

Abstract
During the course of screening for the agents that activate transforming growth factor‐β (TGF‐β) signaling cascade, onnamide A and theopederin B, heterocyclic compounds related to mycalamides from a marine sponge, were found to induce plasminogen activator inhibitor‐1 (PAI‐1) promoter‐derived gene expression in Mv1Lu cells. The maximum induction of the PAI‐1 promoter by onnamide A and theopederin B was observed at the concentrations of 50 nM and 2 nM, respectively. These compounds strongly inhibited protein synthesis at the 50% inhibitory concentrations of 30 nM for onnamide A and 1.9 nM for theopederin B, and induced activation of p38 mitogen‐activated protein kinase and c‐Jun NH2‐terminal protein kinase (JNK). Anisomycin, a well‐known inducer of ribotoxic stress that inhibits protein synthesis and activates both p38 kinase and JNK, also activated PAI‐1 gene expression. Furthermore, PAI‐1 expression by onnamide A, theopederin B, and anisomycin was inhibited by SB202190 and SP600125, specific inhibitors of stress‐activated protein kinases. Onnamide A and theopederin B were cytotoxic to a variety of cell lines and strongly induced apoptosis in HeLa cells within 24 h, which was accompanied by the sustained activation of p38 kinase and JNK. These results suggest that onnamide A and theopedirin B trigger a ribotoxic stress‐like response, thereby inducing p38 kinase and JNK activation, the kinase‐dependent PAI‐1 gene expression, and apoptosis. (Cancer Sci 2005; 96: 357–364)
Transforming growth factor‐β (TGF‐β) regulates the expression of genes that play roles in a large variety of biological phenomena ranging from tissue remodeling to tumor initiation and progression.( 1 , 2 ) Since it has been established that TGF‐β signaling cascade has a tumor suppression function that is lost in many tumor‐derived cell lines, the agents that stimulate the downstream signaling pathway of TGF‐β bear great potential as new drugs for treatment of cancer because of the ability to modulate TGF‐β‐mediated transcription and to induce apoptosis. To obtain potential activators for the TGF‐β signaling pathway, we screened for agents that induce expression of a TGF‐β target gene by using Mv1Lu mink lung epithelial cells stably transfected with a plasminogen activator inhibitor‐1 (PAI‐1) promoter‐luciferase construct.( 3 ) The secondary screening was performed by testing the effect of the agents on the subcellular localization of endogenous Smad2/3, mediators of TGF‐β signaling, which are phosphorylated in response to TGF‐β stimulation, followed by translocation to the nucleus. The latter assay provides the information as to whether the compound that induces PAI‐1 gene expression acts as a stimulator of the relatively upstream pathway including the Smad phosphorylation or the downstream pathway involving transcription of TGF‐β target genes. In the course of our screening, we identified onnamide A and theopedirin B, heterocyclic compounds related to mycalamides from a marine sponge, as active substances that induce the PAI‐1 gene expression without causing nuclear translocation of Smad2/3. This family of compounds share a common core‐chemical structure, and has been reported to have potent cytotoxicity and antitumor activity.( 4 , 5 , 6 , 7 ) However, the molecular mechanism underlying their cytotoxic effect remains elusive. We show that onnamide A and theopedirin B strongly inhibit protein synthesis and activate p38 kinase and c‐Jun N‐terminal kinase (JNK), stress‐activated protein kinases. Our data using specific kinase inhibitors suggest that p38 kinase is involved in the activation of PAI‐1 gene expression by these compounds.
Materials and Methods
Cells, culture conditions, and reagents. A transfected mink lung epithelial cell (TMLEC) line (Mv1Lu) transformed with the firefly luciferase reporter gene at the downstream of the plasminogen activator inhibitor‐1 (PAI‐1) promoter, was kindly provided by Dr M. Abe. To obtain a control cell line, the cytomegalovirus (CMV) enhancer/promoter‐driven luciferase reporter vector, pBC‐Luc, was introduced into Mv1Lu cells and the stably transformed cell line, MCL‐5, was selected with 400 µg/mL geneticin (G418, Sigma‐Aldrich, St. Louis, MO). Clone 6, a rat fibroblast cell line transformed with p53Val135A and activated ras, was kindly provided by Dr S. Khochbin.
TMLEC, MCL‐5, clone 6, CHO C400, and NIH3T3 cells were cultured in Dulbecco's modified Eagles Medium (DMEM) containing 10% heat‐inactivated fetal bovine serum. For luciferase assay, medium without phenol red was used. Cells were grown at 37°C in a 5% CO2 atmosphere and temperature was shifted to 32°C when necessary.
SB202190 and SP600125 were purchased from CALBIOCHEM (San Diego, CA). Anisomycin was purchased from Sigma‐Aldrich. An anti‐Smad2/3 monoclonal antibody was purchased from BD Biosciences (Boston, MA). An anti‐p53 monoclonal antibody (Ab‐1) was purchased from Oncogene Research Products (Boston, MA). Antibodies specific for p38 kinase, phospho‐p38 kinase, JNK, phospho‐JNK, ATF‐2, phospho‐ATF‐2, c‐Jun, and phospho‐c‐Jun lI were purchased from Cell Signaling Technology (Beverly, MA). Alexa Fluor 488‐conjugated antibodies (from goat) against mouse and rabbit Ig were purchased from Molecular Probes (Eugene, OR). FITC‐conjugated or horseradish peroxidase‐linked secondary antibodies were purchased from Amersham Phamacia Biotech UK (Buckinghamshire, UK).
