

Abstract
Sprouty proteins have been shown to negatively regulate a variety of receptor tyrosine kinase (RTK) signaling pathways and are considered to be tumor suppressor proteins. The pathophysiological functions of Sproutys in vivo remain to be investigated. In this study, we examined the physiological function of Sprouty4 as an angiogenic regulator, using Sprouty4 knockout (KO) mice and cells. We found that transplanted tumor cells grow much faster in Sprouty4 KO mice than in wild type (WT) mice, which we associate with enhanced neovascularization in the tumors transplanted into Sprouty4 KO mice. Moreover, vascular endothelial growth factor (VEGF)‐A‐induced angiogenesis and vascular permeability in vivo were enhanced in Sprouty4 KO mice compared with WT mice. Ex vivo angiogenesis, which we induced by VEGF‐A, basic fibroblast growth factor (bFGF), and sphingosine‐1‐phosphate (S1P), was also enhanced in the aortas of Sprouty4 KO mice. We demonstrated that Sprouty4 suppresses Ras‐independent VEGF‐A and S1P signaling, while it does not affect Ras‐dependent VEGF‐C signaling. These data indicate that Sprouty4 selectively suppresses Ras‐independent angiogenic factor signals and is an important negative regulator of pathophysiological angiogenesis. (Cancer Sci 2009; 100: 1648–1654)
Mitogen‐activated protein (MAP) kinases, including extracellular signal‐regulated kinases (ERK), play important roles in many facets of cellular regulation.( 1 , 2 , 3 ) Recently, a family of novel membrane‐bound molecules were identified as negative regulators for growth factor‐induced ERK activation; these are known as the Sprouty (Spry) and Spred proteins.( 1 , 2 , 4 , 5 ) There are four Sprouty orthologs (Spry1–4) and three Spred orthologs (Spred1–3) in mammals. Sproutys and Spreds share a well‐conserved cysteine‐rich domain at the C‐terminus.
While Spreds inhibit ERK activation induced by any growth factor stimuli, Sproutys inhibit basic fibroblast growth factor (bFGF)‐ and vascular endothelial growth factor (VEGF)‐A‐induced ERK activation, though they do not inhibit epidermal growth factor (EGF)‐induced ERK activation.( 6 , 7 , 8 ) Spreds bind to Ras and Raf, thereby suppressing receptor tyrosine kinase (RTK) signaling by inhibiting the activation of Raf.( 4 ) In contrast, several mechanisms have been proposed for the Sprouty‐mediated inhibition of the RTK pathway, including the interruption of Grb2–Sos recruitment( 9 , 10 ) and the inhibition of Raf.( 7 , 11 ) Recently, we reported that Sprouty4 suppresses phosphatidyl inositol‐(4,5) P2 (PIP2) breakdown and protein kinase C (PKC) activation, not only when these processes are induced by VEGF‐A but also when they are induced by lysophosphatidic acid (LPA), a G‐protein coupled receptor (GPCR) ligand.( 12 ) Therefore, multiple mechanisms must be involved in Sprouty‐mediated ERK suppression.
In various cancers, the expressions of Sprouty/Spred‐family genes are repressed; consequently, these families of genes are considered to be tumor suppressor genes.( 13 , 14 ) The mechanism of suppression is thought to consist mainly of DNA methylation and loss of heterozygosity. On the other hand, the microenvironment in which a tumor originates plays a critical role in tumor initiation and progression, and may be an important factor in developing therapeutic approaches. Although there is no doubt that Sproutys play important roles in cancer cells themselves, it remains unclear whether they are also important factors in the tumor microenvironment, perhaps influencing tumor angiogenesis.
Most tumors recruit an adequate blood supply to ensure that they get sufficient oxygen and nutrients.( 15 ) Thus, angiogenesis is required for tumor growth beyond a certain size. For tumor angiogenesis to occur, endothelial cells must properly process both autocrine and paracrine signals from vascular growth factors.( 3 ) Vascular growth factors, such as VEGF‐A, bFGF, and angiopoietins, bind to their cognate RTKs on cell surfaces, thereby activating a myriad of complex intracellular signaling pathways whose outputs include endothelial cell growth, migration, and morphogenesis.( 16 , 17 ) In addition, sphingosine‐1‐phosphate (S1P), which activates GPCRs, has also been implicated in angiogenesis.( 18 ) Improved understanding of how these cascades are precisely controlled will provide insight into both physiological and pathological angiogenesis.
