

Abstract
We recently reported that heparanase, one of the extracellular matrix‐degrading enzymes, which plays a critical role in cancer progression, is located not only in the cytoplasm but also in the nucleus. Here we identified nuclear translocation of heparanase as a key step in cell differentiation. We applied an in vitro differentiation model of HL‐60 cells with 12–0‐tetradecanoylphorbol‐13‐acetate (TPA), in which nuclear translocation of heparanase was observed using immunohistochemical analysis. In this system, nuclear translocation of heparanase was abolished by inhibitors of heat shock protein 90 (HSP90), suggesting the involvement of HSP90 in translocation of heparanase. We further confirmed that overexpression of active form of heparanase induced differentiation of HL‐60 cells, although the catalytic negative form of heparanase did not. Therefore we speculate that nuclear translocation of enzymatically active heparanase may be involved in cellular differentiation. Our results suggest that a novel function of heparanase upon cell differentiation would raise a potential new strategy for cancer therapy of promyeloid leukemia and other types of cancer. (Cancer Sci 2007; 98: 535–540)
Heparanase is an endo‐β‐glucuronidase that specifically cleaves carbohydrate chains of heparan sulfate proteoglycans (HSPG), which are important components of the extracellular matrix (ECM).( 1 , 2 ) Heparanase is present in most malignant tumors such as stomach, esophagus, bladder, pancreas, liver, prostate, melanoma, breast and colon cancers.( 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 ) In addition, heparanase expression in tumor cells correlates with the degree of tumor invasiveness and poor prognosis of cancer patients.( 3 , 4 ) Furthermore, nuclear localization of heparanase suggests that the enzyme may fulfil non‐traditional functions.( 12 ) Recently, we reported that heparanase is located in the nucleus as well as in the cytoplasm in cancer cells and that nuclear heparanase appears to be related to differentiation while cytoplasmic heparanase reveals its well known enzymatic function and can be one of the key factors affecting the fate of cancer patients.( 4 )
Cell differentiation is one of the irreversible mechanisms by which cells terminate their growth.( 13 ) Therefore, induction of differentiation is thought to be one of the effective strategies for cancer therapy. Promyelocytic leukemia is known as a clinical target for differentiation‐inducible therapy.( 14 ) HL‐60, a promyelocytic leukemia cell line differentiates into monocytes and macrophages upon stimulation with 12–0‐tetradecanoylphorbol‐13‐acetate (TPA) and retinoic acid.( 15 , 16 ) Furthermore, these cells are known to express heparanase in the cytosol.( 17 ) In this study, we aimed to determine the role of heparanase in HL‐60 cell differentiation taking into consideration its subcellular localization.
Materials and Methods
Cells and reagents. HL‐60 leukemic cells obtained from Dainippon Pharmaceutical Co. (Osaka, Japan) were maintained in a medium consisting of RPMI1640 supplemented with 10% heat‐inactivated fetal calf serum (FCS), 100 units/mL of penicillin G and 100 mg/mL of streptomycin, in a humidified 5% CO2/air atmosphere at 37°C. 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA), geldanamycin (GA), and 17‐(allylamino)‐17‐demethoxygeldanamysin (17‐AAG) were purchased from Sigma Chemical Co. (St. Louis, MO, US). Anti‐human heparanase mouse monoclonal antibody was raised against recombinant human heparanase. Characterization and specificity of the antiheparanase monoclonal antibody was described in our previous report.( 3 ) Anti‐human heat shock protein 90 rat monoclonal antibody was purchased from StressGen Biotechnologies Corp. (Vancouver, BC, Canada) and fluorescein isothiocyanite (FITC)‐conjugated anti‐mouse IgG goat polyclonal antibody, rhodamine‐conjugated anti‐rat IgG goat polyclonal antibody, FITC‐conjugated anti‐rat IgG goat polyclonal antibody and rhodamine isothiocyanite (RITC)‐conjugated anti‐mouse IgG goat polyclonal antibody were from Chemicon International, Inc. (Temecula, CA, USA). Phycoerythrin (PE)‐conjugated anti‐human CD13 mouse monoclonal antibody was purchased from Exalpha Biologicals Co. (Boston, MA, USA). TPA and GA and 17‐AAG were prepared in a 1 mM and 10 mM stock solution in DMSO, respectively. All other reagents were of analytical grade and obtained from standard suppliers.