Assay for luciferase. TMLEC or MCL‐5 cells were plated 8.5 × 105/mL in 96‐well culture plates (Packard BioScience, Boston, MA) in medium without phenol red. After 6 h incubation for attachment, cells were incubated with screening samples for 16–18 h, and then luciferase activity was measured using the LucLite substrate (Packard BioScience) and a luminometer. All of the readouts were compared with control, and a number reflecting the relative increase in luciferase activity was calculated for each chemical.
Indirect immunofluorescence microscopy. TMLEC or NIH3T3 cells were grown to 70% confluence in 6‐well plates (Falcon Labware) on glass coverslips in complete medium and were then serum‐starved (0.2% fetal bovine serum) overnight prior to incubation with drugs for 1 h. Clone 6 cells grown on glass coverslips to 70% confluence were incubated with or without drugs for 12 h, or incubated at 32°C for 12 h. They were then rinsed with PBS and fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. The fixed cells were washed with PBS and permeabilized with 0.1% Triton X‐100 in PBS for 10 min at room temperature. For minimizing non‐specific binding, the samples were preincubated with 2% fetal bovine serum in buffer containing 150 mM NaCl, 10 mM Tris‐HCl (pH 7.5), 0.1% (v/v) Tween 20 for 1 h at room temperature, and then treated with the primary antibody against Smad2/3 (1:50), phospho‐ATF‐2 (1:50), phospho‐c‐Jun II (1:50), or p53 (1:50) for 1 h, followed by incubation with the secondary antibody Alexa Fluor 488 (1:600, antimouse IgG), Alexa Fluor 488 (1:600, antirabbit IgG) or FITC (1:100, antimouse IgG) for 45 min. The coverslips were washed, mounted with 1 µg/mL DAPI in Vectashield (Vector Laboratories, Burlingame, CA), and observed under a fluorescent microscope (Axiophoto 2, Carl Zeiss).
Analysis of protein synthesis in vivo. The in vivo effects of the active compounds on protein synthesis were determined by measuring the incorporation of [35S]methionine into cellular proteins. The cells were grown to confluence in 35‐mm culture dishes, washed with PBS, transferred to 10% DMEM with [35S]methionine (10 µCi/mL) but without methionine and cystine, and cultured with various concentrations of drugs. After incubation the cells were quickly washed twice with ice‐cold PBS, and lyzed with ice‐cold lysis buffer containing 50 mM HEPES (pH 8.0), 150 mM NaCl, 1% Triton X‐100, 10% glycerol, 0.5 mM DTT, 0.2% Sarkosyl, 1 mM NaF, 1 mM sodium vanadate, 0.5 mM PMSF, and 0.5 µg/mL leupeptin, 1 µg/mL aprotinin, 1 µg/mL pepstatin. Trichloroacetic acid (10%) was added to the lysates, and the acid insolubule fractions were taken for determination of radioactivity.
Western blot analysis. 5 × 105/mL NIH3T3 cells or 2.5 × 105/mL HeLa cells treated with each compound were lyzed with ice‐cold lysis buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 1 mM DTT, 0.1% Tween 20, 10% (v/v) glycerol, 0.1 mM PMSF, 10 mM β‐glycerophosphate, 1 mM NaF, 0.1 mM sodium vanadate, and 10 mM leupeptin. The lysates were centrifuged at 17 000 × g for 15 min at 4°C, and protein concentrations of the supernatants were determined by the Bradford method. An equal amount of protein was separated by sodium dodecyl sulfate‐10% polyacrylamide gel electrophoresis (SDS‐PAGE), followed by western blotting, and visualized with an ECL western blotting detection kit (Amersham Pharmacia Biotech).
In vitro kinase assay. NIH3T3 cells grown to 80% confluent in 10 cm‐diameter dishes were incubated with or without drugs. The ability to phosphorylate ATF‐2 or c‐Jun in the cells treated with the compounds was measured in vitro by using a p38 kinase or SAPK/JNK assay kit (Cell Signaling Technology) according to the manufacturer's instruction.
In vitro proliferation assay. Cells were seeded at 5000 per well in 96‐well flat‐bottomed plates and incubated for 24 h. After treatment with drugs for an additional 3 days, cell viability was assessed by the XTT assay (Cell Proliferation Kit II, Roche) according to the manufacturer's instruction.
Measurement of apoptotic cells. Cells (2.0 × 104/well) were seeded in 96‐well flat‐bottomed chimney‐well plate (Greiner Bio‐One) and incubated for 24 h. After treatment with drugs for an additional 24 h, cells were fixed with 3.7% paraformaldehyde in PBS for 10 min. Following washing once with PBS, cells were treated with Hoechst‐33342 in PBS (0.5 µg/mL). Hoechst‐stained DNA was observed using a conventional fluorescence microscopy (Olympus) with a DAPI filter.
