

Abstract
The purpose of this paper is to determine the efficacy of combining radiation therapy with endostar, a recombined humanized endostatin, in human nasopharyngeal carcinoma and human lung adenocarcinoma xenografts. Tumor xenografts were established in the hind limb of male athymic nude mice (BALB/c‐nu) by subcutaneous transplantation. The tumor‐bearing mice were assigned into four treatment groups: sham therapy (control), endostar (20 mg/kg, once daily for 10 days), radiation therapy (6 Gray per day to 30 Gray, once a day for 1 week), and endostar plus radiation therapy (combination). The experiment was repeated and mice were killed at days 3, 6, and 10 after initiation therapy, and the tumor tissues and blood samples were collected to analyze the kinetics of antitumor, antiangiogenesis, and antivascularization responses of different therapies. In human nasopharyngeal carcinoma and human lung adenocarcinoma xenografts, endostar significantly enhanced the effects of tumor growth inhibition, endothelial cell and tumor cell apoptosis induction, and improved tumor cell hypoxia of radiation therapy. Histological analyses demonstrated that endostar plus radiation also induced a significant reduction in microvascular density, microvascular area, and vascular endothelial growth factor and matrix metalloproteinase‐2 expression compared with radiation and endostar alone respectively. We concluded that endostar significantly sensitized the function of radiation in antitumor and antiangiogenesis in human nasopharyngeal carcinoma and human lung adenocarcinoma xenografts by increasing the apoptosis of the endothelial cell and tumor cell, improving the hypoxia of the tumor cell, and changing the proangiogenic factors. These data provided a rational basis for clinical practice of this multimodality therapy. (Cancer Sci 2009)
Cancer is a major public health problem in China and many other parts of the world. Currently, one in four deaths in the USA is due to cancer.( 1 ) Cancer has been the second leading cause of death in China since 1990, and its annual mortality rate was 106/100 000 in the year 2000, and cancer studies are strongly supported by the government in China.( 2 ) Various treatments for patients with cancer have emerged in succession; however, most patients are diagnosed at an intermediate to advanced stage, and few meaningful therapeutic options are available at that point. RT is an effective adjunct to the palliative treatment of cancer. Local control failure and distant metastasis is common in cases of advanced‐stage primary tumors. Therefore, determination of the mechanisms of redioresistance in cancers could lead to advances in the treatment of cancer. If the mechanisms involved in radioresistance are known, it may be possible to use strategies to inhibit it, which has become the focus of several areas of research.
Induction of the sprouting of new vessels from existing ones (angiogenesis) is a fundamental process in a wide variety of physiological conditions, and all of these processes depend on the tightly regulated growth of blood vessels. When blood vessels grow unabated, angiogenesis becomes a pathological condition and sustains the progression of neoplastic and other diseases.( 3 ) Unlike normal blood vessels, the angiogenesis of tumors is structurally and functionally abnormal, such as poor organization, hyper‐permeability, and impaired flow.( 4 ) Considerable evidence has demonstrated that angiogenesis plays a crucial role in the growth and metastasis of tumors.( 5 , 6 ) The research of antiangiogenic therapy for cancer began in earnest in 1971 with the publication of Folkman's imaginative hypothesis that ‘tumor growth and progression depends on tumor angiogenesis’.( 7 ) Based on the hypothesis, targeting tumor angiogenesis is a very promising approach for controlling tumor growth, and several classes of angiogenesis inhibitors for cancer treatment were developed. Although preclinical studies suggested that monotherapy with antiangiogenic agents is not sufficient for the treatment of patients with advanced cancer given that tumors typically progress before they respond to therapy and microscopic residual disease persists even after prolonged therapy with these agents,( 8 ) the therapeutic potential existed when used in combination with chemotherapy or RT.( 9 )
Radiation therapy is a well‐established treatment for suitable malignancies in a wide variety of anatomical areas. In the USA, approximately 1.2 million new cancers are diagnosed annually, and over half of all patients with cancer receive RT during their course of treatment.( 10 ) For most cancers, RT can provide sustained local control, but cancer tends to reside, recur within organs away from the irradiated volume, or increase metastatic propensity, and salvage options for these patients are limited. Increasing evidence has demonstrated that hypoxic cell‐mediated radiation resistance is the main reason for tumor recurrence and metastasis,( 11 , 12 ) which provides the rationale for combining RT with other therapies.
Endostatin is a 20‐kDa internal fragment of the carboxyterminus of collagen XVIII, first produced by hemangioendothelioma,( 13 ) capable of inhibiting endothelial cell proliferation, migration or invasion, tube formation, and inducing endothelial cell apoptosis. Consequently, endostatin has been reported as one of the most potent endothelial cell inhibitors of angiogenesis and tumor growth without displaying toxic side effects and drug resistance.( 14 ) ES is a recombinant humanized endostatin purified in an Escherichia coli system with an additional nine‐amino acid sequence of soluble protein.( 15 ) In a phase III clinical study, it was demonstrated that ES significantly improved overall and progression‐free survival when used in combination with the first‐line chemotherapy regime in patients with advanced non‐small‐cell lung cancer.( 16 ) Based on systemic preclinical and clinical studies, ES was approved by the State Food and Drug Administration in China for the treatment of non‐small‐cell lung cancer in September 2005.( 17 )
Considerable experimental and clinical evidence has indicated that combined use of RT with angiogenesis inhibitors could be of benefit to overcome the limits of angiogenesis inhibitors and improve tumor response to RT.( 18 , 19 , 20 ) However, additional work is still needed to provide a rational basis for the mechanism of multimodality therapy and identify potential problems that might arise from ES plus RT so that ES will be successfully incorporated into RT in a timely and rational manner. Therefore, we carried out a study to evaluate the radioresponse enhancing effect of ES combined with RT on HNE and SPC‐A1 xenografts in the legs of nude mice.