Plasmids and transfection pShooterTM Vector (pCMV/mycC vectors) were purchased from (Tokyo, Japan). To target the heparanase specifically into the nucleus, we constructed heparanase pShooter™ nuclear form (pCMV/myc/Nuc). pCMV/myc/Nuc vector consisted of triplicate of nuclear localization signal of SV40 large T antigen. All coding sequences of heparanase were amplified by PCR with a forward primer Hep‐NcoI‐S (5‐CACCATGGCCATGCTGCTGC GCTCGAAG‐3) and a reverse primer Hep‐XhoI‐AS (5‐ATCTCGAGGATGCAAGCAGCAACTTTGG‐3), which included adaptors of NcoI (sense) and XhoI (antisense) restriction endonuclease sites from the heparanase vector. Stop codon was removed to efficiently target heparanase protein to the nucleus in the pCMV/myc/HeparanaseNuc vector. After transformation into DH5α competent cells, many colonies with an insert were selected and plasmid DNA was extracted. Sequencing was performed for all selected colonies and a clone without mutation was selected and designated as pShooter/nuc‐hpa. Twenty milligrams of pShooter/nuc‐hpa plasmid were mixed with 8 × 106 cells for 5 min and then electroporated at 250 V and 1000 uF with Gene Pulser II (Bio‐Rad, Hercules, CA, US). Cells were immediately transferred to complete medium prewarmed to 37°C and plated into chamber slides. After 24 h, the medium was changed. The catalytic negative form of heparanase, which is the active center mutant of heparanase was reported by Hulett et al.( 18 , 19 ) A nuclear expression vector encoding the catalytic negative form of heparanase is also constructed and transfected in the same manner.
Flow cytometry. Two × 105 HL‐60 cells were plated onto 25 cm2 tissue culture flasks and treated with TPA, geldanamycin, both of them, or equal concentrations of the vehicle. For longer drug exposure, the medium containing the test drug or vehicle was replaced after 72 h. To determine the CD13 antigen distribution, the adherent cells were removed from the culture surface with trypsin and the harvested supernatant cells were stained with PE‐conjugated anti‐human CD13 mouse monoclonal antibody, and analyzed using a Becton Dickinson FACSCalibur with WinMDI version 2.8 system.
To measure the level of CD13 expression, HL‐60 cells were grown for 7 days in the presence or absence of different drugs as indicated, then incubated with PE‐conjugated anti‐human CD13 mouse monoclonal antibody (diluted 1:100) for 15 min at room temperature and analyzed in a Becton Dickinson FACSCalibur. The peak fluorescence represented the fluorescence channel with the highest number of cells. Peak fluorescence was plotted on the channel units of logarithmic fluorescence.
Invasion assay. Two × 105 cells were plated onto 4 cm2 tissue culture plates, and then treated with TPA, geldanamycin, both, or equal concentrations of the vehicle. Briefly, the in vitro invasion assay system comprised 13 mm polyvinylpyrolidone‐free polycarbonate filters of 8 µm pore size (BD Biosciences, Mississauga, ON, Canada). The Matrigel was reconstituted with serum‐free medium for 1 h at 37°C. The bottom compartments of the blind‐well chambers were filled with medium supplemented with 0.1% bovine serum albumin (BSA). Cells (5 × 105) suspended in 0.2 mL medium supplemented with 0.1% BSA were added to the upper compartment, and the chambers were incubated for 20 h in a humidified 5% CO2/air atmosphere at 37°C. After 24 h incubation, the contents of the lower compartments were collected and the invasive cells were counted. The rate of invasion was expressed in cell numbers. Filters were fixed in 70% ethanol for 30 min, stained with 0.4% trypan blue stain (0.4%, Life Technologies, Gaithersburg, MD, US) for 30 min, and then taken off. The lower surface was inspected microscopically for the presence of attached cells. For comparison, passive migration of cells to the lower compartment was assessed in experiments in which filters without previous coating with Matrigel were used. Results of at least triplicates within one experiment or of at least three comparable experiments were expressed as the mean ± standard deviation (SD), and Student's t‐test was used to determine the significance of the differences (P < 0.05) between the mean values.