In this study, we found that tumor growth and angiogenesis were enhanced in Sprouty4 knockout (KO) mice. VEGF‐A‐induced angiogenesis and vascular permeability in vivo were also augmented in Sprouty4 KO mice. Sprouty4 deficiency promoted not only VEGF‐A‐ but also bFGF‐ and S1P‐induced angiogenesis. Moreover, we showed that Sprouty4 is a negative regulator for VEGF‐A signaling rather than VEGF‐C signaling in vitro. Sprouty4 inhibited not only VEGF‐A and bFGF signals but also S1P signals. These data suggest that Sprouty4 is a physiologically important negative regulator of various signals in angiogenesis and that it plays a key function in the tumor microenvironment.
Materials and Methods
Mice. Sprouty4 KO mice have been described previously.( 19 ) Sprouty4 KO mice were generated with a 129/C57BL/6J mixed background, and then backcrossed into C57BL/6J at least five times. Male Sprouty4 KO mice and wild type (WT) littermates were used for all experiments. All experiments using mice were approved by and performed according to the guidelines of the Animal Ethics Committee of Kyushu University, Fukuoka, Japan.
Plasmids. Myc‐Spred‐1, Myc‐Sprouty4, Flag‐Sprouty4, VEGF receptor (VEGFR)‐2, VEGR‐3, green fluorescent protein (GFP)‐ERK, yellow fluorescent protein (YFP)‐ERK, and N17‐Ras plasmids have been described previously.( 7 , 8 )
Cell culture. Primary mouse embryonic fibroblasts (MEFs) were obtained using standard protocols.( 19 ) MEFs and human embryonic kidney (HEK) 293T cells were cultured in Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin. MEFs stably expressing VEGFR‐2 or VEGFR‐3 were generated as described previously.( 8 )
Antibodies and reagents. Antibodies used in this experiment were as follows: anti‐phospho‐ERK1/2 (#9106), anti‐phospho‐Akt (#4058), anti‐Akt (#9272), anti‐phospho‐PKD/PKC (#2054), anti‐PKD/PKC (#2052) (Cell Signaling Technology, Danvers, MA, USA); anti‐VEGFR‐2 (A‐3) and anti‐ERK2 (C‐14) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti‐VEGFR‐3 (AFL4) (eBioscience, San Diego, CA, USA); and 4G10 (Upstate Biotechnology, Billerica, MA, USA). Human VEGF‐A and bFGF were purchased from PeproTech (London, UK). Human VEGF‐C C156S was purchased from R&D Systems (Minneapolis, MN, USA). S1P was purchased from BioMol (Polymouth Meeting, PA, USA). Bisindolylmaleimide I (Bis I), was purchased from Calbiochem (San Diego, CA, USA).
Luciferase assay and western blot analysis. Elk‐1 reporter assay and western blot analysis were performed as described previously.( 8 , 20 ) The inferior vena cava of each WT or Sprouty4 KO mouse was dissected and incubated with or without 100 ng/mL VEGF‐A or 50 ng/mL bFGF or 1 µM S1P for 30 min. Band intensities were measured using Image J software (http://rsb.info.nih.gov).
Tumor formation assay. The tumorigenicity of murine Lewis lung carcinoma (LLC) cells and B16F10 melanoma cells was assayed by injecting 2 × 105 cells of one of these cell types subcutaneously into the backs of 8‐week‐old‐male WT or Sprouty4 KO mice, as described elsewhere.( 21 ) Tumor volume was monitored, as determined by the following formula: volume = 0.52 × (width)2 × (length). Intra‐tumor vessels of LLC cells or B16F10 melanomas in frozen sections were immunostained with anti‐PECAM‐1/CD31 (MEC13.3; BD Pharmingen, Franklin Lakes, NJ, USA). At least ten 10X micrographs from regions of uniform staining intensity were acquired, and vascular surface area was quantified using Image J software, as described elsewhere.( 22 )
In vivo angiogenesis assay. Directed in vivo angiogenesis assay (DIVAA) was performed using a DIVAA starter kit (Trevigen, Gaithersburg, MD, USA) and following the instructions provided in the kit manual.
Miles vascular permeability assay. Miles assays were performed as described elsewhere.( 23 ) Evans blue dye (100 µl of a 1.0% solution in 0.9% NaCl) was then injected into the orbital veins of WT and Sprouty4 KO mice. After 10 min, 50 µl of either phosphate‐buffered saline (PBS) or PBS with recombinant VEGF‐A (10, 50 ng) was injected intradermally into the dorsal skin. Twenty minutes later, the animals were sacrificed and the area of skin that was visibly blue from the extravasated dye was excised. Evans blue dye was extracted from the pieces of skin by incubating them in formamide at room temperature for 5 days, and the absorbance of the extracted dye was measured at 620 nm using a spectrophotometer.