Materials and Methods
Cell culture, animals, and reagents. HNE and SPC‐A1 cells (State Key Laboratory of Biotherapy, Sichuan University, Sichuan, China) were maintained in RPMI‐1640 medium (Hyclone, Logan, UT, USA) with 10% heat‐inactivated fetal bovine serum (F. Hoffmann‐La Roche, Basel, Switzerland) and kept in a humidified 5% CO2 atmosphere incubator at 37°C. All athymic nude mice (BALB/c nu) were male, matched for age (5–6 weeks old) and weight (24 ± 2.5 g), and were purchased from the Laboratory Animal Research Center (Sichuan University, China) and housed in the rooms of the University Laboratory Animal Research Center with specified pathogen‐free conditions. The room was maintained at 22 ± 1°C under a 12:12 L : D cycle. Food and water were provided ad libitum. All of the experiments were carried out in accordance with guidelines approved by the Laboratory Animal Care Committee of Sichuan Province (licence no. SYXK 2007‐008). ES was provided by Simcere Pharmece Group (Yantai, Shandong Province, China).
Therapeutic experiments with ES and RT. The right proximal hind legs of the mice were subcutaneously injected with 3 × 106 HNE or SPC‐A1 cells. When tumors reached a volume of 250–300 mm3, mice were randomly assigned to receive four treatments: sham treatment, ES alone, RT alone, or ES plus RT. Tumor volume including length (longest diameter of tumor), width (vertical diameter to length of tumor), and depth (depth of tumor) was evaluated by direct measurement with calipers and was calculated using the following formula (length × width × depth)/2, as in previous studies.( 21 ) RT (6 Gy per day to 30 Gy, once a day for 1 week) was delivered via precision 6‐MV photon beam from a linear accelerator (ELEKTA 1232 Medical Linear Accelerator, Stockholm, Sweden) using custom‐designed mouse jigs. These jigs immobilized the mice and specifically exposed the right hind legs (harboring tumor xenografts) for RT without exposing non‐tumor‐bearing normal tissues. The previous animal study showed that recombinant human endostatin can induce regression and prevent growth of subcutaneous experimental tumors when administered in daily doses as high as 20 mg/kg.( 13 ) So ES treatment was initiated 2 days before RT to allow for an effective plasma concentration before the delivery of RT in the combination group and was administered by caudal vein injection at the specified dose (20 mg/kg) and interval (once daily for 10 days). Sham treatment (control) mice received caudal vein injections of 0.9% saline. Each treatment group contained eight mice. Tumor volume was measured every 3 days. Regression and regrowth of tumors were measured up to 78 days after initiation of therapy in order to explore the antitumor efficacy of each group. Subsequently, the treatment experiment was repeated, and mice from each group were killed at days 3, 6, and 10 from initiation of the treatment (three mice from each group for each of the three time points) to harvest tumor tissues and blood samples for further analyses.
Histology and IHC. Tumor tissues from mice from each group were formalin‐fixed and embedded in paraffin, then sectioned and stained with hematoxylin–eosin or immunostained. Sections were used to detect the expression of VEGF, bFGF, CD31, and MMP‐2. The samples were incubated with rabbit antimouse monoclonal VEGF (1:200), rabbit antimouse monoclonal bFGF (1:200), rabbit antimouse polyclonal MMP‐2 (1:50), or rat antimouse monoclonal CD31 (1:400) antibodies (Invitrogen, Carlsbad, CA, USA).
For quantification of IHC reaction intensity, MVD was counted under a light microscope after immunostaining for CD31. Areas containing the highest number of capillaries and small venules were identified by scanning the tumor sections at high power (×100). Individual vessels were counted in 10 random fields at ×100 magnification (0.159 mm2/field). MVA was determined by summing the whole vascular areas in 10 random 0.159‐mm2 fields at ×100 magnification. The immunoreactivity of VEGF‐, bFGF‐, and MMP‐2‐positive cells from each of the differently treated tumor tissue sections was measured in 10 random fields at ×200 magnification by absorbance using a light microscope. The absorbance of positive cells was evaluated by computer‐assisted image analysis and was expressed as a ratio of tumor cell expression to normal muscle cell expression multiplied by 100.
TUNEL staining for endothelial cell and tumor cell apoptosis. The apoptosis of endothelial and tumor cells was detected using the TUNEL assay kit (F. Hoffmann‐La Roche). Endothelial cells were stained with CD31 simultaneously as reported previously.( 21 ) The number of apoptotic endothelial cells with TUNEL and CD31 doublestaining and apoptotic tumor cells with TUNEL staining were counted in nine random fields at ×400 magnification for five consecutive sections per tumor tissue.
Enzyme‐linked immunosorbent assay for proangiogenic factors. Blood samples were collected in nonanticoagulation unfertile tubes from all mice at days 3, 6, and 10 from the initiation of treatment. The samples were centrifuged at 1 000 g for 5 min at 4°C, and the separated serum samples were stored at –20°C until use. The serum VEGF and bFGF levels were measured using the monoclonal antimouse capture antibody (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. Calibration curves were established with known concentrations of the angiogenic factors, with a standard curve for each microtiter plate. The colorimetric reaction was read with a Benchmark Microplate Reader (Benchmark Electronics, Angleton, TX, USA).