Immunohistochemistry and confocal microscopy. Cells were plated onto 4 cm2 tissue culture plates at a density of 2 × 105 and then treated with TPA, geldanamycin, both of them, or equal concentrations of the vehicle. To determine the intracellular distribution of heparanase and heat shock protein 90 (HSP90) proteins, HL‐60 cells were grown for 0, 1, 1.5 or 2 h in the presence of TPA as indicated. The cells were first collected using a cytospin and fixed in 20% formalin for 10 min. After blocking of non‐specific reactivity with rabbit serum for 10 min at room temperature, sections were incubated overnight at 4°C with the antihuman heparanase mouse monoclonal antibody and antihuman heat shock protein 90 rat monoclonal antibody. Cells were washed with PBS and then incubated with RITC‐conjugated antimouse IgG1 rat polyclonal antibody (diluted 1:200) and FITC‐conjugated antirat IgG goat polyclonal antibody (diluted 1:200) for 30 min at 4°C. Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI). After that, stained cells were analyzed using a Carl Zeiss LSM 510, and localization of heparanase and HSP90 protein in each cell was determined. Heparanase staining using 3,3′‐diaminobensidine (DAB)/H2O2 solution was described in our previous report.( 3 )
Western blot. Nuclear extracts were prepared by using NE‐PER Nuclear and Cytoplasmic Extraction Reagents Kit (PIERCE Biotechnology, Inc, Chicago, IL, US). The protein concentration of the supernatant was determined using the Bio‐Rad Protein Assay (Bio‐Rad, Hercules, CA, US). Equal amounts (40 µg) of proteins were electrophoresed under reducing conditions on 12% (w/v) polyacrylamide gels. Proteins were electrophoretically transferred to Hybond‐PVDF transfer membranes (Amersham, Arlington Heights, IL, US) and incubated with primary antibodies against heparanase, and then peroxidase‐linked secondary antibody. An Amersham Enhanced Chemiluminescence Western System (Amersham, Tokyo, Japan) was used to detect secondary probes.
Results
Differentiation of HL‐60 cells induced by TPA. In the present study, we examined the role of nuclear heparanase in differentiation by using a promyelocytic leukemia cell line, HL‐60, and identified nuclear translocation of heparanase as a key step in cell differentiation. We further showed that only an active form of heparanase existed in the nucleus. Figure 1 shows differentiation of HL‐60 cells into macrophages and monocytes by low‐dose (1 nM) and long‐term (7 days) TPA treatment. TPA treatment also enhanced the ability of attachment of HL‐60 cells (Fig. 1). Morphological changes of HL‐60 cells were observed in 75.5 ± 6.3% in TPA‐treated cells. We observed similar morphological changes using high‐dose (100 nM) and short‐term (1 h) TPA treatment (data not shown).
Figure 1.
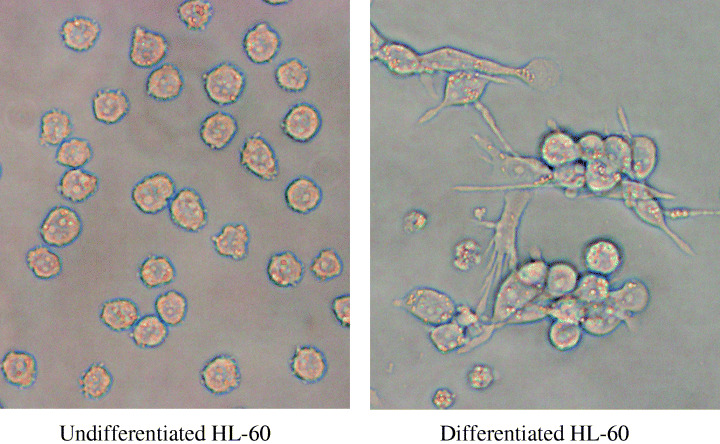
Morphological changes evaluated in HL‐60 cells grown on 4‐cm2 tissue culture plates. The cells were incubated with 1 nM tetradecanoylphorbol‐13‐acetate for 7 days. Cells were attached to a culture plate surface and their shapes (e.g. round, stellate, and linear) were determined.
Low‐dose/long‐term TPA treatment increased the expression of CD13, a macrophage/monocyte differentiation marker, as detected by flow cytometry, suggesting the differentiation of HL‐60 cells (Fig. 2a). We also examined the invasive properties of HL‐60 cells after both low‐dose/long‐term TPA treatment (Fig. 2c) and high‐dose/short‐term TPA treatment (Fig. 2d) by Matrigel invasion assay. HL‐60 cells showed a three‐fold higher invasion ability into Matrigel upon TPA stimulation compared with untreated cells, confirming the differentiation of HL‐60 cells into macrophages and monocytes with invasive properties.
Figure 2.
