mM 2‐mercaptoethanol and antibiotics. Wild‐type and p15INK4B–/– MEF were plated at a density of 4 × 104 cells in 96‐well plates. After incubation with JTP‐70902 for 4 days, cell viability was assessed using Cell Counting Kit‐8 (Dojindo, Kumamoto, Japan).
Western blot analysis and mRNA quantitation. Western blot analysis of HT‐29 and COLO320 DM cell lysates was carried out according to a standard method. Peroxidase‐based chemiluminescence was developed by addition of ECL and the bands were detected using an LAS 3000 Image Analyzer (Fuji Film, Tokyo, Japan). Tumor tissues were homogenized in lysis buffer (1% Nonidet P‐40, 50 mM Tris‐HCl [pH 7.4], 150 mM NaCl, 0.2 mM Na3VO4, 1 mM EDTA) containing the protease inhibitor cocktail Complete (Roche). The resultant homogenate was centrifuged (20 000g, 10 min) and the supernatant was used for western blot analysis. p15INK4b and p27KIP1 mRNA levels in tumor tissues were assessed by quantitative reverse transcription–polymerase chain reaction. Total RNA was extracted from the tumor tissue using a GenElute Mammalian Total RNA Miniprep Kit (Sigma‐Aldrich). The reverse transcription reaction using 2.5 µg of the total RNA was carried out using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA), and quantitative polymerase chain reaction was carried out using Applied PCR mixture (Applied Biosystems) and an ABI PRISM 7700 Sequence Detector (Perkin‐Elmer Biosystems, CA, USA). Oligonucleotide primers and TaqMan probes were designed using Primer Express version 1.5 software (Applied Biosystems), as follows: p15INK4b forward primer 5′‐CCGCCCACAACGACTTTATT‐3′, reverse primer 5′‐CAGCCTTCATCGAATTAGGTG‐3′, probe 5′‐(FAM)TCTTACCCAATTTCCCACCCCCAC(TAMRA)‐3′; p27KIP1 forward primer 5′‐AAAGACTGATCCGTCGGACAG‐3′, reverse primer 5′‐GTAGAAGAATCGTCGGTTGCA‐3′, probe 5′‐(FAM)AGCAATGCGCAGGAATAAGGAAGCGAC(TAMRA)‐3′.
Cell cycle analysis. Unsynchronized HT‐29 cells were exposed to JTP‐70902 for 24 h. The nuclear DNA was stained with propidium iodide using a Cycle Test Plus kit (Becton Dickinson, Mountain View, CA, USA). The propidium iodide fluorescence was measured using a FACSort (Becton Dickinson) flow cytometer, and the data were analyzed using FlowJo software (Tree Star, San Carlos, CA, USA). The proportions of cells in G0/G1, S and G2/M phases were determined using the Watson pragmatic algorithm.
Compound‐immobilized affinity chromatography. JTP‐70902 was chemically modified by adding a spacer that bridged the compound and surface of the resin. Activated CH Sepharose 4B (GE Healthcare Bio‐Science) suspended in coupling buffer (0.25 mM NaHCO3, 50% dioxane [pH 8.0]) was incubated with the modified JTP‐70902 overnight at room temperature, and then stirred in 0.1 mM Tris‐HCl (pH 8.0) for 1 h to block the unreacted active group. After blocking, the resin was washed twice alternately with acetate buffer (pH 4.0) and 0.1 M Tris‐HCl (pH 8.0). The HT‐29 cytosol fraction was prepared in accordance with the method of Hiramoto et al.( 21 ) One milliliter of the cytosol fraction (containing 0.1–1% Tween‐20) was added to 10 µL of the compound‐immobilized resin and incubated for 1 h at 4°C with gentle mixing. The resin was collected and washed with wash buffer (20 mM HEPES [pH 7.9], 20% glycerol, 0.1 M KCl, 0.5 mM EDTA, 0.1% Tween‐20, 1 mM DTT), then resuspended in 2× SDS‐PAGE sample buffer. After heating for 5 min at 95°C, the resin was loaded onto a 4/20% SDS‐PAGE gel and the gel was stained using the 2D Silver‐Staining Reagent ‘Daiichi’ (Daiichi Pure Chemicals, Tokyo, Japan). The protein bands were excised from the gel and provided for protein mass spectrometry analysis (Apro Life Science Institute, Tokushima, Japan).
Protein kinase assays. A Raf–MEK–ERK cascade kinase assay consisting of active Raf (c‐Raf or B‐Raf), inactive MEK (MEK1 or MEK2) and inactive ERK2 was carried out in accordance with the method of Mallon et al.( 22 ) with some modifications. Two micrograms of dephosphorylated MBP (Upstate) in 0.2 M sodium carbonate (pH 9.4) was coated onto 96‐well plates (Immulon 1B ‘U’‐bottom plates; Thermolabsystems, Franklin, MA, USA) at 4°C overnight. After blocking with 1% BSA, the inactive forms of MEK1 (2 ng) or MEK2 (4 ng) with inactive ERK2 (50 ng) diluted in assay buffer (20 mM MOPS [pH 7.4], 0.1% 2‐mercaptoethanol, 0.1% BSA) were preincubated in the plates with test compounds at room temperature for 10 min. The kinase reaction was started by the addition of active B‐Raf (V599E) or c‐Raf, with 10 µM ATP and 12.5 mM MgCl2. After incubation at 30°C for 30 min, the phosphorylation of MBP by ERK2 was detected with peroxidase‐labeled antiphosphorylated MBP antibody.