Results
Activation of PAI‐1 promoter‐dependent transcription by onnamide A and theopederin B. Random screening of more than 20 000 samples containing microbial broths and marine sponge metabolites identified onnamide A and theopederin B as active substances that induce the activation of the PAI‐1 promoter in MV1Lu cells (Fig. 1). These compounds, members of the pederin family isolated from a marine sponge, share the common heterocyclic core structure with different side chains. As shown in Fig. 1(B), onnamide A and theopederin B greatly stimulate the PAI‐1 gene expression in concentration ranges from 10 to 100 and 1 to 10 nM, respectively, while they did not affect the CMV promoter‐directed gene expression in MV1Lu cells stably transfected with the CMV promoter‐luciferase construct. As a secondary test, we examined whether onnamide A and theopederin B causes nuclear translocation of endogenous Smad2/3, the mediators of TGF‐β signaling. As a control experiment, we also examine the effect of these compounds on the subcellular localization of temperature‐sensitive p53 (tsp53), of which nuclear translocation is induced by incubating the cells at 32°C or treating with protein synthesis inhibitors at the non‐permissive temperature 37°C.( 8 , 9 ) As shown in Fig. 2, no obvious accumulation of Smad2/3 in the nuclei was observed in cells treated with 50 ng/mL onnamide A or 5 ng/mL theopederin B, although 10 ng/mL TGF‐β effectively induced the nuclear accumulation of Smad2/3. Therefore, these compounds may activate PAI‐1 gene expression by stimulating the pathway downstream of the Smad2/3 in the TGF‐β signaling cascade or by a mechanism independent of TGF‐β. Surprisingly, both compounds caused a dramatic change in the subcellular localization of tsp53 from the cytoplasm to the nucleus at 37°C (Fig. 2).
Figure 1.

Activation of PAI‐1 promoter by onnamide A and theopederin B. (A), Chemical structures onnamide A and theopederin B and related compounds, pederin and mycalamide A. (B), Effect of onnamide A and theopederin B on PAI‐1 and CMV promoters. TMELC cells for PAI‐1 or MCL‐5 for the CMV promoter were incubated with various concentrations of drugs for 16 h, and then luciferase activity in the cells was measured.
Figure 2.

Effect of onnamide A and theopederin B on nuclear translocation of Smad2/3. TMELC cells (A) were treated with 10 ng/mL TGF‐β (B), 50 ng/mL of onnamide A (C), and 5 ng/mL of theopederin B (D) for 1 h. As a control, rat fibroblast clone 6 cells transformed with temperature sensitive p53 (p53Val135) were cultured for 12 h at 37°C (E), 32°C (F), and with 50 ng/mL of onnamide A (G) or 5 ng/mL of theopederin B (H) at 37°C. Localization of Smad2/3 and p53 was determined by immunofluorescent microscopy. (I), effects of onnamide A, theopederin B, and anisomycin on protein synthesis in cells.
Inhibition of protein synthesis by onnamide A and theopederin B. Tsp53 was relocalized to the nucleus, even at the non‐permissive temperature (37°C) by treatment with protein synthesis inhibitors( 8 ) and mycalamides, structurally related marine natural products, were reported to inhibit protein synthesis.( 4 ) Therefore, it seemed possible that onnamide A and theopederin B also inhibit protein synthesis. To test this possibility, we analyzed the effect of these compounds on the incorporation of labeled methionine into the cells. As shown in Fig. 2(I), low concentrations of onnamide A and theopederin B inhibited the methionine incorporation as did anisomycin, a peptidyltransferase inhibitor. Dose‐response curves indicate that the IC50 values of onnamide A and theopederin B were 30 nM and 1.9 nM, respectively, while that of anisomycin was 200 nM (Fig. 2I).
Activation of JNK and p38 by onnamide A and theopederin B. In addition to Smads, stress‐activated protein kinase pathways including p38( 10 , 11 , 12 , 13 , 14 ) and JNK( 15 , 16 ) have been shown to contribute to the TGF‐β–induced gene expression. We therefore tested whether onnamide A and theopederin B can induce activation of p38 and JNK by immunoblotting using antibodies specific for phosphorylated forms of p38 and JNK. As shown in Fig. 3(A), both compounds increased the amount of phosphorylated p38 and JNK in dose‐dependent manners. Anisomycin has also been reported to activate p38 kinase and JNK.( 17 , 18 ) Anisomycin treatment enhanced the phosphorylation of the kinases to an extent similar to those of onnamide A and theopederin B (Fig. 3B). However, some other protein synthesis inhibitors such as puromycin and cycloheximide failed to increase the phosphorylation of the proteins (Fig. 3B). ATF‐2 and c‐Jun are well‐characterized substrates for p38 kinase and JNK, respectively. We next determined the kinase activity in the cells treated with onnamide A or theopederin B by in vitro assay using ATF‐2 and c‐Jun as the substrates. Western blotting using the antibodies specific for phosphorylated ATF‐2 and phosphorylated c‐Jun demonstrated that both onnamide A and theopederin B enhanced the cellular activity to phosphorylate ATF‐2 and c‐Jun (Fig. 4). Immunofluorescent microscopy showed that the activated, phosphorylated ATF‐2 and c‐Jun were accumulated in the nuclei in the cells treated with not only anisomycin but also onnamide A or theopederin B (not shown). These results clearly show that onnamide A and theopederin B activate the stress‐activated protein kinase pathways.