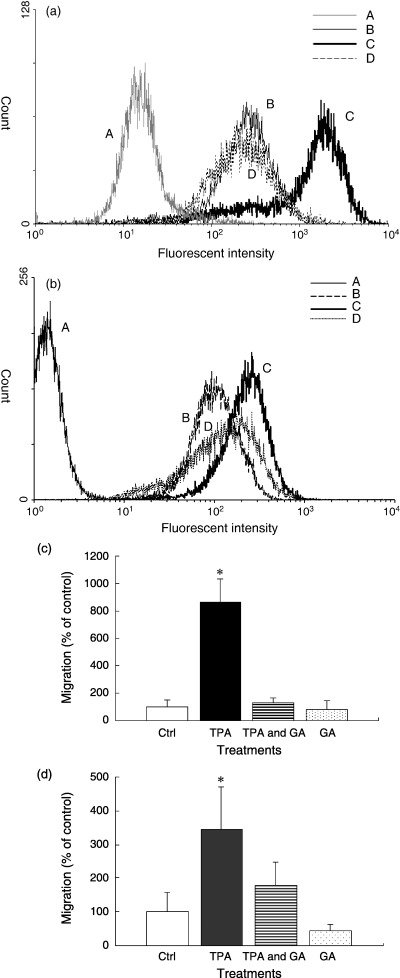
Characteristic evaluation of differentiation in HL‐60 cells. (a) Flow cytometric analysis of CD13 in HL‐60 cells. To measure CD13 expression, HL‐60 cells (A and B: untreated cells, C: tetradecanoylphorbol‐13‐acetate (TPA)‐treated cells, D: TPA + geldanamycin‐treated cells) were grown for 7 days in the presence or absence of different drugs (TPA: 1 nM, geldanamycin: 200 nM). Cells (B, C, and D) were then labeled with phycoerythrin‐conjugated anti‐human CD13 mouse monoclonal antibody or control IgG (A) and analyzed by a Becton Dickinson FACSCalibur. (b) Effect of 17‐(allylamino)‐17‐demethoxygeldanamysin (17‐AAG) on TPA‐induced CD13 expression (A and B: untreated cells, C: TPA‐treated cells, D: TPA +17‐AAG‐treated cells) were grown for 48 h in the presence or absence of different drugs (TPA: 10 nM, 17‐AAG: 20 nM). Cells (B, C, and D) were then labeled with PE‐conjugated anti‐human CD13 mouse monoclonal antibody or control IgG (A) and analyzed. (c) Invasiveness of cultured HL‐60 cells with low‐dose/long‐term TPA treatment through Matrigel. Untreated HL‐60 cells, TPA‐treated cells (1 nM, 7 days), TPA + geldanamycin‐treated cells (TPA 1 nM, geldanamycin 200 nM, 7 days), and geldanamycin‐treated cells (200 nM, 7 days) were compared. (d) Invasiveness of cultured HL‐60 cells with high‐dose/short‐term TPA treatment through Matrigel. Untreated HL‐60 cells, TPA‐treated cells (100 nM, 1 h), TPA + geldanamycin‐treated cells (TPA 100 nM, geldanamycin 2 mM, 1 h), and geldanamycin‐treated cells (2 mM, 1 h) were compared. The results were expressed as a percentage of the control. Data are mean ± SD. *P < 0.05, compared with the control. The results were expressed as percent of the control. Data are mean ± SD. *P < 0.05, compared with the control.
Differentiation of HL‐60 cells is involved in nuclear translocation of heparanase accompanied by HSP90. Immunofluorescence staining using FITC‐labeled anti‐heparanase monoclonal antibody showed that treatment with high‐dose TPA resulted in increased levels of heparanase both in the cytoplasm and nucleus, suggesting the translocation of part of the cytosolic heparanase into the nucleus upon differentiation of HL‐60 cells (Fig. 3b).
Figure 3.