Nude mouse HT‐29 xenograft model. Female BALB/c‐nu/nu mice obtained from Charles River Japan (Astugi, Japan) were used. On day 0, HT‐29 cells suspended in ice‐cold Hanks’ Balanced Salt Solution (HBSS) (–) were inoculated subcutaneously into the right flank of mice at 5.0 × 106 cells/100 µL/site. JTP‐70902 or vehicle (0.5% methylcellulose) was administered orally twice daily for 21 days from day 5. The tumor length (L [mm]) and width (W [mm]) were measured using a microgauge twice a week after commencement of dosing, and tumor volume was calculated using the following formula:
| tumor volume (mm3) = L × W × W/2. |
The tumor volume in treated groups was calculated as a percentage of the tumor volume in the vehicle groups and expressed as treated/control (%). All procedures relating to the use of animals in this study were reviewed and approved by the Institutional Animal Care and Use Committee at Japan Tobacco.
Statistical analysis. Results were expressed as mean ± SD. In the HT‐29 xenograft model, the significance of difference between the vehicle and JTP‐70902‐treated groups was analyzed by one‐way analysis of variance (anova) followed by Dunett's test. In the MEF growth inhibition assay, the significance of difference between the wild‐type MEF and p15INK4b‐deficient MEF was analyzed using Student's t‐test. Differences were considered to be statistically significant when P < 0.05.
Results
JTP‐70902, identified as a potent p15INKb inducer, modulates p27KIP1, cyclin D1, cyclin A and c‐Myc protein levels and induces G1 arrest in HT‐29 cells. We established a high‐throughput branched‐DNA assay and screened a total of 160 000 compounds for their ability to induce the expression of p15INK4b mRNA. As a result, a pyrido‐pyrimidine derivative, JTP‐70902, was identified to have a high p15INK4b‐inducing activity (Fig. 1). In a time‐course analysis using human colon cancer HT‐29 cells, 100 nM JTP‐70902 slightly induced expression of the p15INK4b protein after 8 h exposure, and the effect was pronounced after 24 h exposure (Fig. 2a). In analysis of other cell cycle regulatory proteins, p27KIP1 was upregulated after 8 h exposure, and cyclin D1 and cyclin A were found to be downregulated 4 and 24 h, respectively, after the addition of 100 nM JTP‐70902 (Fig. 2a). The expression of p18INK4c, CDK4, CDK6, CDK2 and cyclin E was not significantly affected. We next examined the effect of JTP‐70902 on c‐Myc, which has been reported to repress expression of p15INK4b and p27KIP1,( 23 , 24 , 25 ) and found that JTP‐70902 drastically reduced its level after 2 h of exposure, preceding the other events observed (Fig. 2a). When HT‐29 cells were exposed to various concentrations of JTP‐70902 for 24 h, the alterations in p15INK4b, p27KIP1 and cyclin A protein levels were observed at a minimum concentration of 10 nM, whereas obvious reduction in the level of cyclin D1 was detected at 100 nM or more (Fig. 2b, top panel). The protein level of c‐Myc was also reduced at a concentration of 10 nM after 2 h exposure (Fig. 2b, bottom panel).
Figure 1.
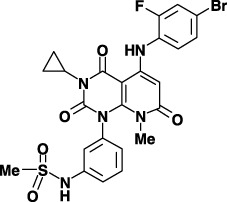
Chemical structure of JTP‐70902.
Figure 2.

Effects of JTP‐70902 on the expression of cell cycle regulatory proteins and cell proliferation. (a) HT‐29 cells were treated with 0.1% dimethyl sulfoxide alone or 100 nM of JTP‐70902 for the indicated time, and expression of cyclin‐dependent kinase (CDK)‐inhibitor, cyclins, CDK and c‐Myc and the phosphorylation of retinoblastoma protein (pRb) were analyzed by western blotting. α‐Tubulin was chosen as a control. (b) HT‐29 cells were treated with various concentrations of JTP‐70902 for 24 h (top panel) or 2 h (bottom panel). The expression level of each protein and the phosphorylation status of pRb were analyzed by western blotting. (c) Asynchronized HT‐29 cells were treated with various concentrations of JTP‐70902 for 24 h and the DNA content of the cells was determined by flow cytometry. (d) HT‐29 cells were treated with JTP‐70902 at various concentrations, and the number of viable cells was determined by Trypan‐blue dye exclusion test. The symbols indicate mean ± SD values (n = 3): closed circle, 0 nM; open triangle, 1 nM; open circle, 5 nM; open square, 25 nM; cross, 100 nM.