Figure 3.

Phosphorylation of p38 kinase and JNK by onnamide A and theopederin B. The amounts of phosphorylated p38 and phosphorylated JNK in cells treated with onnamide A or theopederin B (A) and those in cells treated with other protein synthesis inhibitors (B) were determined by western blot analysis. The total cellular levels of p38 and JNK in cells treated with onnamide A or theopederin B (A) and those in cells treated with other protein synthesis inhibitors (B) were also determined.
Figure 4.
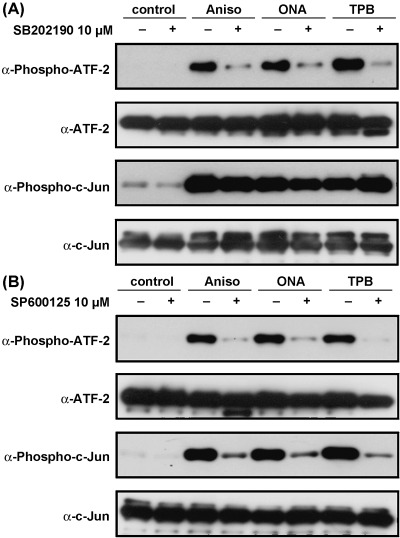
Effect of stress‐activated protein kinase inhibitors on drug‐induced phosphorylation of ATF‐2 and c‐Jun Effects of SB202190 (A) and SP600125 (B) on the phosphorylation of ATF‐2 and c‐Jun. After in vitro kinase reaction, the amounts of phosphorylated or unphosphorylated proteins were determined by western blotting.
Effect of kinase inhibitors on PAI‐1 promoter activation. To test whether the PAI‐1 promoter activation by onnamide A or theopederin B requires the kinase‐mediated signal transduction pathways, we analyzed the effect of the inhibitors of p38 kinase and JNK on the PAI‐1 gene expression. Pretreatment of SB202190, a specific inhibitor for p38 kinase, greatly reduced the drug‐induced phosphorylation of ATF‐2 but not c‐Jun (Fig. 4A). On the other hand, SP600125, an inhibitor of JNK, blocked both p38 and JNK and inhibited the ATF‐2 phosphorylation by p38 as well as c‐Jun phosphorylation by JNK in our experiments (Fig. 4B). Either SB202190 (Fig. 5A) or SP600125 (Fig. 5B) reduced the PAI‐1 promoter activity induced by anisomycin, onnamide A, or theopederin B in a dose‐dependent manner. These results strongly suggest that these compounds induce the PAI‐1 gene expression via the stress‐activated protein kinase pathways. In particular, p38 kinase may play a major role in activating the PAI‐1 gene promoter, since SB202190 sufficiently reduced the drug‐induced gene expression.
Figure 5.
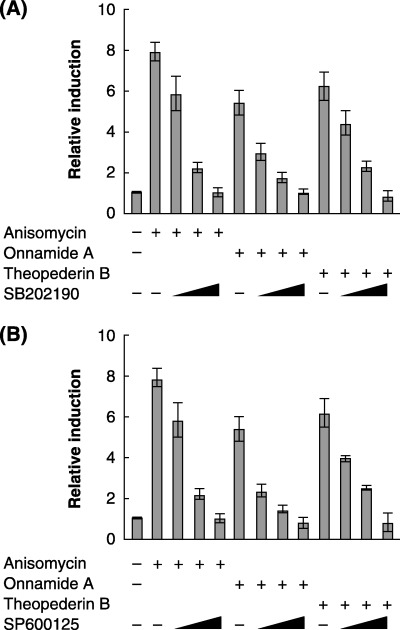
p38 kinase and/or JNK‐dependent activation of PAI‐1 gene expression by onnamide A and theopederin B. The levels of PAI‐1 promoter activation by anisomycin (380 nM), onnamide A (63 nM), and theopederin B (8.7 nM) were determined by the luciferase reporter assay in the presence or absence of various concentrations (1, 5, and 10 M) of SB202190 (A) and SP600125 (B).
Effects of onnamide A or theopederin B on cell proliferation and apoptosis. Anisomycin has been shown to induce strong cytotoxicity compared to other protein synthesis inhibitors probably through its ability to induce JNK activation.( 19 , 20 ) To test the possibility that onnamide A and theopederin B also induce cytotoxicity, we analyzed the dose‐response of NIH3T3 and three human cancer cell lines to onnamide A and theopederin B 72 h after drug challenge and determined the IC50 values for the four cell lines (Fig. 6). As shown in Table 1, theopederin B was more potent than onnamide A and other drugs in inhibiting the cell growth, and its IC50 values were at the low nanomolar level, which coincided well with the effective concentrations for the PAI‐1 gene activation and protein synthesis inhibition. The IC50 values of onnamide A were several fold higher than those of theopederin B. This difference was comparable to that in the activity to inhibit protein synthesis (Fig. 2). On the other hand, anisomycin inhibited cell proliferation more strongly than protein synthesis.