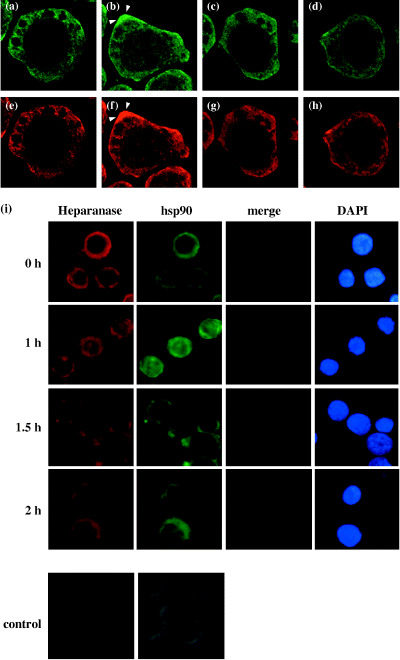
Redistribution of heparanase and heat‐shock protein 90 (HSP90) during tetradecanoylphorbol‐13‐acetate (TPA)‐induced differentiation of HL‐60 cells. The cells were incubated with vehicle (a, e), 100 nM TPA (b, f), 100 nM TPA +2 mM geldanamycin (c, g), or 2 mM geldanamycin (d, h) for 1 h. Note the localization of heparanase expression in the cytoplasm in a, c, and d, and in the nucleus in B. HSP90 expression was also localized in the cytoplasm in e, g, and h, and in the nucleus in F. Heparanase and HSP90 were detected with FITC‐labeled anti‐heparanase monoclonal antibody and with RITC‐labeled anti‐heat shock protein 90 antibody, respectively, and analyzed using a confocal microscopy (× 100). Note the dense localization of heparanase and HSP90 in lamellipodia in TPA‐treated cells (arrows, b, f). (i), Time‐dependent nuclear localization of heparanase and HSP90 induced by TPA. The nuclei were stained with DAPI to be compared with the immunofluorescence from heparanase and HSP90. The lowest panel shows a negative control using non‐specific mouse IgG1 with FITC‐conjugated anti‐mouse IgG antibody.
We investigated how heparanase translocates into the nucleus. Since heparanase itself does not possess a nuclear translocation signal domain,( 20 , 21 ) we proposed that a kind of chaperone might bring heparanase into the nucleus. Thus we tried to assess the effect of geldanamycin (GA), which had already been reported as a specific inhibitor of the most well known chaperone, heat shock protein 90 (HSP90).
When the cells were treated with TPA, we observed a similar enhanced expression and translocation of HSP90 from the cytoplasm into the nucleus in TPA‐treated cells (Fig. 3f). We also noted dense localization of heparanase and HSP90 in lamellipodia (arrows in Fig. 3b,f). Since previous studies have not yet reported the kinetics of heparanase, our report shows for the first time the cyto‐nuclear shuttling of heparanase. Heparanase and HSP90 translocated to the nucleus 1 h after treatment with 100 nM TPA and later returned to the cytoplasm (Fig. 3i). HSP90 showed the similar dynamic distribution in the cell. Since heparanase translocated into the nucleus after TPA treatment, we were interested in the role of heparanase in cell differentiation. Therefore, we first focused on cyto‐nuclear shuttling and examined the relationship between heparanase and HSP90.
Next, we examined the effect of GA, an inhibitor of HSP90, on TPA‐induced changes in heparanase cellular distribution. GA administered with TPA blocked the transfer of heparanase as well as HSP90 from the cytoplasm to the nucleus (Fig. 3c,g). If heparanase plays a role in cell differentiation, then we should be able to examine cell differentiation by adjusting the localization of heparanase using TPA and GA. In the next series of experiments, we blocked the differentiation of TPA‐treated cells by GA. Differentiation of HL‐60 cells by TPA treatment is well known.( 15 ) However, the effect of combined treatment of TPA and GA on differentiation has not been investigated. As shown in Fig. 2a, CD13 expression did not change after combined treatment of TPA and GA, suggesting the inhibition of HL‐60 cell differentiation. We have tried a less toxic analog of GA: 17‐AAG had partial inhibitory effect on TPA‐induced CD13 expression (Fig. 2b).
Furthermore, no morphological changes were noted in such cells (data not shown). Moreover, we have confirmed that GA inhibited the invasive capacity of HL‐60 cells (Fig. 2c,d). This finding suggested that GA suppressed the TPA‐induced cell differentiation. Inhibition of the nuclear transfer of heparanase and the blockade of differentiation of TPA‐treated cells by GA indicate the potentially important role of heparanase in cell differentiation. As a control, GA alone had no effect on HL‐60 cell differentiation as determined by a low invasive capacity similar to untreated cells (Fig. 2c). Moreover, cells treated with GA alone showed no sign of nuclear transfer of heparanase or HSP90 compared with untreated cells (Fig. 3d,h). These results suggested that GA specifically inhibited the effect of TPA on cell differentiation and nuclear translocation of heparanase and HSP90.
In summary, cell differentiation was associated with nuclear translocation of heparanase and this differentiation was suppressed by inhibiting heparanase and HSP90 translocation into the nucleus with GA.
