

Abstract
The gastrointestinal stromal tumor cell line, GIST‐T1, has a heterogenic 57‐base pair deletion in exon 11 of the c‐kit mutation, and the c‐KIT protein in the GIST‐T1 cells constitutively activated. We report that STI571 (Glivec; Novartis, Basel, Switzerland), a specific inhibitor of c‐KIT, inhibits the clustering of c‐KIT at the cell membrane of the GIST‐T1 cells. Furthermore, STI571 prevents the interaction between c‐KIT and the molecular chaperone, heat shock protein 90 (Hsp90). Geldanamycin, an inhibitor of Hsp90, also prevents interaction between c‐KIT and Hsp90, and inhibits tyrosine phosphorylation of c‐KIT. Our results indicate that c‐KIT molecules are assembled on the cell surface of the GIST‐T1 cells, and that the interaction between c‐KIT and Hsp90 plays an important role in c‐KIT activation. (Cancer Sci 2005; 96: 116–119)
Gastrointestinal stromal tumor (GIST) is characterized by the expression of both c‐KIT and CD34 on the cell surface membrane. Recent studies reveal that mutations of c‐kit are associated with oncogenic activation of GIST cells. 1 , 2
C‐Kit, a type III receptor tyrosine kinase, binds to its ligand, stem cell factor (SCF) to initiate signals that are critical for the growth and development of mast cells, melanocytes, hematopoietic stem cells, and the interstitial cells of Cajal. 3 , 4 Gain‐of‐function mutations in c‐kit are associated with a number of cancers in humans. 1 , 5 , 6 , 7 The mechanism of constitutive c‐KIT phosphorylation in the majority of the GIST involves a gain of function mutations in exon 11 (cytoplasmic juxtamembrane domain), and other mutations in exon 9 (extracellular membrane domain), exon 13 (first part of the split tyrosine kinase domain) and exon 17 (phosphotransferase domain). (8)
STI571 (Glivec; Novartis, Basel, Switzerland) is a specific tyrosine kinase inhibitor that acts with BCR‐ABL, platelet‐derived growth factor receptor (PDGFR) and c‐KIT. STI571 has been used successfully in unresectable or metastatic GIST patients with constitutive activation of c‐KIT. (9)
The molecular chaperone, heat shock protein (Hsp90), plays an important role in stress tolerance, protein folding, and the post translational control of the stability, as well as in the function of many key regulators of cell growth, differentiation and apoptosis. (10) Hsp90 interacts with mutated p53, BCR‐ABL, RAF‐1, AKT, and hypoxia‐inducible factor‐1α. (10) Recently, microbial fermentation products have been identified that bind Hsp90 with high affinity and selectively alter its function. These Hsp90‐binding agents have proven useful in a role with ATP in Hsp90 chaperone activity. 11 , 12 A role has also been established for Hsp90 in regulating the function, stability and degradation of multiple signal transduction molecules relevant to oncogenic transformation. Hsp90 inhibitors, by interacting specifically with a single molecular target, cause the destabilization and eventual degradation of the proteins that are associated with Hsp90, and have shown promising antitumor activity. (10) One such inhibitor, geldanamycin, can block dissociation from glucocorticoid receptors, while disrupting the formation of a heteroprotein complex between Hsp90 and the cystic fibrosis transmembrane conductance regulator. 13 , 14 , 15 In the present study, we analyzed the behavior of c‐KIT in the GIST cell line, GIST‐T1, treated with or without STI571.
GIST‐T1, established from a metastatic GIST patient, (16) has the c‐kit mutation in exon 11, and c‐KIT is constitutively activated in these cells. Furthermore, we analyzed whether activated c‐KIT could interact with Hsp90 using geldanamycin.
Materials and Methods
Cell and cell culture. The human GIST cell line, GIST‐T1, has been characterized in detail by Taguchi et al. (16) GIST‐T1 was cultured in Dulbecco's modified Eagle's medium supplemented with penicillin, streptomycin and 8% fetal bovine serum, and maintained in an atmosphere consisting of 95% air and 5% CO2 at 37°C in a humidified incubator.