Figure 6.
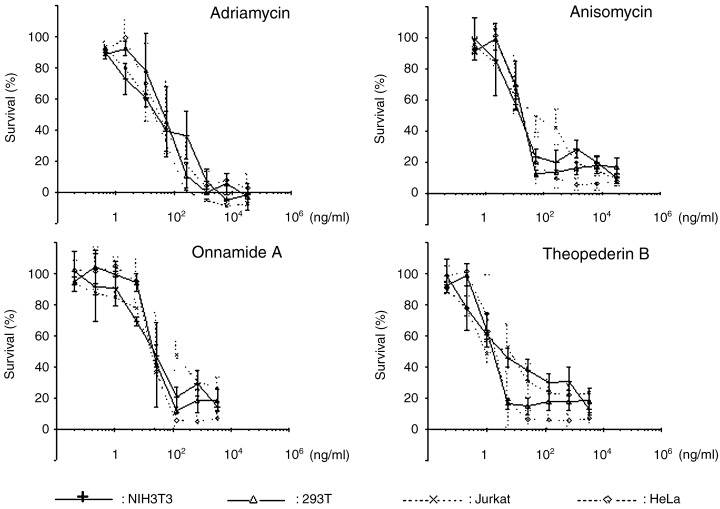
Effects on cell proliferation. Effects of onnamide A, theopederin B, anisomycin, and adriamycin on cell proliferation were measured by XTT assay. The survival is shown as the mean ± SD percentage of control culture in three independent experiments. Each experiment was carried out in duplicate.
Table 1.
Effect of onnamide A and theopederin B on cell growth
| Cell line | IC50 (nM) | |||
|---|---|---|---|---|
| Andriamycin | Anisomycin | Onnamide A | Theopederin B | |
| NIH3T3 | 42 | 49 | 27 | 7.0 |
| 293T | 81 | 72 | 14 | 3.1 |
| Jurkat | 39 | 190 | 120 | 7.5 |
| HeLa | 57 | 75 | 24 | 3.7 |
We next examined whether these inhibitors induce apoptosis in the early stage of drug challenge by determining the proportion of apoptotic cells of NIH3T3 and HeLa cells 24 h after drug addition (Fig. 7). In NIH3T3 cells, only a small fraction of cells underwent apoptosis in all the drugs tested (Fig. 7A). However, more than 40% of the HeLa cells treated with onnamide A or theopederin B for 24 h showed apoptotic changes in the morphology of the nuclei in a dose‐dependent manner. Anisomycin was very weak in inducing apoptosis in HeLa cells. To obtain a clue to understanding the difference between onnamide A/theopederin B and anisomycin, we tested whether anisomycin as well as onnamide A and theopederin B can also induce p38 kinase and JNK activation in HeLa cells. As shown in Fig. 7(B), anisomycin induced phosphorylation of these kinases to an extent similar to theopederin B but more effectively than onnamide A. In addition, the activation occurred earlier than that by onnamide A or theopederin B. However, the phosphorylated p38 kinase and JNK rapidly decrease after peaking at 30 min in anisomycin‐treated cells, while the induced kinase activation was sustained at least until 180 min in the cells treated with onnamide A or theopederin B.
Figure 7.
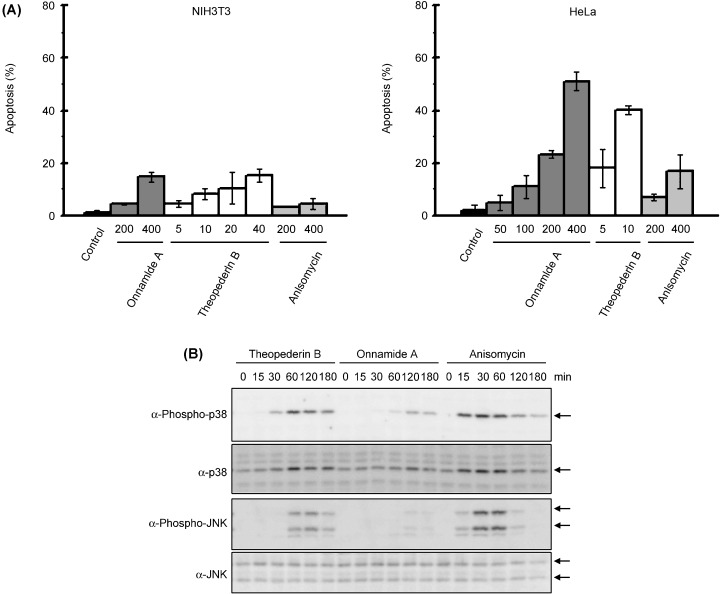
Effects on apoptosis. (A), Apoptosis induction by treatment of onnamide A, theopederin B, and anisomycin was determined by counting apoptotic cells under microscopic observation. Results are expressed as the mean ± SD of triplicate wells. (B), Phosphorylation of p38 and JNK in HeLa cells. Adhered HeLa cells were treated with onnamide A (100 nM), theopederin B (10 nM), or anisomycin (100 nM) for indicated period.