Translocation of active form of heparanase into nucleus. We speculated that the differentiation and nuclear transfer of heparanase and HSP90 by TPA occurred in the same cell. However, which of the events occurred first was not clear. To resolve this issue, we examined the cytoplasmic as well as nuclear heparanase protein in TPA‐treated and untreated HL‐60 cells by Western blot analysis (Fig. 4). Our results showed the transfer of heparanase into the nucleus first, followed by differentiation of TPA‐treated cells. A 53 kDa active form of heparanase protein is produced after post‐translational modification from a 65 kDa proform.( 20 , 21 ) The active form of heparanase protein was detected in the nuclei of TPA‐treated HL‐60 cells only, while it was observed in the cytoplasm of both TPA‐treated and untreated cells. These results suggested that upon TPA treatment, heparanase translocated into the nucleus first and forced the cells into differentiation.
Figure 4.
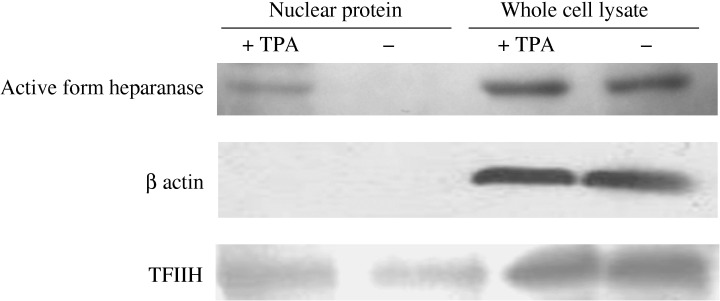
Translocation of active form of heparanase into nucleus. (a) Detection of heparanase protein by Western blot analysis. The cells were incubated with vehicle or 100 nM tetradecanoylphorbol‐13‐acetate (TPA) for 1 h and then the nuclei were fractioned as described in Materials and Methods. Heparanase protein was transported into the nucleus after treatment with TPA, and appeared as a 50 kDa active form. The nuclear fraction did not contain β‐actin, verifying the purity of the nuclear fraction without contamination of cytoplasmic proteins.
Overexpression of heparanase in the nucleus of Hl‐60 cells induces CD13 expression. It is still not clear whether the active form of heparanase alone is involved in the differentiation process of TPA‐treated cells. To study this, we constructed an expression vector, which specifically directs heparanase into the nucleus (pShooter nuclear heparanase, p‐shooter‐nuc). Heparanase was successfully overexpressed in the nuclei of HL‐60 cells by using the p‐shooter nuc vector transfection in which nuclear localization signals are attached. (Fig. 5).
Figure 5.
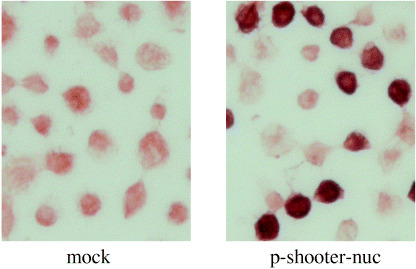
Transfected heparanase was observed in the nucleus of HL‐60 cells using nuclear expression vector; p‐shooter‐nuc.
To investigate the enzymatic activity of nuclear heparanase, we constructed a new expression vector, which encodes the active center mutant of heparanase protein: catalytic negative form of heparanase. It has also been reported in a previous article published by Hulett et al.( 18 , 19 ) As shown in Fig. 6a, the transfection of HL‐60 cells with p‐shooter vectors for nuclear heparanase expression resulted in a remarkable increase of a differentiation marker, CD13, while the same experiment with the transfection of an active center mutant (inactive) heparanase did not show any change in CD13 expression in the flow cytometric analysis in Fig. 6b, suggesting the nuclear localization of active heparanase leads the differentiation of the cells.
Figure 6.
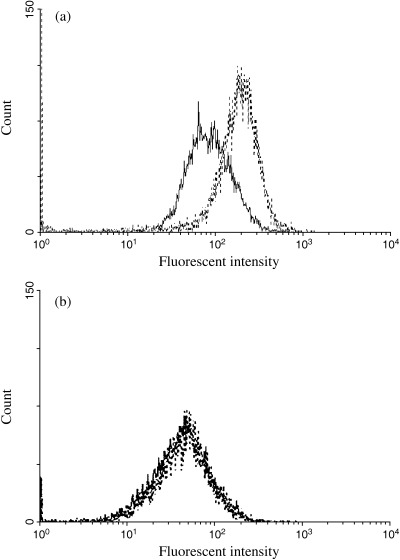
(a) Flow cytometric analysis of CD13 in nuclear heparanase‐transfected HL‐60 cells. HL‐60 cells were grown for 4 days after transfection with control vector (continuous line) or heparanase‐p‐shooter vector (dotted line). Cells were then labeled with phycoerythrin‐conjugated anti‐human CD13 mouse monoclonal antibody and analyzed in a Becton Dickinson FACSCalibur. (b) Flow cytometric analysis of CD13 in catalytic negative form of heparanase‐transfected HL‐60 cells. HL‐60 cells were grown for 4 days after transfection with control vector (continuous line) or catalytic negative heparanase‐p‐shooter vector (dotted line). Cells were then labeled with PE‐conjugated anti‐human CD13 mouse monoclonal antibody and analyzed in a Becton Dickinson FACSCalibur.