Ex vivo aortic ring assay. We performed aortic ring assays as described elsewhere.( 24 ) One‐mm thoracic aortic rings were placed between two layers of 50 µL growth factor‐reduced Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) supplemented with 20 U/mL heparin, and overlaid with 100 µL of RPMI‐1640 (Sigma‐Aldrich, St Louis, MO, USA) containing 1% FBS with or without VEGF‐A (50 ng/mL), bFGF (20 ng/mL), or S1P (100 nM). Microvessel outgrowth was visualized with phase microscopy and the number of vessels growing from each aortic ring was counted each day using a microscope.
Statistical analysis. Statistical significance was tested with an unpaired two‐tailed Student's t‐test. The differences were considered to be significant if P < 0.05.
Results
Accelerated tumor angiogenesis in Sprouty4 KO mice. First, to investigate whether Sprouty4 deficiency had any effect on adult tumor angiogenesis, WT and Sprouty4 KO mice were injected subcutaneously with murine LLC cells. The sizes of LLC tumors were enhanced significantly in Sprouty4 KO mice compared to WT mice (Fig. 1a,b). Sections of tumors were immunostained with anti‐PECAM‐1/CD31 antibody, and the blood‐vessel area was quantified by measuring CD31‐positive area per unit area across entire tumor sections (Fig. 1c). This procedure revealed that Sprouty4 KO mice had significantly elevated intratumor CD31‐positive blood‐vessel areas compared to WT controls (Fig. 1c). Peritumor blood vessels of LLC tumors were also increased in Sprouty4 KO mice (Fig. 1b). Similar accelerated tumor growth and angiogenesis in Sprouty4 KO mice were observed when WT and Sprouty4 KO mice were injected subcutaneously with murine B16F10 melanoma cells (data not shown). Our data thus indicate that deficiency of Sprouty4 can enhance tumor growth and angiogenesis.
Figure 1.
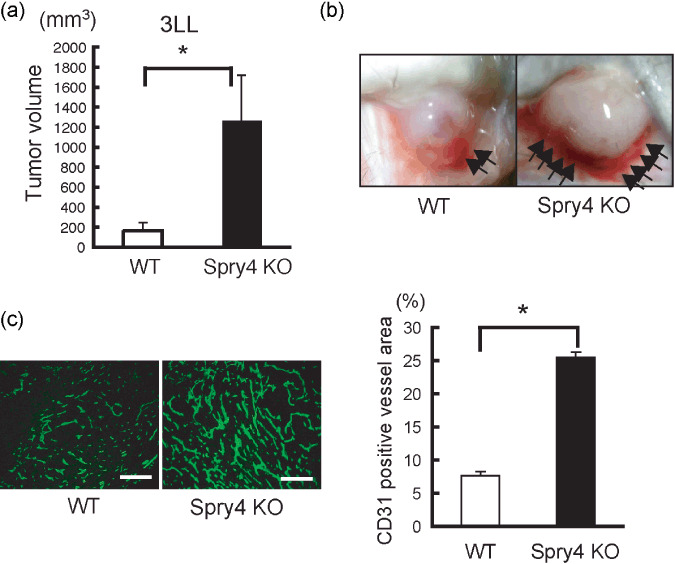
Sprouty4 knockout (KO) mice show accelerated tumor growth and angiogenesis. (a) Tumor volumes at 9 days after transplantation of Lewis lung carcinoma (LLC) cells (n = 5–6 mice/genotype). Data shown are means ± SEM. *P < 0.05. (b) Representative photos of LLC cells at 9 days post‐implantation in wild‐type (WT) and Sprouty4 KO mice (8 weeks old). The arrows indicate peritumor angiogenesis. (c) Anti‐PECAM‐1/CD31 Ab staining of sections of LLC cells. Intra‐tumor CD31‐positive vessel area (n = 5–6 mice/genotype) was quantified. Data shown are means ± SEM. *P < 0.05. Scale bars (c): 100 µm.