Reagents. STI571, Glivec capsule (Novartis), was dissolved in water (5 µg/mL) and stored at − 20°C. Geldanamycin was dissolved in dimethyl sulfoxide (0.1 µg/mL) and stored at −20°C. Antibodies were used to detect the c‐KIT (K963, rabbit polyclonal immunoglobulin G [IgG]; Immuno‐Biological Laboratories, Japan), phosphotyrosine [PY20, mouse monoclonal IgG; Zymed Laboratories, USA], and Hsp90 [D‐19, goat polyclonal IgG; Santa Cruz Biotechnology, USA).
Tyrosine phosphorylation of c‐KIT was determined by Western blot analysis. Tyrosine phosphorylation of c‐KIT was assessed by Western blot using PY20. Cells were washed three times with ice‐cold phosphate‐buffered saline (PBS) and then lyzed in RIPA buffer (Upstate Biotechnology, USA) containing 20 mM sodium pyrophosphate, 20 mM NaF, 1 mM orthovandate, 2 mM pyrophosphate, 1 mM phenylmethylsulfonyl fluoride, 10 µg/mL aprotinin, and 10 µg/mL leupeptin. Cell lysates containing comparable amounts of protein, estimated by a Bradford assay (Bio‐Rad, Munich, Germany), were subjected to direct Western blotting.
Interaction of c‐KIT with Hsp90 was assessed by immunoprecipitation analysis. Cells lysates in RIPA buffer were subjected to immunoprecipitation with c‐KIT antibody. The immunoprecipitates were reacted with protein A‐agarose and washed with Tris‐buffered saline (TBS). They were then finally resuspended in 3 × sodium dodecylsulfate (SDS) sample buffer containing 30 × dithiothreitol (DTT), and boiled at 95°C for 5 min. Samples were separated by 7.5% SDS‐polyacrylamide gel electrophoresis and transferred to a membrane for immunoblot analysis.
Immunogold electron microscopy to localized c‐KIT and Hsp90 in GIST‐T1 cells. Cells were treated with/without the indicated reagent for 6 h. Adherent cells were washed twice with ice‐cold PBS and scraped. Scraped cells were collected by centrifugation and the collected cells were fixed with 1% glutaraldehyde for 2 h on ice. Fixed cells were dehydrated through graded ethanols, embedded in Lowicryl K4M (Electron Microscopy Science, USA), polymerized in an Ultraviolet Polymerizer Tur 200 (Dosaka EM Co., Kyoto, Japan) for 24 h at −35°C and 3 days at room temperature. For immunogold electron microscopy, thin sections mounted on collodion‐carbon‐coated nickel grids were immersed in 0.01 M PBS and then treated with anti‐c‐KIT or anti‐Hsp90 for 12 h. The sections were then treated with 15 nm colloidal gold (goat antirabbit IgG; Chemicon, USA) for c‐KIT, or 5 nm colloidal gold (mouse antigoat IgG; Chemicon) for Hsp90, diluted at an adequate concentration in 0.01 M PBS containing 3% skim milk for 30 min in a moist chamber. The sections were rinsed in 0.01 M PBS and stained with uranyl acetate and lead citrate. Control staining was performed without primary antibody and then treated with a secondary antibody. We did not detect the gold particles on control staining.
MTT assay. In a 96‐well plate, 1 × 104 cells/100 µL of cell suspension were used to seed each well. After 24 h with or without STI571 and/or geldanamycin treatment, 100 µL of a 2.5 mg/mL solution in PBS of the tetrazolium substrate, MTT (3,‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide; Sigma‐Aldrich, Tokyo, Japan), was added and incubated for 4 h at 37°C. The resulting violet formazan precipitate was solubilized by the addition of 100 µL of a 50% N,N,‐Dimethyl formamide/10% SDS solution, and incubated for 4 h at room temperature. The plate was then analyzed on a plate reader at 570 nm to determine the absorbance of the samples.