Flow cytometry. Pimonidazole hydrochloride (PIM) (Millipore, Billerica, MA, USA) was dissolved in double‐distilled water and the final concentration was 20 mg/mL. Ninety minutes before death, mice were injected via the caudal vein with 100 mg/kg pimonidazole hydrochloride that labeled to hypoxic cells through metabolism within the tumor.( 22 ) Tumor single‐cell suspensions were prepared and stained as previously described:( 23 ) (i) fluorescein isothiocyanate‐conjugated antipimonidazole antibody (1:50 dilution) for 1 h at 4°C; (ii) dead cells were excluded by 7‐AAD (BD PharMingen, San Jose, CA, USA) staining. Samples were analyzed on an EPICS ELITE‐ESP flow cytometer (BD FACS Array, San Jose, CA, USA), and data were analyzed with FACS express, version 2 (De Novo Software, BD San Jose, CA, USA). Typically, 500 000 events for tumor single‐cell suspensions were acquired and the frequency of positive cells was measured.
Data analysis. All descriptive statistics, including mean ± SD, were carried out. We applied repeated measures one‐way analysis of variance for tumor growth, the rank sum test for the percentage of hypoxic cells, and one‐way analysis of variance for the other data to evaluate differences between each treatment group in our studies. Bilateral P‐values < 0.05 were considered to be significant. Data analyses were done with a statistical software package (SPSS 13.0 for Windows; SPSS, Chicago, IL, USA).
Results
Endostar plus RT increased tumor growth delay. We found that the ES plus RT treatment regimen led to a significant decrease in tumor growth compared with RT alone, ES alone, and the control group in HNE and SPC‐A1 xenografts. For the RT alone and ES alone treatment regimens, the tumor growth‐inhibiting effects were significant higher than that of the control, but significantly lower than that of ES plus RT. The inhibiting effects reached a peak at 24 days after treatment initiation in the ES plus RT group in SPC‐A1 xenografts. Moreover, two of eight HNE and SPC‐A1 xenografts were all nearly completely inhibited in the ES plus RT therapy groups at the end of the experiment (Fig. 1).
Figure 1.
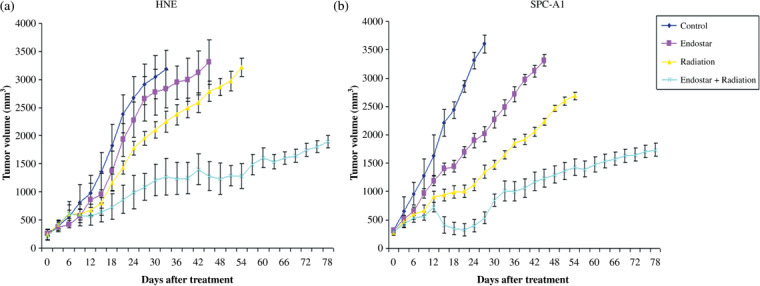
The growth curves of (a) human nasopharyngeal carcinoma (HNE) and (b) human lung adenocarcinoma (SPC‐A1) tumor xenografts. The right proximal hind legs of the nude mice (BALB/c‐nu) subcutaneously were injected with HNE and SPC‐A1 tumor cells respectively. When xenografts reached a volume of 250–300 mm3, mice were randomly assigned to receive four treatments: sham treatment, endostar alone (20 mg/kg), radiation alone (6 Gy per day in a week, to 30 Gy), and endostar plus radiation. Each group consisted of eight mice. Data are expressed as mean ± SD.
To further quantify the antitumor effects of the different treatments, we calculated the time taken for tumors to quadruple in volume. We found that this time was significantly shorter for the ES plus RT group than those for the ES alone, RT alone, and control groups in both HNE and SPC‐A1 xenografts (Table 1).
Table 1.
Effect of endostar on the radioresponse of human nasopharyngeal carcinoma (HNE) and human lung adenocarcinoma (SPC‐A1) tumor xenografts
| Tumor type | HNE tumor xenografts | SCP‐A1 tumor xenografts | ||||||
|---|---|---|---|---|---|---|---|---|
| Time required for tumor to grow four times volume (days) (mean ± SD) | Absolute growth delay (days) † | Normalized growth delay (days) ‡ | Enhancement factor § | Time required for tumor to grow four times volume (days) (mean ± SD) | Absolute growth delay (days) | Normalized growth delay (days) | Enhancement factor | |
| Sham | 10 ± 1.7 | 9 ± 1.6 | ||||||
| Endostar | 17 ± 3.1 | 7 | 15 ± 2.3 | 6 | ||||
| Radiation therapy | 27 ± 7.5 | 17 | 29 ± 5.5 | 20 | ||||
| Endostar plus radiation therapy | 44 ± 4.5 | 34 | >27 | 1.6 | 47 ± 7.6 | 38 | >32 | 1.61 |
Absolute tumor growth delay caused by radiation, endostar, or both agents is defined as the time in days that tumors required to reach four times the volume of the time of treatment initiation minus the time in days required by control tumors to grow to four times the volume.
Normalized tumor growth delay is defined as the time in days for tumors to reach four times the volume in mice treated with the combination of endostar and radiation therapy minus the time in days to reach four times the volume in mice treated with endostar only.
Enhancement factors were obtained by dividing normalized tumor growth delay in mice treated with endostar plus radiation therapy with the absolute growth delay in mice treated with radiation only.
Endostar plus RT increased endothelial and tumor cell apoptosis. We next investigated the effect of each treatment on endothelial and tumor cell apoptosis at days 3, 6, and 10 after treatment initiation. In HNE and SPC‐A1 xenografts, both ES plus RT and RT alone led to a significant increase in endothelial cell apoptosis compared with the control group at days 3, 6, and 10, with the combination therapy group being the most highly effective. A significant increase in endothelial cell apoptosis counts was also noted in ES plus RT compared with RT alone at day 10 in the two models. In addition, there was a significant increase in endothelial cell apoptosis in ES plus RT compared with ES alone at days 3, 6, and 10 in HNE xenografts and at days 6 and 10 in SPC‐A1 xenografts. However, ES alone did not show a change in endothelial cell apoptosis compared with the control group at the three time points in the two models (Fig. 2a,b).
Figure 2.