As expected from the induction of p15INK4b and p27KIP1 and reduction of cyclin D1 and cyclin A, conversion of hyperphosphorylated pRb to the hypophosphorylated form was detected after 8 h exposure to 100 nM JTP‐70902, coincident with the timing of alterations in p15INK4b and p27KIP1 levels (Fig. 2a). The phosphorylation of pRb at Ser780 by CDK4/6 was slightly diminished after 8 h exposure, and became undetectable thereafter. These changes in the phosphorylation status of pRb occurred with 10 nM JTP‐70902 (Fig. 2b). In accordance with these observations, cell cycle analysis by flow cytometry after 24 h exposure revealed that JTP‐70902 induced G1 arrest at a concentration of 10 nM (Fig. 2c). It is likely that the cell cycle was arrested after the induction of p15INK4b and p27KIP1 as the increment in cell number in G1 phase was not detected after 8 h exposure to even 100 nM JTP‐70902 (data not shown). In addition, JTP‐70902 exerted growth‐inhibitory effects from a concentration of 5 nM in a proliferation assay of HT‐29 cells, and showed complete inhibition at 100 nM (Fig. 2d). Overall, these results suggest that JTP‐70902 induces p15INK4b and p27KIP1 expression and reduces c‐Myc, cyclin D1 and cyclin A levels, and consequently inhibits the growth of HT‐29 cells by arresting the cell cycle at G1 phase.
JTP‐70902 binds specifically to MEK1/2. The rapid downregulation of c‐Myc led us to investigate whether JTP‐70902 influences growth‐signaling pathways. In the evaluation of the phosphorylation status of ERK1/2, p38MAPK and Akt in HT‐29 cells, only the phosphorylation of ERK1/2 was inhibited by JTP‐70902 at a concentration of 10 nM or more, whereas phosphorylation of p38MAPK and Akt was not affected even at 1000 nM (Fig. 3a). This inhibition of ERK1/2 signaling occurred within 1 h of exposure to JTP‐70902, prior to the downregulation of c‐Myc, cyclin D1 and cyclin A and induction of p15INK4b and p27KIP1 (2, 3). These results indicate that the molecular target of JTP‐70902 may be involved in the Raf–MEK–ERK pathway.
Figure 3.
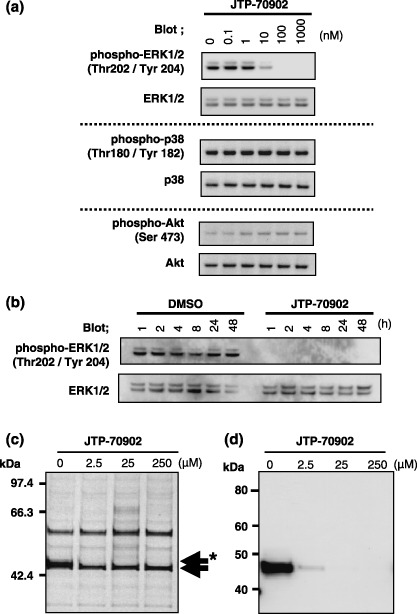
Effects of JTP‐70902 on the mitogen‐activated protein kinase/extracellular signal‐regulated kinase (ERK) kinase (MEK)–ERK pathway and identification of MEK1/2 as a JTP‐70902‐binding protein. (a) HT‐29 cells were treated with various concentrations of JTP‐70902 for 2 h, and the phosphorylation of ERK1/2, p38MAPK and Akt was analyzed by western blotting. (b) HT‐29 cells were treated with 0.1% dimethyl sulfoxide alone or 100 nM of JTP‐70902 for the indicated time, and phosphorylation of ERK1/2 was analyzed by western blotting. (c,d) The HT‐29 cytosolic fraction was supplemented with increasing concentrations of JTP‐70902 prior to loading on the resin, on which a JTP‐70902 analog was immobilized. The proteins bound to the resin were analyzed by sodium dodecylsulfate–polyacrylamide gel electrophoresis followed by silver staining (c) or western blotting with anti‐MEK1/2 antibody (d). (c) The 46‐kDa protein (asterisked arrow) and a slightly smaller non‐specific protein (arrow) are indicated.
To identify the molecular target of JTP‐70902 directly, we carried out compound‐immobilized affinity chromatography. The HT‐29 cytosol fraction was loaded onto JTP‐70902‐immobilized resin. Among the proteins recovered from the resin, only the band representing the 46‐kDa protein clearly disappeared when increasing concentrations of JTP‐70902 were added to the cytosolic fraction prior to loading (Fig. 3c, indicated by asterisked arrow), indicating specific binding of the 46‐kDa protein to JTP‐70902. Liquid chromatography–tanden mass spectrometry (LC‐MS/MS) analysis of the excised 46‐kDa band presented several peptide sequences of MEK1 and MEK2 (data not shown). Furthermore, western blot analysis showed that the 46‐kDa protein was stained with anti‐MEK1/2 antibody, and the band intensity detected was also reduced by addition of JTP‐70902 into the cytosolic fraction (Fig. 3d). These results strongly suggest that MEK1/2 is the intracellular molecular target of JTP‐70902.