Discussion
Onnamide A and theopederin B are members of the pederin family including pederin, the insect chemical defense agent isolated from beetles, and mycalamides isolated from marine sponges, both of which are suggested to be produced by the symbiont bacteria (Fig. 1A).( 21 , 22 ) These compounds exhibit strong cytotoxicity and antitumor activity.( 4 , 5 , 6 , 7 ) Unfortunately, detailed studies on the clinical potential of these compounds have been hampered because they are available in only minute quantities from natural sources. To overcome this problem, several groups have recently synthesized the related compounds.( 23 , 24 , 25 ) However, the mechanism by which the compounds of this family induce cell death in tumor cells is poorly understood. Mycalamides were shown to inhibit protein synthesis.( 4 ) Recently, Hood et al. reported that mycalamide A induces apoptosis,( 7 ) but the mechanism is still unknown. In the present study, we identified onnamide A and theopederin B as inducers of the expression of PAI‐1 gene, a well‐characterized TGF‐β‐responsive gene. These compounds did not activate Smad2/3 but strongly inhibited protein synthesis. Furthermore, we showed that onnamide A and theopederin B activated p38 kinase and JNK, like anisomycin, an inhibitor of protein synthesis, which induces ribotoxic stress response. As p38 kinase and JNK inhibitors suppressed the PAI‐1 gene expression by these compounds as well as anisomycin, these protein kinases are suggested to be involved in the induction of PAI‐1 gene expression by these compounds. TGF‐β also activates p38 and JNK, which are involved in activation of TGF‐β‐responsive genes such as PAI‐1.( 10 , 11 , 13 , 14 , 15 , 16 ) Indeed, a specific p38 kinase inhibitor has been shown to inhibit activation of TGF‐β‐responsive genes including PAI‐1.( 13 , 26 ) Therefore, onnamide A and theopederin B can activate part of the downstream of the TGF‐β signaling pathway.
JNK and p38 are primarily activated by not only TGF‐β but also various environmental stresses, including toxins, physical stresses and inflammatory cytokines, and constitute a pivotal signaling pathway in cytokine‐ and stress‐induced apoptosis.( 27 ) Specific inhibitors of p38 kinase and JNK pathways, or dominant‐negative mutants of p38 and JNK suppress various types of stress‐induced apoptosis. Studies on fibroblasts with targeted disruptions of all the JNK genes established an essential role for JNK in UV‐induced and other stress‐induced apoptosis. These kinase pathways in response to stress are thought to be important in controlling cancer, as many primary tumors, as well as transformed cells, display higher sensitivity than normal cells to chemotherapeutic drugs because of the potentiation of the stress‐activated kinase pathways. Indeed, we showed that onnamide A and theopederin B inhibited proliferation of cells including human cancer cell lines at the nanomolar concentrations (Fig. 6, Table 1). This is probably a result of their apoptosis‐inducing activity in addition to the protein synthesis‐inhibitory activity (Fig. 7A). Therefore, our data suggest that the cytotoxicity and antitumor activity of onnamide A and theopederin B are ascribed to their ability to induce p38 kinase and JNK activation.
Anisomycin induces rapid apoptosis in human lymphoid cells in contrast to the delayed apoptosis induced by many other protein synthesis inhibitors that do not activate JNK and p38 kinase,( 19 , 20 ) suggesting an important role of the kinases in anisomycin‐induced apoptosis in tumor cells. To our surprise, however, anisomycin was weak in inducing apoptosis in HeLa cells compared with onnamide A and theopederin B, although the p38 kinase and JNK were highly activated. Phosphorylation of these kinases in anisomycin‐treated cells was transient, while that in cells treated with onnamide A or theopederin B was sustained, suggesting that the transient activation of these kinases is insufficient to induce apoptosis in HeLa cells. It is still unclear why the time course of kinase activation is different between the drugs. It seems possible that the binding to the different site on ribosomes generates a different mode of the signal that activates downstream kinases. Further studies are needed for understanding the mechanism of apoptosis by the drugs inducing p38 kinase and JNK activation.