Results
Tyrosine phosphorylation of c‐KIT. In non‐treated GIST‐T1 cells, c‐KIT was tyrosine phosphorylated (Fig. 1). STI571 inhibited tyrosine phosphorylation of the c‐KIT. The level of tyrosine phosphorylation of the c‐KIT was reduced by treatment of the cells with geldanamycin (0.1 and 1 µg/mL). Equal amounts of c‐KIT were loaded in each lane (Fig. 1, lower panel).
Figure 1.
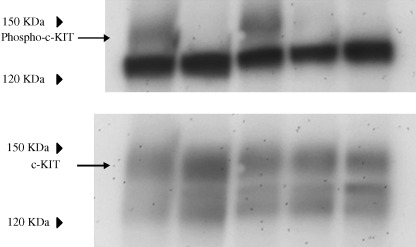
Cells, 3 × 106, were seeded on a 10‐cm diameter dish. After 24 h, cells were treated with/without STI571 (1 µg/mL) or geldanamycin (0.01, 0.1, 1 µg/mL) for 5 h. The cells were scraped and collected by centrifugation and then lyzed in radioimmunoprecipitation assay buffer as described in the Materials and Methods section. Membranes were reacted with antiphosphotyrosine (1:250) or anti‐c‐KIT (1:500) overnight at 4C°. Lanes 1–5: non‐treated, STI571 1 µg/mL, geldanamycin 0.01 µg/mL, 0.1 µg/mL and 1 µg/mL, respectively.
Interaction between c‐KIT and Hsp90 after treatment with STI571 or geldanamycin. The accumulation of Hsp90 was decreased by treatment with STI571 or geldanamycin compared with the non‐treated cells (Fig. 2). It was indicated that STI571 or geldanamycin prevented the interaction between c‐KIT and Hsp90. These results suggest that tyrosine‐phosphorylated c‐KIT can bind to Hsp90.
Figure 2.
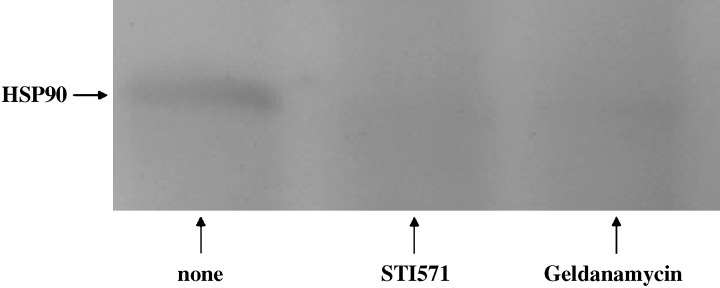
Cells, 3 × 106, were treated with/without STI571 (1 µg/mL) or geldanamycin (0.1 µg/mL). Immunoprecipitated with anti‐c‐KIT (1:100) overnight at 4C°, and immunoprecipitates were protein A agarose as described in the Materials and Methods section. Anti‐heat shock protein 90 antibody (1:250) was used for Western blot analysis.
Observation of c‐KIT molecules on the cell surface membrane of GIST‐T1. C‐KIT molecules were assembled on the cell surface of non‐treated GIST‐T1 cells. (Fig. 3a). STI571 or geldanamycin inhibited the clustering of c‐KIT on the cell surface membrane (Fig. 3b,c, respectively). These results suggest that tyrosine‐phosphorylated c‐KIT is involved in the cluster formation at the cell membrane.
Figure 3.