JTP‐70902 is a potent inhibitor of MEK1/2 kinase. The direct inhibition by JTP‐70902 against the kinase activity of MEK1/2 was examined using an ELISA‐based Raf–MEK–ERK cascade kinase assay consisting of active Raf (c‐Raf or B‐Raf), inactive MEK (MEK1 or MEK2) and inactive ERK2. JTP‐70902 inhibited the phosphorylation of MBP by the cascade kinase reaction regardless of the isotype of Raf or MEK used, and the inhibitory activity was much more potent than that of the known MEK inhibitor U0126 (Table 1). JTP‐70902 did not inhibit the activity of c‐Raf, B‐Raf or ERK1/2 even at 10 µM, indicating that the cascade kinase reactions were inhibited as a consequence of MEK1/2‐inhibition (Supplementary Table S1). In addition, JTP‐70902 had no inhibitory activities to the other 23 Tyr kinases and 13 Ser/Thr kinases (Supplementary Table S1). As previous studies have shown that HT‐29 cells express an active mutant of B‐Raf (V599E) that is responsible for constitutive activation of the MEK–ERK pathway and tumorigenic proliferation,( 26 ) it was concluded that the direct inhibition of MEK1/2 by JTP‐70902 results in growth inhibition in HT‐29 cells. Furthermore, it was observed that known MEK inhibitors, U0126 and PD98059, also induced p15INK4b and p27KIP1 expression in HT‐29 cells as well as JTP‐70902, corroborating that inhibition of MEK causes p15INK4b and p27KIP1 induction (data not shown).
Table 1.
Inhibition of mitogen‐activated protein kinase/extracellular signal‐regulated kinase (ERK) kinase (MEK) enzyme activity by JTP‐70902
| MEK | Activator | IC50 value (nM) | |
|---|---|---|---|
| JTP‐70902 | U0126 | ||
| MEK1 | B‐Raf | 11 | 1200 |
| c‐Raf | 47 | 2200 | |
| MEK2 | B‐Raf | 7.2 | 130 |
| c‐Raf | 6.0 | 220 | |
Raf–MEK–ERK cascade assays were carried out as described in Materials and Methods. IC50 values of JTP‐70902 and U0126 in various combinations of Raf and MEK are shown.
JTP‐70902 shows a unique inhibition profile in comparison with conventional anticancer drugs in vitro, and antitumor activity in vivo. We next investigated the in vitro growth inhibition profile of JTP‐70902 on a panel of 39 human cancer cell lines, for comparison with those listed in a drug sensitivity database.( 19 , 27 , 28 , 29 , 30 ) It has been reported that agents targeting the same molecule or sharing the same mechanism of action produce a similar antitumor spectrum with high correlation coefficient in COMPARE analysis.( 30 , 31 , 32 ) A growth inhibition parameter, log GI50, of JTP‐70902 is shown in Fig. 4a. The mean log GI50 value of JTP‐70902 was –5.86, which lay in the middle of the value range of standard anticancer drugs in the panel.( 19 ) Most of the colorectal cancer cell lines and some of the breast, lung, melanoma, ovarian and stomach cancer cell lines were sensitive to JTP‐70902. The effective growth inhibition in colorectal cancer cell lines caused by JTP‐70902 agreed with frequent activation of the Ras–Raf–MEK–ERK pathway in these cells and their sensitivities to known MEK inhibitors.( 33 , 34 , 35 , 36 ) The COMPARE analysis revealed that there was no conventional anticancer drug showing a high correlation coefficient with JTP‐70902. These results are in accordance with the findings that the molecular target of JTP‐70902 is MEK1/2.
Figure 4.

Antitumor activities of JTP‐70902 in vitro and in vivo. (a) Log GI50 values for 39 human cancer cell lines treated with JTP‐70902 for 48 h are presented. Columns extending to the right or left indicate the sensitivity or resistance of the cells to JTP‐70902, respectively. MG‐MID, the mean of the logarithm of the 50% growth inhibition values for 39 cell lines; Delta, the logarithm of the difference between the MG‐MID and GI50 of the most sensitive cell line; Range, the logarithm of the difference between the GI50 of the most resistant cell line and that of the most sensitive one. (b) HT‐29 cells were inoculated subcutaneously into the right flank of nude mice, and JTP‐70902 or vehicle was administered orally twice daily for 21 days starting from 5 days after the inoculation (closed circle, vehicle; open triangle, 10 mg/kg twice daily; open circle, 30 mg/kg twice daily; open square, 100 mg/kg twice daily). Tumor volumes were measured twice a week. The results are expressed as mean ± SD (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle (Dunnett's test). (c,d) Single oral dose of 100 mg/kg JTP‐70902 or vehicle was administered to nude mice bearing the HT‐29 xenograft (n = 3). The tumor tissues were excised, the phosphorylation level of ERK1/2 at 4 h after dosing was analyzed by western blotting (c), and the expression levels of p15INK4b and p27KIP1 mRNA at 4 and 8 h after dosing were determined by quantitative reverse transcription–polymerase chain reaction (d). (d) The data are expressed as mean ± SD. White column, vehicle treatment group; black column, JTP‐70902 treatment group.