Discussion
Previous studies demonstrated that heparanase is expressed in the cytoplasm of cells at the invasion front of various cancers and that this expression was associated with poor prognosis in many types of carcinomas.( 3 , 4 ) However, in all these cancer types, heparanase expression was detected in the cytoplasm only, and thus, the cyto‐nuclear shuttling of heparanase and its role in differentiation is not known in cancer cells. Here we show that heparanase is involved in cell differentiation in addition to its known role in cell invasion and metastasis.
We demonstrated that the initial process noted after TPA‐induced differentiation of HL‐60 cells was the translocation of heparanase into the nucleus as examined morphologically. Invasion assay and flow cytometric analysis showed that HL‐60 cells differentiated upon treatment with TPA. Immunofluorescence staining using antiheparanase monoclonal antibody showed that treatment with TPA resulted in an increase of heparanase both in the cytoplasm and nucleus, suggesting the translocation of the cytosolic heparanase into the nucleus upon differentiation of HL‐60 cells.
Previous reports show that heparanase does not possess a remarkable nuclear translocation signal domain.( 20 , 21 ) Thus we proposed a new hypothesis that heat shock protein 90 (HSP 90) may work as a chaperone for nuclear translocation of heparanase. As shown in Fig. 3, each nuclear translocation of heparanase was accompanied with that of HSP90. Furthermore, TPA‐induced cellular differentiation assessed by flow cytometric analysis and Matrigel invasion assay was blocked by the inhibitors of HSP90. In our study, nuclear translocation of heparanase induced by TPA stimulation occurs in just 1 h. Such a rapid event may be explained by the hypothesis that HSP90 may work as a chaperone for nuclear translocation of heparanase. Although the precise role of HSP90 as a chaperone for nuclear translocation of heparanase is still unknown, this novel concept will be a critical event of cellular differentiation or oncogenic signal transduction.
We have already reported nuclear translocation of heparanase was mimicked by nuclear expression of heparanase using pShooterTM vector targeting heparanase specifically into the nucleus.( 22 ) In this study, we tried to achieve nuclear expression of heparanase in HL‐60 cells, resulting in cellular differentiation assessed by CD13 expression. The catalytic negative form of heparanase does not possess the ability to induce CD13. These findings suggest that the active form of nuclear heparanase may play a critical role in cellular differentiation.
Since our findings in this study were noted in a leukemia cell line, there is a need to examine the role of heparanase in the differentiation of other cell types. In fact, we have recently published our observation of differentiation by nuclear translocation of heparanase in breast( 22 ) and esophageal epithelial cells.( 23 ) We believe that this novel concept will be applied to investigate a universal oncogenic signal. Our findings emphasized the significant role of heparanase in cancer differentiation and could be potentially useful in the design of therapeutic strategies aimed at the prevention of invasion and metastasis of human cancers by translocation of heparanase from the cytoplasm to the nucleus.
We thank Yoshiko Shirakiya and Tae Yamanishi for their excellent technical assistance and our colleagues for valuable scientific advice.