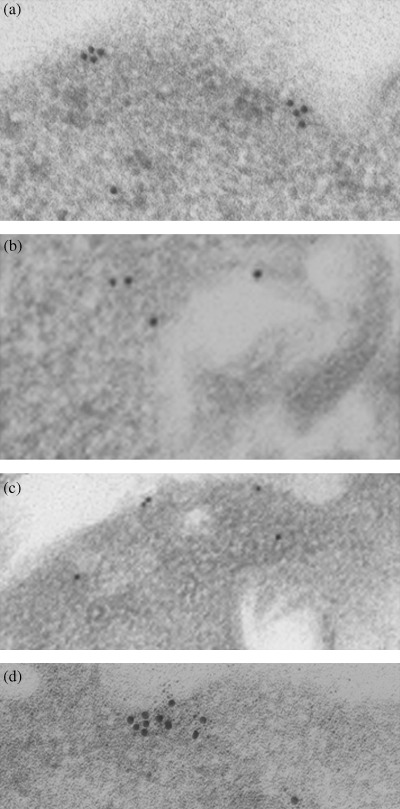
Immunoelectromicroscopic analysis. Cells were treated with/without STI571 (1 µg/mL) or geldanamycin (0.1 µg/mL). Samples were prepared as described in the Materials and Methods section. (a) Non‐treated, (b) STI571 treated, (c) geldanamycin‐treated, and (d) non‐treated, but samples were reacted with anti‐c‐KIT and 15‐nm‐gold labeled secondary antibody, and then they were reacted with anti‐heat shock protein 90 antibody and 5‐nm‐gold labeled secondary antibody.
Localization of Hsp90 and c‐KIT on the cell surface of GIST‐T1. Immunoprecipitation analysis revealed that tyrosine‐phosphorylated c‐KIT could interact biochemically with Hsp90. This interaction was shown to occur at the plasma membrane using immunogold labeling for electron microscopy. Hsp90 (5 nm gold) was diffused throughout the cytoplasm and also co‐localized in the cluster formation of c‐KIT (15 nm gold) at the plasma membrane (Fig. 3d).
GIST‐T1 cell viability following treatment with STI571 or geldanamycin. Cell viability of GIST‐T1 cells was measured by the MTT assay. STI571 reduced the cell viability to: 74.3 ± 2.1%, 60.3 ± 0.5%, 36.2 ± 1.5% and 33.7 ± 2.1% at the concentrations of 0.01, 0.05, 0.1, and 1 µg/mL, respectively (Fig. 4). Geldanamycin also reduced the cell viability to: 79.7 ± 1.0%, 71.9 ± 0.64%, 53.3 ± 1.2%, 22.1 ± 0.8% and 18.3 ± 0.5% at the concentrations of 0.01, 0.02, 0.05, 0.1 and 1 µg/mL, respectively. In this study, geldanamycin was therefore a more effective reagent compared with STI571. The combination of both reagents diminished the effect of the geldanamycin treatment.
Figure 4.
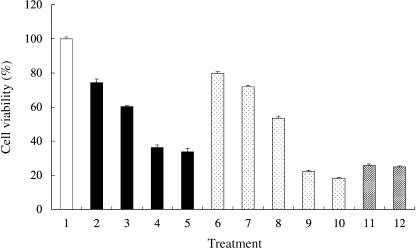
Cell viability was measured by (3,‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide assay. 1, non‐treated; 2, STI571 0.01 µg/mL; 3, STI571 0.05 µg/mL; 4, STI571 0.1 µg/mL; 5, STI571 1 µg/mL; 6, geldanamycin 0.01 µg/mL; 7, geldanamycin 0.02 µg/mL; 8, geldanamycin 0.05 µg/mL; 9, geldanamycin 0.1 µg/mL; 10, geldanamycin 1 µg/mL; 11, STI571 1 µg/mL + geldanamycin 0.1 µg/mL; 12, STI571 1 µg/mL + geldanamycin 1 µg/mL. The survival percentages are shown as the mean ± standard deviation of triplicate experiments. The y‐axis presents the percentage viability of gastrointestinal stromal tumor cell line (GIST)‐T1.