To confirm the in vivo efficacy of JTP‐70902, we evaluated the antitumor activity of JTP‐70902 in the nude mouse HT‐29 xenograft model. HT‐29 cells were inoculated subcutaneously into the right flank of nude mice, and JTP‐70902 was administered orally twice daily for 21 days starting from 5 days after the inoculation. Tumor growth was significantly suppressed in mice treated with 100 mg/kg JTP‐70902 during the course of the treatment. The treated/control values at the end of the observation period (day 25) were 100, 80 and 43% in the 10, 30 and 100 mg/kg groups, respectively (Fig. 4b). Neither significant changes in bodyweight nor gross lesions at autopsy were observed in the mice treated with the maximum dose of JTP‐70902. Furthermore, by single oral dosing of 100 mg/kg JTP‐70902, the phosphorylation of ERK1/2 in the established tumor tissues was completely inhibited (Fig. 4c), and both p15INK4b and p27KIP1 mRNA levels were upregulated in parallel (Fig. 4d). These results indicate that JTP‐70902 exerts an antitumor effect in vivo by the mechanisms of action that were demonstrated in the in vitro studies.
JTP‐70902 cannot inhibit cell proliferation independently of the MEK–ERK pathway, and requires induction of p15INK4b for its full activity. Lastly, we examined the correlation between MEK1/2 inhibition and growth inhibition by JTP‐70902, and the involvement of p15INK4b in its effect. We found that JTP‐70902 could not inhibit the growth of human colon cancer COLO320 DM cells even at 10 µM (Fig. 5a). Western blot analysis revealed that constitutive phosphorylation of ERK1/2 did not occur in COLO320 DM cells, although the abundance of ERK1/2 in COLO320 DM cells was higher than in HT‐29 cells (Fig. 5b). Moreover, the induction of p15INK4b and p27KIP1 by JTP‐70902 was not observed in COLO320 DM cells (Fig. 5b). These results suggest that COLO320 DM proliferation is independent of the MEK–ERK pathway and this is the reason for the insensitivity to JTP‐70902. These observations also support the idea that the antitumor activity and p15INK4b‐ and p27KIP1‐inducing activities of JTP‐70902 are dependent on MEK1/2 inhibition. To reveal whether p15INK4b contributes to the growth‐inhibitory activity of JTP‐70902, we compared the sensitivities of p15INK4b–/– and wild‐type MEF to JTP‐70902. The p15INK4b–/– MEF were more resistant to the growth‐inhibitory effect of JTP‐70902 than wild‐type MEF (Fig. 5c), suggesting that p15INK4b at least partially contributes to the growth‐inhibitory effect of JTP‐70902.
Figure 5.

Growth‐inhibitory effects of JTP‐70902 against COLO320 DM cells and p15INK4b‐deficient mouse embryonic fibroblasts (MEF). (a) HT‐29 cells (closed circle) and COLO320 DM cells (open circle) were treated with JTP‐70902 for 72 h, and cell viability was measured by Sulforhodamine B assay. The results are shown as percentage of dimethyl sulfoxide (DMSO) treatment (mean ± SD, n = 3). (b) HT‐29 and COLO320 DM cells were treated with JTP‐70902 at the concentrations indicated for 24 h. The phosphorylation of ERK1/2 and the expression of p15INK4b and p27KIP1 were analyzed by western blotting. (c) Wild‐type (gray column) or p15INK4b‐deficient MEF (white column) were treated with JTP‐70902 at the concentrations indicated for 4 days, and cell viability was determined as described in Materials and Methods. The results are shown as percentage of DMSO (mean ± SD, n = 3). *P < 0.05 versus p15INK4b wild type (Student's t‐test).