References
- 1. Nakajima M, Irimura T, Nicolson GL. Heparanase and tumor metastasis. J Cell Biochem 1988; 36: 157–67. [DOI] [PubMed] [Google Scholar]
- 2. Vlodavsky I, Eldor A, Haimovitz‐Friedman A et al. Expression of heparanase by platelets and circulating cells of the immune system: possible involvement in diapedesis and extravasation. Invasion Metastasis 1992; 12: 112–27. [PubMed] [Google Scholar]
- 3. Takaoka M, Naomoto Y, Ohkawa T et al. Heparanase expression correlates with invasion and poor prognosis in gastric cancers. Lab Invest 2003; 83: 613–22. [DOI] [PubMed] [Google Scholar]
- 4. Ohkawa T, Naomoto Y, Takaoka M et al. Localization of heparanase in esophageal cancer cells: respective roles in prognosis and differentiation. Lab Invest 2004; 84: 1289–304. [DOI] [PubMed] [Google Scholar]
- 5. Gohji K, Okamoto M, Kitazawa S et al. Heparanase protein and gene expression in bladder cancer. J Urol 2001; 166: 1286–90. [PubMed] [Google Scholar]
- 6. Rohloff J, Zinke J, Schoppmeyer K et al. Heparanase expression is a prognostic indicator for postoperative survival in pancreatic adenocarcinoma. Br J Cancer 2002; 86: 1270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. El‐Assal ON, Yamanoi A, Ono T, Kohno H, Nagasue N. The significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin Cancer Res clinicopathological 2001; 7: 1299–305. [PubMed] [Google Scholar]
- 8. Kosir MA, Quinn CC, Zukowski KL, Grignon DJ, Ledbetter S. Human prostate carcinoma cells produce extracellular heparanase. J Surg Res 1997; 67: 98–105. [DOI] [PubMed] [Google Scholar]
- 9. Nakajima M, Irimura T, Di Ferrante N, Nicolson GL. Metastatic melanoma cell heparanase. Characterization of heparan sulfate degradation fragments produced by B16 melanoma endoglucuronidase. J Biol Chem 1984; 259: 2283–90. [PubMed] [Google Scholar]
- 10. Maxhimer JB, Quiros RM, Stewart R et al. Heparanase‐1 expression is associated with the metastatic potential of breast cancer. Surgery 2002; 132: 326–33. [DOI] [PubMed] [Google Scholar]
- 11. Nobuhisa T, Naomoto Y, Ohkawa T et al. Heparanase expression correlates with malignant potential in human colon cancer. J Cancer Res Clin Oncol 2005; 131: 229–37. [DOI] [PubMed] [Google Scholar]
- 12. Schubert SY, Ilan N, Shushy M, Ben‐Izhak O, Vlodavsky I, Goldshmidt O. Human heparanase nuclear localization and enzymatic activity. Laboratory Invest 2004; 84: 535–44. [DOI] [PubMed] [Google Scholar]
- 13. Hristov M, Weber C. Endothelial progenitor cells: characterization, pathophysiology, and possible clinical relevance. J Cell Mol Med 2004; 8: 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Batista EL Jr, Warbington M, Badwey JA, Van Dyke TE. Differentiation of HL‐60 cells to granulocytes involves regulation of select diacylglycerol kinases (DGKs). J Cell Biochem 2005; 94: 774–93. [DOI] [PubMed] [Google Scholar]
- 15. Zheng X, Ravatn R, Lin Y et al. Gene expression of TPA induced differentiation in HL‐60 cells by DNA microarray analysis. Nucl Acids Res 2002; 30: 4489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL‐60) by retinoic acid. Proc Natl Acad Sci USA 1980; 77: 2936–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yahalom J, Fibach E, Bar‐Tana R, Fuks Z, Vlodavsky I. Differentiating human leukemia cells express heparanase that degrades heparan sulfate in subendothelial extracellular matrix. Leuk Res 1988; 12: 711–7. [DOI] [PubMed] [Google Scholar]
- 18. Hulett MD, Freeman C, Hamdorf B, Baker RT, Harris MJ, Parish CR. Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nat Med 1999; 5: 803–9. [DOI] [PubMed] [Google Scholar]
- 19. Hulett MD, Hornby JR, Ohms SJ et al. Identification of active‐site residues of the pro‐metastatic endoglycosidase heparanase. Biochemistry 2000; 39: 15659–67. [DOI] [PubMed] [Google Scholar]
- 20. Vlodavsky I, Friedmann Y, Elkin M et al. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis. Nat Med 1999; 5: 793–802. [DOI] [PubMed] [Google Scholar]
- 21. Toyoshima M, Nakajima M. Human heparanase. Purification, characterization, cloning, and expression. J Biol Chem 1999; 274: 24153–60. [DOI] [PubMed] [Google Scholar]
- 22. Nobuhisa T, Naomoto Y, Takaoka M et al. Emergence of nuclear heparanase induces differentiation of human mammary cancer cells. Biochem Biophys Res Commun 2005; 331: 175–80. [DOI] [PubMed] [Google Scholar]
- 23. Kobayashi M, Naomoto Y, Nobuhisa T et al. Heparanase regulates esophageal keratinocyte differentiation through nuclear translocation and heparan sulfate cleavage. Differentiation 2006; 74: 235–43. [DOI] [PubMed] [Google Scholar]