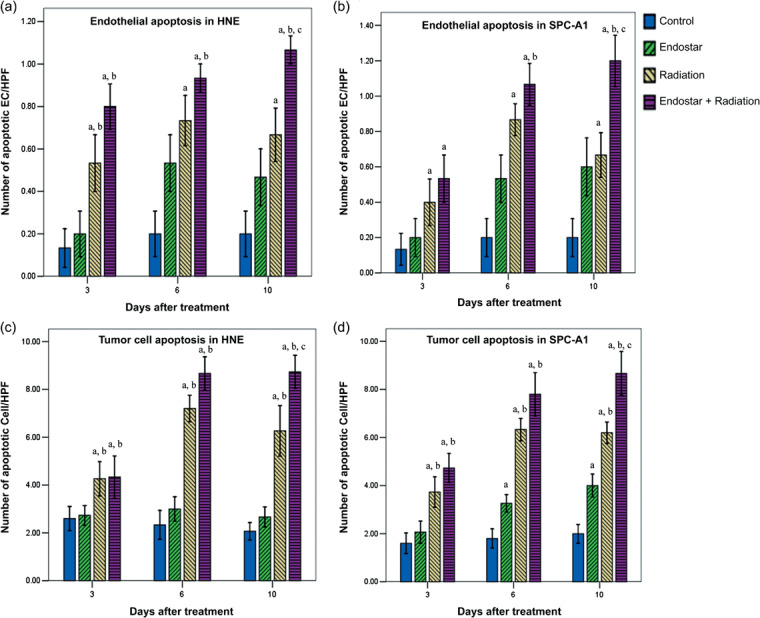
Time course analyses of endothelial cell apoptosis and tumor cell apoptosis. Endothelial cell apoptosis was determined by platelet/endothelial cell adhesion molecule (CD31)–TUNEL double staining assay. Tumor cell apoptosis was evaluated by TUNEL staining assay. The number of apoptotic endothelial cells and tumor cells was counted in nine random fields at ×400 magnification. Data were obtained from tumor tissues from mice in each treatment group and are plotted as mean ± SD. a P < 0.05 versus control; b P < 0.05 versus endostar; c P < 0.05 versus radiation.
The tumor cell apoptosis evaluation in the current study deserves further attention. In HNE and SPC‐A1 xenografts, ES plus RT and RT alone caused a significant increase in tumor cell apoptosis compared with the control group and ES alone, with the combination therapy group being the most highly effective. At day 10 a significant increase in tumor cell apoptosis was also observed in ES plus RT compared with RT alone in the two models (Fig. 2c,d).
Endostar improved tumor cell hypoxia after RT. Tumor cell hypoxia was assessed by sequential quantification of the percentage of hypoxic cells in HNE and SPC‐A1 xenografts. Figure 3(a,b) shows the representative quantification percentage of hypoxic cells using flow cytometry. For the two tumor models, ES plus RT and RT alone were significantly effective in improving hypoxia, as compared with the control at days 3, 6, and 10. ES plus RT caused markedly improved tumor cell hypoxia compared with RT alone at days 3, 6, and 10 in HNE xenografts and at days 6 and 10 in SPC‐A1 xenografts. However, ES alone conferred no significant improvement in tumor cell hypoxia compared with the control group in the two tumor models (Fig. 3c,d).
Figure 3.

Tumor cell hypoxia using flow cytometry for (a) human nasopharyngeal carcinoma (HNE) and (b) human lung adenocarcinoma (SPC‐A1) xenografts. Analysis gates were used to enumerate pimonidazole (PIM) + hypoxia tumor cells and exclude platelets, dead cells, and debris for HNE and SPC‐A1 xenografts. Representative staining for PIM + tumor cells in HNE and SPC‐A1 tumor tissue harvested from mice killed at 10 days after treatment initiation. The percentage of hypoxic tumor cells was evaluated in (c) HNE and (d) SPC‐A1 xenografts. Data are expressed as mean ± SD. a P < 0.05 versus control; b P < 0.05 versus endostar; c P < 0.05 versus radiation.
Endostar plus RT decreased MVD and MVA. To confirm the findings, MVD was assessed in HNE and SPC‐A1 xenografts by counting the number of CD31‐positive cells present in 10 random high‐power fields. The representative IHC stains of CD31 are shown in Figure 4(a). All three treatment regimens led to a significant decrease in MVD compared with the control group at days 6 and 10. For ES plus RT compared with RT alone, a significant decrease was observed only at days 6 and 10 in SPC‐A1 xenografts (Fig. 4b,c). The results of MVA were same with MVD (Fig. 4d,e).
Figure 4.
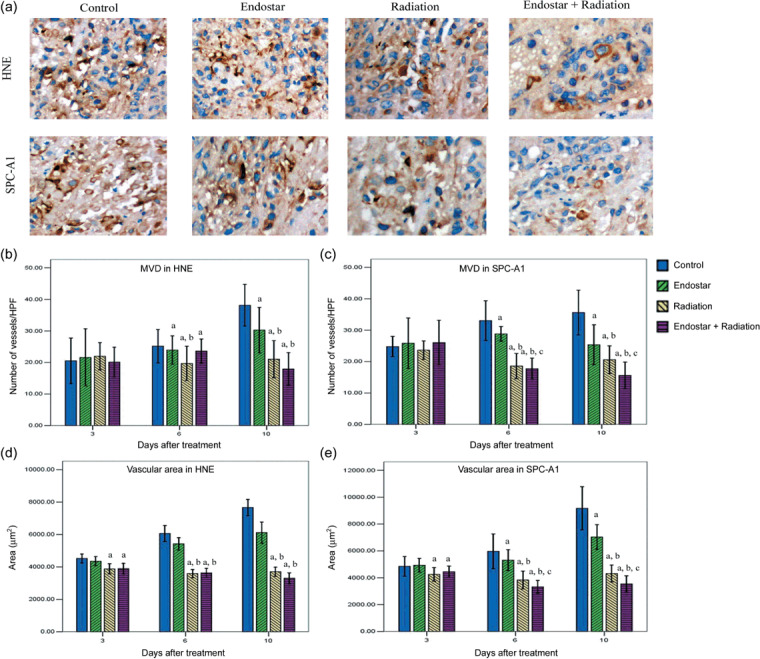
Time course analyses of tumor microvessel density (MVD) and microvascular area (MVA) by platelet/endothelial cell adhesion molecule (CD31) staining. (a) Representative CD31 staining in tumor tissue harvested from mice killed at day 10 after treatment initiation in human nasopharyngeal carcinoma (HNE) and human lung adenocarcinoma (SPC‐A1) xenografts (×400). (b,c) MVD was quantified in 10 random 0.159‐mm2 fields at ×100 magnification. (d,e) MVA was determined by summing the whole vascular areas in 10 random 0.159‐mm2 fields at ×100 magnification. Data are expressed as mean ± SD. a P < 0.05 versus control; b P < 0.05 versus endostar; c P < 0.05 versus radiation.