Discussion
Our present work is based on the findings that p15INK4b has similar functions to p16INK4a as a tumor suppressor,( 7 ) but its mutation or defect in tumor cells is less frequent than in p16INK4a.( 10 ) These reports suggest that p15INK4b may act as a replacement for p16INK4a, and a p15INK4b‐inductive agent might enable development of a new strategy for the prevention or therapy of malignancies.( 13 ) Previous studies demonstrating that several antiproliferative agents, such as trichostatin‐A, indole‐3‐carbinol and 15‐deoxy‐Δ12,14‐PGJ2, inhibit cell proliferation accompanied by p15INK4b induction also support this view.( 14 , 15 , 16 )
We identified JTP‐70902, which has p15INK4b‐inducing activity and inhibits the growth of HT‐29 cells. Using compound‐immobilized affinity chromatography, the molecular target of JTP‐70902 was identified as MEK1/2, and JTP‐70902 inhibited its kinase activity. Because HT‐29 cells were reported to have a transcriptional defect in p16INK4a due to promoter methylation,( 17 , 18 ) it is possible for p15INK4b induced by JTP‐70902 to complement the CKI function of p16INK4a. Indeed, we found that the p15INK4b–/– MEF were more resistant to the growth‐inhibitory effect of JTP‐70902 than wild‐type MEF (Fig. 5c), suggesting that p15INK4b at least partially plays a role in growth arrest by a MEK inhibitor. As the possible mechanism of the partial growth suppression by JTP‐70902 in p15INK4b–/– MEF cells, there is the possibility that JTP‐70902 may have p27KIP1‐inducing activity or cyclin D‐ and c‐Myc‐reducing activities, as previously reported for other known MEK inhibitors.( 37 , 38 , 39 ) As other possible mechanisms, unknown pathways might exist that have growth‐inhibitory effects on MEF cells. A recent report showed that the upregulation of p27KIP1 was necessary for cell cycle arrest of pancreatic cell lines by the MEK inhibitor U0126, as determined by a knockdown study using antisense DNA and small interfering RNA.( 37 ) In contrast, it has been demonstrated that p27KIP1 induction and inhibition of CDK2 were dispensable, but CDK4/6 inhibition was required for growth arrest by U0126 in several human colon cancer cell lines,( 38 ) indicating that p15INK4b induction may be necessary for CDK4/6 inhibition in these cells. Furthermore, our observations of activities in a panel of 10 human colorectal cancer cell lines revealed that either p15INK4b or p27KIP1 or both were upregulated in nine cell lines in which proliferation was repressed by JTP‐70902 analogs, but not in COLO320 DM cells (data not shown). COLO320 DM cells were insensitive to JTP‐70902, and there was no alteration in the expression of p15INK4b or p27KIP1 in the cells (Fig. 5b). These findings indicate that inhibition of MEK1/2 may elicit cell cycle arrest by distinct mechanisms involving p15INK4b or p27KIP1, depending on the cancer cell lines. The precise contribution of each CKI induction to the antitumor effects should be further investigated.
The current study is the first report demonstrating that p15INK4b can be induced by MEK1/2 inhibition. In contrast, although the MEK–ERK pathway was originally regarded to promote cellular proliferation and downregulation of CKI, such as p21WAF1 and p27KIP1, several studies reported that activation of the ERK pathway was required for induction of p15INK4b.( 40 , 41 , 42 ) The ERK signaling pathway triggered by TPA and Saikosaponin A was involved in p15INK4b induction and growth inhibition in HepG2 cells,( 41 ) and further evidence demonstrated that sustained ERK activation is associated with cell cycle arrest.( 43 ) Together with our observations in the panel of colorectal cancer cell lines described above, it is most likely that induction of p15INK4b depends on the intensity and duration of MEK–ERK activation: a robust and persistent activation of ERK by agents such as TPA promotes p15INK4b induction, whereas in the case of the MEK–ERK pathway involved in autonomous cell proliferation, inhibition of the MEK–ERK pathway leads to induction of p15INK4b. Furthermore, JTP‐70902 downregulated c‐Myc, which is one of the target proteins of the MEK–ERK pathway. Phosphorylation by ERK1/2 was reported to enhance the stability of c‐Myc protein,( 44 , 45 ) and the expression of p15INK4b and p27KIP1 is repressed by c‐Myc.( 23 , 24 , 25 ) These findings raise the possibility that inactivation of ERK1/2 causes rapid degradation of c‐Myc protein and results in the induction of p15INK4b and p27KIP1 in HT‐29 cells.
Although mutation or defects in MEK1/2 itself were rarely detected in human malignant tumors, constitutive activation of the upstream molecules of MEK1/2, such as K‐Ras or B‐Raf, was often observed.( 4 ) A recent report demonstrated that cells harboring the B‐Raf mutation have higher sensitivity to the MEK inhibitor CI‐1040 (also called PD 184352) than either Ras mutant cells or Ras/B‐Raf wild‐type cells, implying that B‐Raf mutant cells are much more reliant on the MEK–ERK pathway.( 39 ) Our data were in agreement with this, showing that HT‐29 expressing mutant B‐Raf was one of the most sensitive cell lines to JTP‐70902. With respect to Ras mutant cell lines, we found a discrepancy in the sensitivities because human colon cancer HCT 116 cells displayed the same sensitivity as HT‐29 cells, but A549 was resistant to JTP‐70902 (Fig. 4a). Further studies are required to establish the relationship between Ras mutation and c‐Myc reduction, p15INK4b‐ or p27KIP1‐induction by MEK inhibitors.
In conclusion, we discovered a p15INK4b‐inductive compound, JTP‐70902, that exerts potent antitumor effects in vitro and in vivo, and found that its molecular target is MEK1/2. JTP‐70902 also induces expression of p27KIP1 and downregulates cyclin D1, cyclin A and c‐Myc in tumor cells, as previously reported for other known MEK inhibitors.( 37 , 38 , 39 ) We also found that p15INK4b at least partially contributes to the growth‐inhibitory effect of JTP‐70902, and p15INK4b induction is dependent on MEK1/2 inhibition. These findings suggest that restoration of CKI‐mediated cell cycle control is implicated in growth‐inhibitory effects of MEK inhibitors and would be a potentially beneficial approach for clinical treatment of malignancies.