Ribotoxic stress response is a conserved cellular reaction to cytotoxic interference with the function of the large ribosomal (23S or 28S) RNA.( 28 ) In mammalian cells, the ribotoxic stress response involves activation of JNK and p38 kinase, and subsequent transcriptional induction of immediate early genes such as c‐fos and c‐jun. The best known chemical inducer of the ribotoxic stress response is anisomycin. However, inhibition of protein synthesis per se does not stimulate the JNK and p38 kinase. Pactamycin and emetine, which block translational initiation and ribosomal translocation, respectively, have been reported to fail to activate JNK. In the present study, we also showed that cycloheximide and puromycin had no activity, or if any, a very weak activity to induce activation of these kinases (Fig. 3B). Efficient kinase activation was achieved with concentrations of anisomycin that inhibited protein synthesis by less than 50%; it was therefore concluded that anisomycin activates protein kinases independently of its ability to inhibit protein synthesis.( 29 , 30 ) In contrast, ricin A chain and α‐sarcin, ribotoxic enzymes that catalyze sequence‐specific RNA damage in the 28S rRNA, stimulated the ribotoxic stress response.( 31 ) UV radiation causes specific damage to the 28S rRNA, thus inducing inhibition of protein synthesis and JNK activation in the presence of active ribosomes.( 32 ) However, the mechanisms by which these agents, including anisomycin, induce the ribotoxic stress response are currently unknown. Present study strongly suggests that the pederin family members including onnamide A and theopederin B are novel, potent ribotoxic stress inducers. Anisomycin has been described to bind to the large subunit of ribosomes and inhibit the peptidyl transfer reaction.( 33 ) Our previous studies on 13‐deoxytedanolide, a marine sponge‐derived macrolide, demonstrated that it bound directly to the 60S large ribosomal subunit and that onnamide A and theopederin B but not anisomycin competed with 13‐deoxytedanolide for the ribosome binding.( 34 ) These results suggest that onnamide A and theopederin B inhibit ribosome function by binding at the site different from that of anisomycin. In addition, there is an indication that the ribosome translocation but not the peptidyl transfer was impaired by pederin,( 35 ) suggesting that the mechanism by which onnamide A and theopederin B induces the ribotoxic stress response is distinct from those of other inducers. Thus, onnamide A and theopederin B will serve as not only candidates for anticancer drugs but also new tools for analyzing the mechanism of the ribotoxic stress response induction.
Acknowledgments
We thank Dr M. Abe and Dr S. Khochbin for supplying the cell lines. This work was supported in part by the CREST Research Project, Japan Science and Technology Agency, a special grant for Advanced Research on Cancer, and Special Coordination Funds for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science and Technology, of the Japanese Government.
References
- 1. Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000; 103: 295–309. [DOI] [PubMed] [Google Scholar]
- 2. Shi Y, Massague J. Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell 2003; 113: 685–700. [DOI] [PubMed] [Google Scholar]
- 3. Keeton MR, Curriden SA, Van Zonneveld AJ, Loskutoff DJ. Identification of regulatory sequences in the type 1 plasminogen activator inhibitor gene responsive to transforming growth factor beta. J Biol Chem 1991; 266: 23048–52. [PubMed] [Google Scholar]
- 4. Burres NS, Clement JJ. Antitumor activity and mechanism of action of the novel marine natural products mycalamide‐A and ‐B and onnamide. Cancer Res 1989; 49: 2935–40. [PubMed] [Google Scholar]
- 5. Ogawara H, Higashi K, Uchino K, Perry NB. Change of ras‐transformed NRK‐cells back to normal morphology by mycalamides A and B, antitumor agents from a marine sponge. Chem Pharm Bull (Tokyo) 1991; 39: 2152–4. [DOI] [PubMed] [Google Scholar]
- 6. Richter A, Kocienski P, Raubo P, Davies DE. The in vitro biological activities of synthetic 18‐O‐methyl mycalamide B, 10‐epi‐18‐O‐methyl mycalamide B and pederin. Anticancer Drug Des 1997; 12: 217–27. [PubMed] [Google Scholar]
- 7. Hood KA, West LM, Northcote PT, Berridge MV, Miller JH. Induction of apoptosis by the marine sponge (Mycale) metabolites, mycalamide A and pateamine. Apoptosis 2001; 6: 207–19. [DOI] [PubMed] [Google Scholar]
- 8. Gannon JV, Lane DP. Protein synthesis required to anchor a mutant p53 protein which is temperature‐sensitive for nuclear transport. Nature 1991; 349: 802–6. [DOI] [PubMed] [Google Scholar]
- 9. Akakura S, Yoshida M, Yoneda Y, Horinouchi S. A role for Hsc70 in regulating nucleo‐cytoplasmic transport of a temperature‐sensitive p53 (p53Val135). J Biol Chem 2001; 276: 14649–57. [DOI] [PubMed] [Google Scholar]
- 10. Hannigan M, Zhan L, Ai Y, Huang CK. The role of p38 MAP kinase in TGF‐beta1‐induced signal transduction in human neutrophils. Biochem Biophys Res Commun 1998; 246: 55–8. [DOI] [PubMed] [Google Scholar]
- 11. Hanafusa H, Ninomiya‐Tsuji J, Masuyama N et al. Involvement of the p38 mitogen‐activated protein kinase pathway in transforming growth factor‐beta‐induced gene expression. J Biol Chem 1999; 274: 27161–2167. [DOI] [PubMed] [Google Scholar]