Sprouty4 deletion enhances VEGF‐A‐stimulated responses. Because VEGF‐A is thought to be the primary factor that stimulates tumor angiogenesis,( 25 ) we hypothesized that VEGF‐A‐stimulated responses in general might be elevated in Sprouty4 KO mice. To examine whether Sprouty4 deficiency accelerated VEGF‐A‐induced angiogenesis in vivo, basement membrane extract implants impregnated with VEGF‐A or PBS were administered subcutaneously to WT and Sprouty4 KO mice, and blood‐vessel infiltration of the implants was quantified. In contrast to PBS controls, VEGF‐A induced angiogenic responses in both WT and Sprouty4 KO mice, and this response was significantly greater in Sprouty4 KO mice than in WT mice (Fig. 2a,b).
Figure 2.
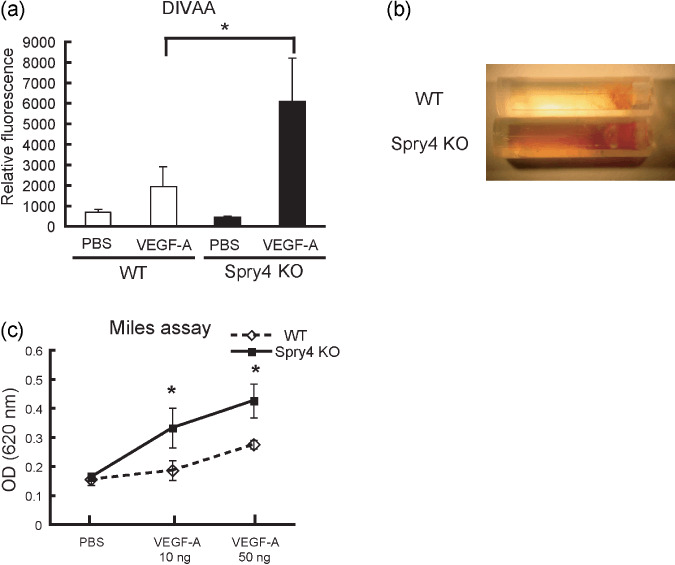
Vascular endothelial growth factor (VEGF)‐A‐stimulated response is enhanced by Sprouty4 deletion. (a) Directed in vivo angiogenesis assay (DIVAA). Angioreactors were pre‐filled with extracellular matrix with or without VEGF‐A (500 ng/mL) and heparin, and then implanted into the dorsal flanks of wild‐type (WT) and Sprouty4 knockout (KO) mice. Angiogenic responses were quantified by staining with FITC‐lectin. Results represent the mean fluorescence ± SEM. (n = 5–6). *P < 0.05. (b) After 15 days, the angioreactors were removed and photographed. (c) Vessel permeability was assessed by Miles assay. Data shown are means ± SEM. *P < 0.05.
Because the color of the implants containing VEGF‐A was more reddish in Sprouty4 KO mice than in WT mice (Fig. 2b), we hypothesized that vascular permeability by VEGF‐A was also enhanced in Sprouty4 KO mice. Thus we next examined the effect of Sprouty4 deficiency on microvessel leakiness in the dorsal skin of adult mice using a Miles vascular hyperpermeability assay. Intravenous injection of Evans blue dye was followed by intradermal injection of recombinant VEGF‐A at various doses. Spectrophotometric measurements of the extravasated dye showed that the increase of vascular permeability in response to VEGF‐A was greater in Sprouty4 KO mice than in WT mice (Fig. 2c).
Further analysis of VEGF‐A‐induced neovascularization responses was carried out using ex vivo aortic ring assays. WT and Sprouty4 KO aortic rings were embedded in Matrigel in the absence or presence of VEGF‐A, and the numbers of vascular sprouts per aortic ring were assessed. In the absence of angiogenic factor(s), such as VEGF‐A, bFGF, or S1P, vessel sprouts of both WT and Sprouty4 KO aortic rings were not observed in this condition (data not shown). In the presence of VEGF‐A, the total number of vessel sprouts was significantly higher for Sprouty4 KO samples than for WT controls (Fig. 3a).
Figure 3.