Endostar attenuated the upregulation of proangiogenic factors induced by RT. It has been proven that growth factors including VEGF, bFGF, and MMP‐2 play critical roles in tumor angiogenesis. To further characterize the inhibition of tumor revascularization, the expression of proangiogenic factors was studied sequentially by optical density analysis. In HNE and SPC‐A1 xenografts, monotherapy with RT on tumors showed a significant increase in VEGF, MMP‐2. and bFGF levels compared with the control after treatment. For ES plus RT treatment, the expression of VEGF and MMP‐2 in tumors was significantly inhibited compared with RT alone at day 10 (Fig. 5a,b). However, no significant difference in bFGF expression level was observed among these two groups (Fig. 5c). Representative IHC stains of VEGF and MMP‐2 are shown in Figure 6. Hence, our results indicate that ES can selectively offset proangiogenic factors in tumors that are increased in response to RT therapy.
Figure 5.

Time course analyses of the expression of proangiogenic factors by immunohistochemistry and ELISA. The expression of vascular endothelial growth factor (VEGF), matrix metalloproteinase (MMP)‐2, and basic fibroblast growth factor (bFGF) as a ratio was determined by the absorbance intensity for each of the differently treated tumor tissues, measured in 10 random fields at ×200 magnification. (a–c) Normal muscle cell expression was multiplied by 100. (d) The VEGF level in serum was evaluated by ELISA. Data are expressed as mean ± SD. a P < 0.05 versus control; b P < 0.05 versus endostar; c P < 0.05 versus radiation.
Figure 6.
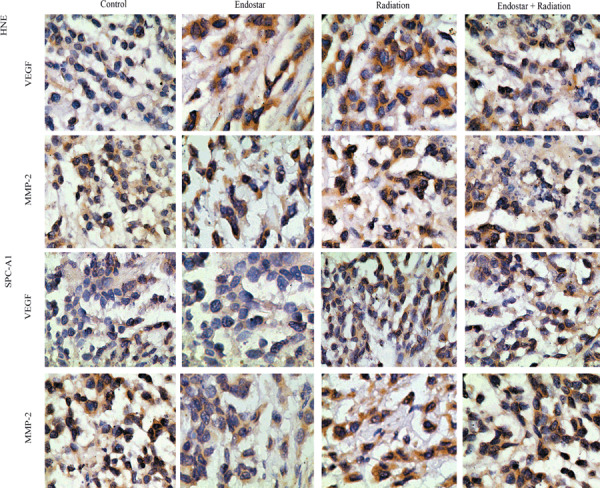
Representative staining for vascular endothelial growth factor (VEGF) and matrix metalloproteinase (MMP)‐2 in human nasopharyngeal carcinoma (HNE) and human lung adenocarcinoma (SPC‐A1) xenograft tissue harvested from mice killed at day 10 after treatment initiation.
Furthermore, the serum concentration of proangiogenic factors was detected by ELISA (Fig. 5d). In HNE and SPC‐A1 xenografts, ES plus RT and RT alone led to a significant increase in serum VEGF concentration compared with the control and ES groups at days 3, 6, and 10. Nevertheless, the serum VEGF level for ES plus RT was not significantly different to RT alone at the three time points. Unfortunately, the serum concentration of bFGF was too low to be detected by ELISA in our study.