Acknowledgments
We thank Y. Watanabe for advice, suggestions and manuscript reading; H. Kawasaki for advice and suggestions; H. Kurachi for fruitful discussion and technical assistance in the screening; and S. Ebihara, T. Matsui, H. Chatani and Y. Ueda for technical assistance.
References
- 1. Sherr CJ. Mammalian G1 cyclins. Cell 1993; 73: 1059–65. [DOI] [PubMed] [Google Scholar]
- 2. Dowdy SF, Hinds PW, Louie K, Reed SI, Arnold A, Weinberg RA. Physical interaction of the retinoblastoma protein with human D cyclins. Cell 1993; 73: 499–511. [DOI] [PubMed] [Google Scholar]
- 3. Dyson N. The regulation of E2F by pRB‐family proteins. Genes Dev 1998; 12: 2245–62. [DOI] [PubMed] [Google Scholar]
- 4. Sebolt‐Leopold JS, Herrera R. Targeting the mitogen‐activated protein kinase cascade to treat cancer. Nat Rev Cancer 2004; 4: 937–47. [DOI] [PubMed] [Google Scholar]
- 5. Roovers K, Assoian RK. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays 2000; 22: 818–26. [DOI] [PubMed] [Google Scholar]
- 6. Mercer KE, Pritchard CA. Raf proteins and cancer: B‐Raf is identified as a mutational target. Biochim Biophys Acta 2003; 1653: 25–40. [DOI] [PubMed] [Google Scholar]
- 7. Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta 1998; 1378: F115–77. [DOI] [PubMed] [Google Scholar]
- 8. Serrano M, Lee H, Chin L, Cordon‐Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996; 85: 27–37. [DOI] [PubMed] [Google Scholar]
- 9. Sharpless NE, Bardeesy N, Lee KH et al . Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001; 413: 86–91. [DOI] [PubMed] [Google Scholar]
- 10. Stone S, Dayananth P, Jiang P et al . Genomic structure, expression and mutational analysis of the P15 (MTS2) gene. Oncogene 1995; 11: 987–91. [PubMed] [Google Scholar]
- 11. Hannon GJ, Beach D. p15INK4B is a potential effector of TGF‐β‐induced cell cycle arrest. Nature 1994; 371: 257–61. [DOI] [PubMed] [Google Scholar]
- 12. Thullberg M, Bartkova J, Khan S et al . Distinct versus redundant properties among members of the INK4 family of cyclin‐dependent kinase inhibitors. FEBS Lett 2000; 470: 161–6. [DOI] [PubMed] [Google Scholar]
- 13. Matsuzaki Y, Sakai T. INK4 family – a promising target for ‘gene‐regulating chemoprevention’ and ‘molecular‐targeting prevention’ of cancer. Environ Health Prev Med 2005; 10: 72–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hitomi T, Matsuzaki Y, Yokota T, Takaoka Y, Sakai T. p15INK4b in HDAC inhibitor‐induced growth arrest. FEBS Lett 2003; 554: 347–50. [DOI] [PubMed] [Google Scholar]
- 15. Matsuzaki Y, Koyama M, Hitomi T, Kawanaka M, Sakai T. Indole‐3‐carbinol activates the cyclin‐dependent kinase inhibitor p15INK4b gene. FEBS Lett 2004; 576: 137–40. [DOI] [PubMed] [Google Scholar]
- 16. Matsuzaki Y, Koyama M, Hitomi T, Takaoka Y, Kawanaka M, Sakai T. 15‐Deoxy‐Δ12,14‐prostaglandin J2 activates the expression of p15INK4b gene, a cyclin‐dependent kinase inhibitor. Int J Oncol 2005; 27: 497–503. [PubMed] [Google Scholar]
- 17. Kubo A, Nakagawa K, Varma RK et al . The p16 status of tumor cell lines identifies small molecule inhibitors specific for cyclin‐dependent kinase 4. Clin Cancer Res 1999; 5: 4279–86. [PubMed] [Google Scholar]
- 18. Fang JY, Lu J, Chen YX, Yang L. Effects of DNA methylation on expression of tumor suppressor genes and proto‐oncogenes in human colon cancer cell lines. World J Gastroenterol 2003; 9: 1976–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamori T, Matsunaga A, Sato S et al . Potent antitumor activity of MS‐247, a novel DNA minor groove binder, evaluated by an in vitro and in vivo human cancer cell line panel. Cancer Res 1999; 59: 4042–9. [PubMed] [Google Scholar]
- 20. Latres E, Malumbres M, Sotillo R et al . Limited overlapping roles of p15INK4b and p18INK4c cell cycle inhibitors in proliferation and tumorigenesis. EMBO J 2000; 19: 3496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hiramoto M, Shimizu N, Sugimoto K et al . Nuclear targeted suppression of NF‐κB activity by the novel quinone derivative E3330. J Immunol 1998; 160: 810–19. [PubMed] [Google Scholar]
- 22. Mallon R, Feldberg LR, Kim SC et al . An enzyme‐linked immunosorbent assay for the Raf/MEK1/MAPK signaling cascade. Anal Biochem 2001; 294: 48–54. [DOI] [PubMed] [Google Scholar]