- 12. Sano Y, Harada J, Tashiro S, Gotoh‐Mandeville R, Maekawa T, Ishii S. ATF‐2 is a common nuclear target of Smad and TAK1 pathways in transforming growth factor‐beta signaling. J Biol Chem 1999; 274: 8949–57. [DOI] [PubMed] [Google Scholar]
- 13. Liao JH, Chen JS, Chai MQ, Zhao S, Song JG. The involvement of p38 MAPK in transforming growth factor beta1‐induced apoptosis in murine hepatocytes. Cell Res 2001; 11: 89–94. [DOI] [PubMed] [Google Scholar]
- 14. Wang L, Ma R, Flavell RA, Choi ME. Requirement of mitogen‐activated protein kinase kinase 3 (MKK3) for activation of p38alpha and p38delta MAPK isoforms by TGF‐beta 1 in murine mesangial cells. J Biol Chem 2002; 277: 47257–62. [DOI] [PubMed] [Google Scholar]
- 15. Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor‐beta‐mediated transcription. J Biol Chem 1999; 274: 37413–20. [DOI] [PubMed] [Google Scholar]
- 16. Hocevar BA, Brown TL, Howe PH. TGF‐beta induces fibronectin synthesis through a c‐Jun N‐terminal kinase‐dependent, Smad4‐independent pathway. EMBO J 1999; 18: 1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hazzalin CA, Cano E, Cuenda A, Barratt MJ, Cohen P, Mahadevan LC. p38/RK is essential for stress‐induced nuclear responses. JNK/SAPKs and c‐Jun/ATF‐2 phosphorylation are insufficient. Curr Biol 1996; 6: 1028–31. [DOI] [PubMed] [Google Scholar]
- 18. Bogoyevitch MA, Ketterman AJ, Sugden PH. Cellular stresses differentially activate c‐Jun N‐terminal protein kinases and extracellular signal‐regulated protein kinases in cultured ventricular myocytes. J Biol Chem 1995; 270: 29710–7. [DOI] [PubMed] [Google Scholar]
- 19. Kochi SK, Collier RJ. DNA fragmentation and cytolysis in U937 cells treated with diphtheria toxin or other inhibitors of protein synthesis. Exp Cell Res 1993; 208: 296–302. [DOI] [PubMed] [Google Scholar]
- 20. Polverino AJ, Patterson SD. Selective activation of caspases during apoptotic induction in HL‐60 cells. Effects of a tetrapeptide inhibitor. J Biol Chem 1997; 272: 7013–21. [DOI] [PubMed] [Google Scholar]
- 21. Piel J. A polyketide synthase‐peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc Natl Acad Sci USA 2002; 99: 14002–7. Epub 2002 Oct 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Piel J, Hofer I, Hui D. Evidence for a symbiosis island involved in horizontal acquisition of pederin biosynthetic capabilities by the bacterial symbiont of Paederus fuscipes beetles. J Bacteriol 2004; 186: 1280–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trost BM, Yang H, Probst GD. A formal synthesis of (‐)‐mycalamide A. J Am Chem Soc 2004; 126: 48–9. [DOI] [PubMed] [Google Scholar]
- 24. Roush WR, Pfeifer LA. Total synthesis of mycalamide A and 7‐epi‐mycalamide A. Org Lett 2000; 2: 859–62. [DOI] [PubMed] [Google Scholar]
- 25. Toyota M, Hirota M, Hirano H, Ihara M. A stereoselective synthesis of the C‐10 to C‐18 (right‐half) fragment of mycalamides employing lewis acid promoted intermolecular aldol reaction. Org Lett 2000; 2: 2031–4. [DOI] [PubMed] [Google Scholar]
- 26. Laping NJ, Grygielko E, Mathur A et al. Inhibition of transforming growth factor (TGF) ‐beta1‐induced extracellular matrix with a novel inhibitor of the TGF‐beta type I receptor kinase activity: SB‐431542. Mol Pharmacol 2002; 62: 58–64. [DOI] [PubMed] [Google Scholar]
- 27. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001; 410: 37–40. [DOI] [PubMed] [Google Scholar]
- 28. Laskin JD, Heck DE, Laskin DL. The ribotoxic stress response as a potential mechanism for MAP kinase activation in xenobiotic toxicity. Toxicol Sci 2002; 69: 289–91. [DOI] [PubMed] [Google Scholar]
- 29. Cano E, Hazzalin CA, Mahadevan LC. Anisomycin‐activated protein kinases p45 and p55 but not mitogen‐activated protein kinases ERK‐1 and ‐2 are implicated in the induction of c‐fos and c‐jun. Mol Cell Biol 1994; 14: 7352–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zinck R, Cahill MA, Kracht M, Sachsenmaier C, Hipskind RA, Nordheim A. Protein synthesis inhibitors reveal differential regulation of mitogen‐activated protein kinase and stress‐activated protein kinase pathways that converge on Elk‐1. Mol Cell Biol 1995; 15: 4930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iordanov MS, Pribnow D, Magun JL et al. Ribotoxic stress response. activation of the stress‐activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence‐specific RNA damage to the alpha‐sarcin/ricin loop in the 28S rRNA. Mol Cell Biol 1997; 17: 3373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Magun BE. Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J Biol Chem 1998; 273: 15794–803. [DOI] [PubMed] [Google Scholar]
- 33. Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J Mol Biol 2003; 330: 1061–75. [DOI] [PubMed] [Google Scholar]
- 34. Nishimura S, Matsunaga S, Lee K‐H et al. 13‐Deoxytedanolide, a marine sponge‐derived antitumor macrolide, binds to 60S large ribosomal subunit. Bioorg Med Chem 2004; 13: 449–54. [DOI] [PubMed] [Google Scholar]
- 35. Carrasco L, Vazquez D. Survey of inhibitors in different steps of protein synthesis by mammalian ribosomes. J Antibiot 1972; 25: 732–7. [DOI] [PubMed] [Google Scholar]