Discussion
In our study, we found that ES significantly enhanced the radioresponse of HNE and SPC‐A1 xenografts in mice. The findings in the present study suggest that ES enhanced tumor growth inhibition, increased endothelial cell and tumor cell apoptosis, and improved tumor cell hypoxia of RT. Further histological analyses demonstrated a reduction in MVD, MVA, VEGF, and MMP‐2, but not bFGF, after ES plus RT compared with RT alone. Hence, ES plus RT significantly enhanced the antitumor, antiangiogenic, and antivascular effects seen in HNE and SPC‐A1 xenografts without systemic toxicity and induction of drug resistance. These data provide a rational basis for clinical practice of the multimodality therapy of ES combined with RT. Previous animal studies also demonstrated that the use of endostatin plus RT could enhance the antitumor efficacy compared with endostatin or RT alone. A study of endostatin plus RT in treatment of the radioresistant SQ‐20B tumor cell line demonstrated significant growth inhibition with the combination when compared with RT alone, and reduction in microvascularity was also seen.( 24 ) A recent animal study combining endostatin with high‐dose conformal radiation demonstrated that endostatin can block tumor revascularization after RT, thereby augmenting radioresponse.( 21 )
In the current study, enhancement of tumor response to ES plus RT has been explained by increases in endothelial and tumor cell apoptosis. At the endothelial and tumor cell level, there was a statistically significant increase in endothelial and tumor cell apoptosis after treatment with ES plus RT compared with treatment with RT or ES alone. Tumor cell apoptosis may occur directly by radiation‐induced DNA damage to tumor cells or indirectly by ES and radiation‐induced damage to endothelial cells. It has been speculated that the molecular mechanism by which damage to endothelial cells results in increased rates of tumor cell apoptosis is ES inhibition of tumor cell‐expressed autocrine growth factors and receptors,( 25 ) or that loss of endothelial‐derived paracrine factors needed for tumor growth contributes to tumor cell cytotoxicity via apoptotic mechanisms.( 26 ) A previous clinical trial also demonstrated that endostatin is associated with increased endothelial cell apoptosis.( 27 )
It had long been assumed that an angiogenesis inhibitor would impair the effect of ionizing radiation by inducing tumor hypoxia, until Teicher and colleagues observed that antiangiogenic agents given in combination with RT improved tumor oxygenation and antitumor efficacy.( 28 ) Indeed, there were some factors that impacted on the efficacy of radiotherapy for tumors. First, experimental and clinical studies have provided evidence for the presence of hypoxic cells in tumors.( 29 , 30 ) Second, some studies showed that primary tumors recurring after inadequate RT showed increased metastatic propensity, which was associated with an increase in the fraction of hypoxic cells and also with hypoxia‐induced upregulation of metastasis‐promoting gene products.( 31 ) In turn, increasing evidence supports the idea that hypoxia is strongly associated with a diminished therapeutic response to RT and with malignant progression in some tumor types.( 32 , 33 ) Therefore, the search is on for new drugs and remedies to enhance the therapeutic effects now that RT has become a hotspot of research. In the current study, ES significantly improved tumor cell hypoxia after 10 days of RT in HNE and SPC‐A1 xenografts. Hence, the pivotal role of tumor hypoxia in determining radiation response serves to emphasize the clinical potential of combing ES with RT.
As early as 1972, Brem and colleagues proposed a grading system for human brain tumors that relied on the analysis of endothelial cell characteristics in conjunction with vessel density measurements.( 34 ) Consequently, MVD counts, as a measure of angiogenesis, have been investigated widely as a cancer prognostic factor.( 35 ) Despite its importance as a prognostic indicator in untreated tumors, it was controversial that the efficacy of antiangiogenic therapy was evaluated by determining MVD.( 36 ) To study the feasibility of using MVD as a surrogate marker for response to combined RT and antiangiogenic therapy, we measured MVD in tumor tissues after each treatment. Our data suggest there was a positive correlation between MVD and the tumor response to ES plus RT.
Furthermore, we evaluated changes in the expression of proangiogenic molecules including VEGF, bFGF, and MMP‐2 in tumor tissue and blood serum. In our studies, the levels of VEGF and MMP‐2, but not bFGF, were higher with RT alone than with ES plus RT in tumor tissue. In our models, bFGF levels were not significantly impacted by RT, suggesting that RT did not induce sustained overexpression of bFGF. However, VEGF and MMP‐2 expression increased substantially after RT. The addition of ES to RT may offset VEGF and MMP‐2 activation and block the RT‐associated overexpression of them.
We know that angiogenesis is driven by tumors and tumor cells secrete a variety of angiogenesis‐promoting factors that induce angiogenesis. Therefore, in theory, antiangiogenic therapy requires joint application of a variety of angiogenesis inhibitors or angiogenesis inhibitors in combination with the cytotoxic effects of chemotherapy or RT. The former can overcome the potential drug resistance of application of a single angiogenesis inhibitor, the latter can reduce the tumor burden, thereby reducing the secretion of angiogenesis factor promoters, in turn increasing the sensitivity of angiogenesis inhibitors. A number of preclinical studies have shown that ES in combination with chemotherapy, RT, or other angiogenesis inhibitors reaches a synergistic effect.( 24 , 37 , 38 ) In clinical studies, the use of angiogenesis inhibitors plus chemotherapy or RT is a convenient and effective method that provides more treatment benefits and will gradually receive recognition. For example, the addition of bevacizumab to fluorouracil‐based combination chemotherapy increases 4.7 months of median survival time and 4.4 months of a median time to progression among patients with metastatic colorectal cancer.( 9 ) Hence, such combination treatment modalities break through the bottleneck of angiogenesis inhibitor monotherapy, which reveals that outlook its antitumor potential will have significant clinical value. However, it should be noted that our study has some limitations. For one thing, a previous study showed that a U‐shaped curve in the efficacy of endostatin is optimal between very low and very high doses depending on the tumor type.( 39 ) Our study also demonstrated that a U‐shaped efficacy curve occurred with ES plus RT in SPC‐A1 xenografts other than in HNE xenografts. Therefore, actively looking for the optimal dose, intervention time, and interval between angiogenesis inhibitor and RT for the various tumor types will help increase the efficacy of the combination modality. Also, the ability to identify biological markers that can predict targeted therapy sensitivity may become clinically relevant, but although researchers have found markers such as VEGF and MVD, they have not found the ideal target.( 40 , 41 , 42 ) Recent studies revealed that endothelial progenitor cells or E‐selectin may be useful biomarkers for ES treatment of patients with cancer.( 43 , 44 ) Nevertheless, ES therapy involves many genes and signaling pathways and needs to further be explored.( 45 ) However, our data were consistent with recent studies( 21 , 24 ) and suggest that VEGF, MMP‐2, and MVD can directly elevate the efficacy of ES when used as biomarkers.
In conclusion, findings from the present study suggest that ES offers the potential for improved RT efficacy by increasing the apoptosis of endothelial and tumor cells, improving the hypoxia of tumor cells, and changing the proangiogenic factors. It is clear from the present study and other previous work in the field that ES in conjunction with RT will provide new hope for increasing the therapeutic efficacy of cancer treatment. However, we would like to carry out studies with other cell lines to determine whether ES can enhance the radioresponse of tumors before ES plus RT can be recommended routinely.