Discussion
GIST‐T1 cells have the c‐kit mutation in exon 11, thereby c‐KIT is constitutively activate in these cells. STI571 was identified as an inhibitor of BCR/ABL, PDGFR and c‐KIT. STI571 has been used successfully in treating unresectable or metastatic GIST patients with constitutive activation of c‐KIT. (9) In the present study, STI571 inhibited tyrosine phosphorylation of the c‐KIT of GIST‐T1 cells. Geldanamycin, an inhibitor of Hsp90, also inhibited the tyrosine phosphorylation of c‐KIT. Little is known of the interaction between Hsp90 and c‐KIT, prompting us to assess whether activated c‐KIT interacted with Hsp90. c‐KIT treated with STI571 could not bind to Hsp90, and geldanamycin also prevented the interaction between Hsp90 and c‐KIT. Theses results suggested that interaction of c‐KIT with Hsp90 is necessary and sufficient for activation of c‐KIT in GIST‐T1 cells. We further investigated the interaction between c‐KIT and Hsp90 in DLD‐1 cells (data not shown), which are of colon cancer origin and express c‐KIT. (17) We stimulated DLD‐1 using SCF (100 ng/mL) for 30 min and cells were lyzed in RIPA buffer, as described previously in Materials and Methods. Immunoprecipitation analysis revealed that Hsp90 bound to c‐KIT strongly in the case of SCF stimulation. This finding raised the question of how geldanamycin can inhibit the activation of c‐KIT. We did not test whether geldanamycin acted on c‐KIT directly. Further research is required to clarify which molecules are involved in the activation of c‐KIT and the regulation by geldanamycin.
Furthermore, we investigated the difference between activated c‐KIT and dephosphorylated c‐KIT by immunogold labeling electron microscopic analysis. In a previous study, Borrello et al. demonstrated that cluster formation of RET on the cell surface of TT cells compared with wild‐type RET expressed in SH‐SY‐5Y cells by immunoelectron microscopic analysis. (18) TT is a medullary thyroid cancer cell line, which expresses a RET allele with a multiple endocrine neoplasma type 2A mutation at codon 634 (Cys to Trp). RET is activated in TT cells without ligand stimulation and the protein localizes in clusters at the plasma membrane. In contrast, wild‐type RET, expressed in the neuroblastoma cell line, SH‐SY‐5Y, shows no cluster formation without ligand stimulation. In our study, activated c‐KIT molecules showed cell‐surface clustering, which was inhibited by STI571 or geldanamycin. Broudy et al. reported that stimulation with SCF induced capping and internalization of c‐KIT. (19) In their experiments, stimulation with a high concentration of SCF (200 ng/mL) strongly and transiently induced the activation of c‐KIT. In our study, the activation of c‐KIT was at the physiological level, hence we could not detect the capping of c‐KIT by electron microscopy. Immunoprecipitation and Western blot analysis revealed that activated c‐KIT could bind to Hsp90, and the immunogold electron microscopy showed c‐KIT localized with Hsp90 in the cell surface clusters. The functional part of the c‐KIT protein is localized on the cell surface, and allows transduction of downstream signaling to the nucleus via phosphorylation of tyrosine residues in the signaling proteins.
Finally, we assessed the viability of cells treated with/without STI571 or geldanamycin. MTT assay demonstrated that both STI571 and geldanamycin reduced the cell viability of GIST‐T1 cells. It is not clear why geldanamycin had more of an effect on cell viability than STI571, or why the effect was a diminished effect in the case of combination of both.
An inhibitor of Hsp90, benzoquinone ansamycin, 17‐allylaminogeldanamycin (17AAG), is undergoing a phase I clinical trial. (10) We did not use 17‐AAG in this study, but our results suggest that an inhibitor of Hsp90 will prove useful agents in therapy for GIST.
Acknowledgments
We thank Motoko Miyata and Yuka Takezaki for their technical assistance.