- 23. Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFβ influences Myc, Miz‐1 and Smad to control the CDK inhibitor p15INK4b . Nat Cell Biol 2001; 3: 400–8. [DOI] [PubMed] [Google Scholar]
- 24. Staller P, Peukert K, Kiermaier A et al . Repression of p15INK4b expression by Myc through association with Miz‐1. Nat Cell Biol 2001; 3: 392–9. [DOI] [PubMed] [Google Scholar]
- 25. Yang W, Shen J, Wu M et al . Repression of transcription of the p27Kip1 cyclin‐dependent kinase inhibitor gene by c‐Myc. Oncogene 2001; 20: 1688–702. [DOI] [PubMed] [Google Scholar]
- 26. Davies H, Bignell GR, Cox C et al . Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–54. [DOI] [PubMed] [Google Scholar]
- 27. Dan S, Tsunoda T, Kitahara O et al . An integrated database of chemosensitivity to 55 anticancer drugs and gene expression profiles of 39 human cancer cell lines. Cancer Res 2002; 62: 1139–47. [PubMed] [Google Scholar]
- 28. Naasani I, Seimiya H, Yamori T, Tsuruo T. FJ5002: a potent telomerase inhibitor identified by exploiting the disease‐oriented screening program with COMPARE analysis. Cancer Res 1999; 59: 4004–11. [PubMed] [Google Scholar]
- 29. Nakatsu N, Yoshida Y, Yamazaki K et al . Chemosensitivity profile of cancer cell lines and identification of genes determining chemosensitivity by an integrated bioinformatical approach using cDNA arrays. Mol Cancer Ther 2005; 4: 399–412. [DOI] [PubMed] [Google Scholar]
- 30. Yamori T. Panel of human cancer cell lines provides valuable database for drug discovery and bioinformatics. Cancer Chemother Pharmacol 2003; 52 (Suppl 1): S74–9. [DOI] [PubMed] [Google Scholar]
- 31. Paull KD, Shoemaker RH, Hodes L et al . Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J Natl Cancer Inst 1989; 81: 1088–92. [DOI] [PubMed] [Google Scholar]
- 32. Yaguchi S, Fukui Y, Koshimizu I et al . Antitumor activity of ZSTK474, a new phosphatidylinositol 3‐kinase inhibitor. J Natl Cancer Inst 2006; 98: 545–56. [DOI] [PubMed] [Google Scholar]
- 33. Lee SH, Lee JW, Soung YH et al . Colorectal tumors frequently express phosphorylated mitogen‐activated protein kinase. APMIS 2004; 112: 233–8. [DOI] [PubMed] [Google Scholar]
- 34. Ikehara N, Semba S, Sakashita M, Aoyama N, Kasuga M, Yokozaki H. BRAF mutation associated with dysregulation of apoptosis in human colorectal neoplasms. Int J Cancer 2005; 115: 943–50. [DOI] [PubMed] [Google Scholar]
- 35. Sebolt‐Leopold JS, Dudley DT, Herrera R et al . Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo . Nat Med 1999; 5: 810–16. [DOI] [PubMed] [Google Scholar]
- 36. Wang Z, Li Y, Liu ET, Yu Q. Susceptibility to cell death induced by blockade of MAPK pathway in human colorectal cancer cells carrying Ras mutations is dependent on p53 status. Biochem Biophys Res Commun 2004; 322: 609–13. [DOI] [PubMed] [Google Scholar]
- 37. Gysin S, Lee SH, Dean NM, McMahon M. Pharmacologic inhibition of ΡΑF→ΜΕΚ→ΕΡΚ signaling elicits pancreatic cancer cell cycle arrest through induced expression of p27Kip1 . Cancer Res 2005; 65: 4870–80. [DOI] [PubMed] [Google Scholar]
- 38. Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 2003; 3: 233–45. [DOI] [PubMed] [Google Scholar]
- 39. Solit DB, Garraway LA, Pratilas CA et al . BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006; 439: 358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu WS, Hsu HY. Involvement of p‐15INK4b and p‐16INK4a gene expression in saikosaponin a and TPA‐induced growth inhibition of HepG2 cells. Biochem Biophys Res Commun 2001; 285: 183–7. [DOI] [PubMed] [Google Scholar]
- 41. Wu WS. ERK signaling pathway is involved in p15INK4b/p16INK4a expression and HepG2 growth inhibition triggered by TPA and Saikosaponin a. Oncogene 2003; 22: 955–63. [DOI] [PubMed] [Google Scholar]
- 42. Malumbres M, Perez De Castro I, Hernandez MI, Jimenez M, Corral T, Pellicer A. Cellular response to oncogenic ras involves induction of the Cdk4 and Cdk6 inhibitor p15INK4b . Mol Cell Biol 2000; 20: 2915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal‐regulated kinase activation. Cell 1995; 80: 179–85. [DOI] [PubMed] [Google Scholar]
- 44. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras‐dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 2000; 14: 2501–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell 1999; 3: 169–79. [DOI] [PubMed] [Google Scholar]