Disclosure Statement
No benefits in any form have been or will be received from a commercial party related directly or indirectly to the subject of this article.
Abbreviations
| bFGF | basic fibroblast growth factor |
| CD31 | platelet/endothelial cell adhesion molecule |
| ELISA | enzyme‐linked immunosorbent assay |
| ES | endostar |
| HNE | human nasopharyngeal carcinoma |
| IHC | immunohistochemistry |
| MMP | matrix metalloproteinase |
| MVA | microvascular area |
| MVD | microvessel density |
| RT | radiation therapy |
| SPC‐A1 | human lung adenocarcinoma |
| VEGF | vascular endothelial growth factor |
Supporting information
Supporting information 1. The bodyweights of mice in each group.
Supporting information 2. A few photos for macroscopic view of HNE xenografts.
Supporting information 3. The histopathology on HNE and SPC‐A1 xenografts (×200).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We thank Professor Jing‐Bo Wu at the Department of Oncology, The Affiliated Hospital of Luzhou Medical College, Luzhou, China, for valuable scientific suggestions and his language editing of this manuscript. We also thank Weibo Feng, MD, at the institute of laboratory medicine, Guangdong Medical College, Dongguan, China, for his assistance and the Simcere Pharmece Group (Yantai, Shandong Province, China) for providing endostar. We also thank the anonymous referee for his/her very helpful comments, which remarkably improved the quality of this paper.
References
- 1. Jemal A, Siegel R, Ward E, et al . Cancer statistics, 2008. CA Cancer J Clin 2008; 58: 71–96. [DOI] [PubMed] [Google Scholar]
- 2. Li L, Lu F, Zhang S. Analyses of variation trend and short‐term detection of Chinese malignant tumor mortality during twenty years. Zhonghua Zhong Liu Za Zhi 1997; 19: 3–9. [PubMed] [Google Scholar]
- 3. Folkman J. Clinical applications of research on angiogenesis. N Engl J Med 1995; 333: 1757–17. [DOI] [PubMed] [Google Scholar]
- 4. Jain RK. Normalizing tumor vasculature with anti‐angiogenic therapy: a new paradigm for combination therapy. Nat Med 2001; 7: 987–9. [DOI] [PubMed] [Google Scholar]
- 5. Sharma RA, Harris AL, Dalgleish AG, Steward WP, O’Byrne KJ. Angiogenesis as a biomarker and target in cancer chemoprevention. Lancet Oncol 2001; 2: 726–32. [DOI] [PubMed] [Google Scholar]
- 6. Carmeliet P, Jain RK. Angiogenesis in cancer and other disease. Nature 2000; 407: 249–57. [DOI] [PubMed] [Google Scholar]
- 7. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971; 285: 1182–6. [DOI] [PubMed] [Google Scholar]
- 8. Jain RK, Duda DG, Clark JW, Leoffler JS. Lessons from phase III clinical trials on anti‐VEGF therapy for cancer. Nat Clin Pract Oncol 2006; 3: 24–40. [DOI] [PubMed] [Google Scholar]
- 9. Hurwitz H, Fehrenbacher L, Novotny W, et al . Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–42. [DOI] [PubMed] [Google Scholar]
- 10. Jemal A, Thomas A, Murray T, Thun M. Cancer statistics, 2002. CA Cancer J Clin 2002; 52: 23–47. [DOI] [PubMed] [Google Scholar]
- 11. Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR. Tumor oxygenation predicts for the likelihood of distant metastasis in human soft tissue sarcoma. Cancer Res 1996; 56: 941–3. [PubMed] [Google Scholar]
- 12. Graeber TG, Osmanian C, Jacks T, et al . Hypoxia‐mediated selection of cells with diminished apoptotic potential in solid tumors. Nature 1996; 379: 88–91. [DOI] [PubMed] [Google Scholar]
- 13. O’Reilly MS, Boehm T, Shing Y, et al . Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997; 88: 277–85. [DOI] [PubMed] [Google Scholar]
- 14. Boehm T, Folkman J, Browder T, O’Reilly MS. Antiangiogenic therapy of experimental cancer dose not induce acquired drug resistance. Nature 1997; 390: 404–7. [DOI] [PubMed] [Google Scholar]
- 15. Han Q, Fu Y, Zhou H, He Y, Luo Y. Contributions of Zn (II)‐binding to the structural stability of endostatin. FEBS Lett 2007; 581: 3027–32. [DOI] [PubMed] [Google Scholar]
- 16. Sun Y, Wang J, Liu Y, et al . Results of phase III trial of rh‐endostatin (YH‐16) in advanced non‐small cell lung cancer (NSCLC) patients. Annual ASCO Meeting 2005. May in FL, USA (Abstract 7134). [Google Scholar]
- 17. State Food and Drug Administration . 2005. Available from http://app1.sfda.gov.cn/datasearch/face3/base.jsp [Accessed 12 September 2008].