References
- 1. Hirota S, Isozaki K, Moriyama Y, Kanakura Y, Nishida T, Ishigro S, Kawano K, Hanada M, Kurata A, Takeda M, Tunio GM, Matsuzawa Y, Shinomura Y, Kitamura Y. Gain‐of‐function mutation of c‐kit in human gastrointestinal stromal tumors. Science 1998; 279: 577–80. [DOI] [PubMed] [Google Scholar]
- 2. Nishida T, Hirota S. Biological and clinical review of stromal tumors in the gastrointestinal tract. Histol Histopathol 2000; 4: 1293–301. [DOI] [PubMed] [Google Scholar]
- 3. Galli SJ, Zsebo KM, Geissler EN. The kit ligand, stem cell factor. Adv Immunol 1994; 55: 1–96. [DOI] [PubMed] [Google Scholar]
- 4. Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1995; 373: 347–9. [DOI] [PubMed] [Google Scholar]
- 5. Longley BJ Jr, Metcalfe DD, Tharp M, Wang X, Tyrell L, Lu S‐Z, Heitjan D, Ma Y. Activating and dominant inactivating c‐KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci USA 1999; 96: 1609–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tian Q, Frierson HF Jr, Krystal GW, Moskaluk CA. Activating c‐kit gene mutations in human germ cell tumors. Am J Pathol 1999; 154: 1643–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beghini A, Peterlongo P, Ripamonti CB, Larizza L, Cairoli R, Morra E, Mecucci C. C‐kit mutations in core binding factor leukemias. Blood 2000; 95: 726–7. [PubMed] [Google Scholar]
- 8. Kitamura Y, Hirota S, Nishida T. Gastrointestinal stromal tumors (GIST): a model for molecule‐based diagnosis and treatment of solid tumors. Cancer Sci 2003; 94: 315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Demetri GD, Von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002; 347: 472–89. [DOI] [PubMed] [Google Scholar]
- 10. Len Neckers. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med 2002; 8: S55–61. [DOI] [PubMed] [Google Scholar]
- 11. Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 1999; 42: 260–6. [DOI] [PubMed] [Google Scholar]
- 12. Grenert JP, Sullivan WP, Fadden P, Haystead TAJ, Clark J, Mimnaugh E, Krutzsch H, Ochel H‐J, Schulte TW, Sausville E, Neckers LM, Toft DO. The amino‐terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem 1997; 272: 2384. 3–50. [DOI] [PubMed] [Google Scholar]
- 13. Young JC, Hartl FU. Polypeptide release by HSP 90 involves ATP hydrolysis and is enhanced by the co‐chaperon p23. EMBO J 2000; 19: 5930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schneider C, Sepp‐Lorenzio L, Nimmesgern E, Ouerfelli O, Danisiieefsky S, Rosen N, Hartl FU. Pharmacological shifting of a balance between protein refolding and degradation mediated by HSP 90. Proc Natl Acad Sci USA 1996; 93: 14536–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loo MA, Jensen TJ, Cui L, Hou Y, Chang XB, Riordan JR. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by proteasome. EMBO J 1998; 17: 6879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taguchi T, Sonobe H, Toyonaga S, Yamasaki I, Shuin T, Takano A, Araki K, Akimaru K, Yuri K. Conventional and molecular cytogenetic characterization of a new human cell line, GIST‐T1, established from gastrointestinal stromal tumor. Lab Invest 2002; 82: 663–5. [DOI] [PubMed] [Google Scholar]
- 17. Bellone G, Carbone A, Sibona N, Bosco O, Tibaudi D, Smirne C, Martone T, Gramigni C, Camandona M, Emanuelli G, Rodeck U. Abberrant activation of c‐KIT protects colon carcinoma cells against apoptosis and enhances their invasive potential. Cancer Res 2001; 61: 200–6. [PubMed] [Google Scholar]
- 18. Borrello MG, Smith DP, Pasini B, Bongarzone I, Greco A, Lorenzo MJ, Arighi E, Miranda C, Eng C, Alberti L, Bocciardi R, Mondellini P, Scopsi L, Romeo G, Ponder BAJ, Pievotti MA. RET activation germline MEN2A MEN2B mutations. Oncogene 1995; 11: 2419–27. [PubMed] [Google Scholar]
- 19. Virginia C, Broudy NL, Lin W, Conrad L, Seth JC, O'Laughlin B, Mou S, Linnekin D. Signaling via src family kinases is required for normal internalization of the receptor c‐KIT. Blood 1999; 94: 1979–86. [PubMed] [Google Scholar]