- 18. Wachsberger P, Burd R, Dicker AP. Tumor response to ionizing radiation combined with antiangiogenesis or vascular targeting agents: exploring mechanisms of interaction. Clin Cancer Res 2003; 9: 1957–71. [PubMed] [Google Scholar]
- 19. Ma BB, Bristow RG, Kim J, Siu LL. Combined‐modality treatment of solid tumors using radiotherapy and molecular targeted agents. J Clin Oncol 2003; 21: 2760–76. [DOI] [PubMed] [Google Scholar]
- 20. O’Reilly MS. Radiation combined with antiangiogenic and antivascular agents. Semin Radiat Oncol 2006; 16: 45–50. [DOI] [PubMed] [Google Scholar]
- 21. Itasaka S, Komaki R, Herbst RS, et al . Endostatin improves radioresponse and blocks tumor revascularization after radiation therapy for A431 xenografts in mice. Int J Radiat Oncol Biol Phys 2007; 67: 870–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Olive PL, Durand RE, Raleigh JA, et al . Comparison between the comet assay and pimonidazole binding for measuring tumour hypoxia. Br J Cancer 2000; 83: 1525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leong KG, Hu X, Li L, et al . Activated notch4 inhibits angiogenesis: role of beta 1‐integrin activation. Mol Cell Bio 2002; 22: 2830–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanna NN, Seetharam S, Mauceri HJ, et al . Antitumor interaction of short‐course endostatin and ionizing radiation. Cancer J 2000; 6: 287–93. [PubMed] [Google Scholar]
- 25. Masood R, Cai J, Zheng T, Smith DL, Hinton DR, Gill PS. Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor‐positive human tumors. Blood 2001; 98: 1904–13. [DOI] [PubMed] [Google Scholar]
- 26. O’Reilly MS, Holmgren L, Chen C, Folkman J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat Med 1996; 2: 689–92. [DOI] [PubMed] [Google Scholar]
- 27. Herbst RS, Mullani NA, Davis DW, et al . Development of biologic markers of response and assessment of antiangiogenic activity in a clinical trial of human recombinant endostatin. J Clin Oncol 2002; 20: 3804–14. [DOI] [PubMed] [Google Scholar]
- 28. Teicher BA, Dupuis N, Kusomoto T, et al . Antiangiogenic agents can increase tumor oxygenation and response to radiation therapy. Rad Oncol Invest 1995; 2: 269–76. [Google Scholar]
- 29. Rockwell S, Moulder JE. Hypoxic fractions of human tumors xenografted into mice: a review. Int J Radiat Oncol Biol Phys 1990; 19: 197–202. [DOI] [PubMed] [Google Scholar]
- 30. Evans SM, Hahn SM, Magarelli DP, Koch CJ. Hypoxic heterogeneity in human tumors: EF5 binding, vasculature, necrosis, and proliferation. Am J Clin Oncol 2001; 24: 467–72. [DOI] [PubMed] [Google Scholar]
- 31. Rofstad EK, Mathiesen B, Kindem K, et al . Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res 2006; 66: 6699–707. [DOI] [PubMed] [Google Scholar]
- 32. Hockel M, Schlenger K, Aral B, et al . Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res 1996; 56: 4509–15. [PubMed] [Google Scholar]
- 33. Brizel DM, Dodge RK, Clough RW, et al . Oxygenation of head and neck cancer: changes during radiotherapy and impact on treatment outcome. Radiother Oncol 1999; 53: 113–17. [DOI] [PubMed] [Google Scholar]
- 34. Brem S, Cotran R, Folkman J. Tumor angiogenesis: a quantitative method for histologic grading. J Natl Cancer Inst 1972; 48: 347–56. [PubMed] [Google Scholar]
- 35. Hlatky L, Hahnfeldt P, Folkman J. Clinical application of antiangiogeneic therapy: microvessel density, what it does and doesn't tell us. J Natl Cancer Inst 2002; 94: 883–93. [DOI] [PubMed] [Google Scholar]
- 36. Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2003; 2: 727–39. [DOI] [PubMed] [Google Scholar]
- 37. Plum SM, Hanson AD, Volker KM, et al . Synergistic activity of recombinant human endostatin in combination with adriamycin: analysis of in vitro activity on endothelial cells and in vivo tumor progression in an orthotopic murine mammary carcinoma model. Clin Cancer Res 2003; 9: 4619–26. [PubMed] [Google Scholar]
- 38. Sun L, Ye HY, Zhang YH, Guan YS, Wu H. Epidermal growth factor receptor antibody plus recombinant human endostatin in treatment of hepatic metastases after remnant gastric cancer resection. World J Gastroenterol 2007; 13: 6115–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Celik I, Surucu O, Dietz C, et al . Therapeutic efficacy of endostatin exhibits a biphasic dose–response curve. Cancer Res 2005; 65: 11 044–50. [DOI] [PubMed] [Google Scholar]
- 40. Thomas JP, Arzoomanian RZ, Alberti D, et al . Phase I pharmacokinetic and pharmacodynamic study of recombinant human endostatin in patients with advanced solid tumors. J Clin Oncol 2003; 21: 223–31. [DOI] [PubMed] [Google Scholar]
- 41. Herbst RS, Mullani N, Davis DW, et al . Development of biologic markers of response and assessment of antiangiogenic activity in a clinical trial of human recombinant endostatin. J Clin Oncol 2002; 20: 3804–14. [DOI] [PubMed] [Google Scholar]
- 42. Davis DW, Shen Y, Mullani NA, et al . Quantitative analysis of biomarkers defines an optimal biological dose for recombinant human endostatin in primary human tumors. Clin Cancer Res 2004; 10: 33–42. [DOI] [PubMed] [Google Scholar]
- 43. Capillo M, Mancuso P, Gobbi A, et al . Continuous infusion of endostatin inhibits differentiation, mobilization, and clonogenic potential of endothelial cell progenitors. Clin Cancer Res 2003; 9: 377–82. [PubMed] [Google Scholar]
- 44. Yu Y, Moulton KS, Khan MK, et al . E‐selectin is required for the antiangiogenic activity of endostatin. Proc Natl Acad Sci USA 2004; 101: 8005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Abdollahi A, Hahnfeldt P, Maercker C, et al . Endostatin's antiangiogenic signaling network. Mol Cell 2004; 13: 649–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information 1. The bodyweights of mice in each group.
Supporting information 2. A few photos for macroscopic view of HNE xenografts.
Supporting information 3. The histopathology on HNE and SPC‐A1 xenografts (×200).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item