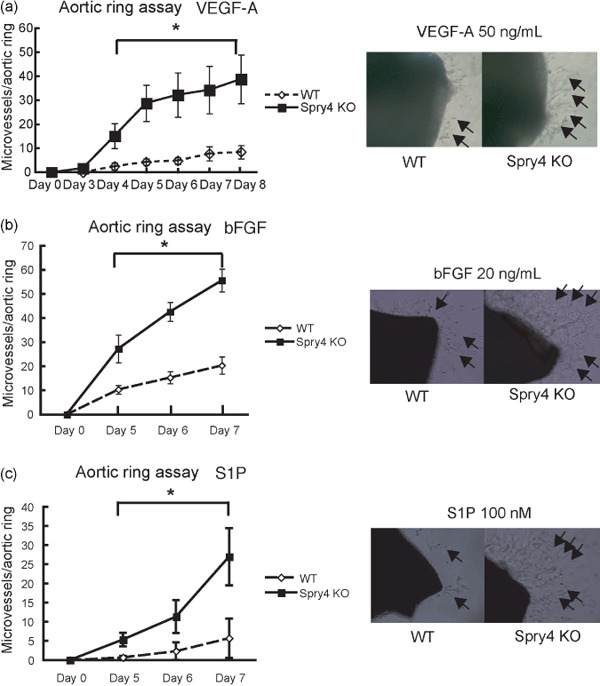
Sprouty4 deficiency accelerates angiogenesis induced by various angiogenic factors. (a–c) Left: In ex vivo aortic ring assays, microvessel numbers were counted from wild‐type (WT) and Sprouty4 knockout (KO) aortic rings in the presence of 50 ng/mL vascular endothelial growth factor (VEGF)‐A (a), 20 ng/mL basic fibroblast growth factor (bFGF) (b), or 100 nM sphingosine‐1‐phosphate (S1P) (c). Data shown are means ± SEM. (n = 3–5 aortic rings/group) *P < 0.05. Right: Low‐power light micrographs of a representative WT and Sprouty4 KO aortic ring grown in the presence of VEGF‐A (a), bFGF (b), or S1P (c). Arrows indicate vessels sprouting from aortic rings.
These data suggest that Sprouty4 is an important regulator of VEGF‐A‐induced angiogenesis and vascular permeability.
Sprouty4 deficiency also accelerates FGF‐ and S1P‐stimulated responses. Angiogenesis is promoted not only by VEGF‐A but also by various other factors including bFGF and S1P.( 16 , 17 , 18 ) We evaluated bFGF‐ and S1P‐induced neovascularization responses using ex vivo aortic ring assays. WT and Sprouty4 KO aortic rings were embedded in Matrigel in the presence of bFGF or S1P, and the numbers of vascular sprouts per aortic ring were counted. In the presence of bFGF or S1P, as in the presence of VEGF‐A, the total number of vessel sprouts was significantly higher for Sprouty4 KO samples than for WT controls (Fig. 3b,c).
These data suggest that Sprouty4 is a negative regulator of bFGF‐ and S1P‐induced angiogenesis as well as VEGF‐A‐induced angiogenesis.
Effect of Sprouty/Spred on VEGF‐A and VEGF‐C signaling. As demonstrated in our previous report, Spred1/Spred2 double KO (DKO) mice show defects of lymphatic vessels but not of blood vessels.( 8 ) Sprouty2 and Sprouty4 single‐deficient mice, in contrast, show defects of blood vessels but not of lymphatic vessels (Koji Taniguchi, unpublished data, 2009).
To uncover the reason for this, we investigated the functions of Sproutys and Spreds in the signal transduction cascades which are most important for angiogenesis and lymphangiogenesis, i.e. VEGF‐A/VEGF receptor (VEGFR)‐2 signaling and VEGF‐C/VEGFR‐3 signaling, respectively. According to our western blot analysis, while Spreds could inhibit both VEGF‐A‐ and VEGF‐C‐induced ERK activation in 293T cells which were expressing VEGFR‐2 or VEGFR‐3, Sproutys could inhibit only VEGF‐A‐induced ERK activation in that cell type, and could not suppress VEGF‐C‐induced ERK activation (Fig. 4a,b). In endothelial cells, VEGF‐A binds to VEGFR‐2 and activates ERK via the phospholipase C (PLC)‐γ/PKC pathway, where PKC phosphorylates Raf1, thereby activating ERK in a Ras‐independent manner.( 26 ) Sprouty4 binds to Raf1 via a highly conserved C terminal domain and inhibits VEGF‐A‐induced ERK activation in a Ras‐independent manner.( 7 ) It is unclear, however, whether VEGF‐C/VEGFR‐3‐induced ERK activation is Ras‐dependent or independent. To find out, we examined whether bisindolylmaleimide I (Bis I), a general PKC inhibitor, could inhibit VEGF‐A or VEGF‐C signaling in 293T cells which were expressing VEGFR‐2 or VEGFR‐3. While Bis I inhibited VEGF‐A‐induced ERK activation as expected, it could not inhibit VEGF‐C‐induced ERK activation, which suggests that VEGF‐C signaling is PKC‐independent (Fig. 4c).
Figure 4.
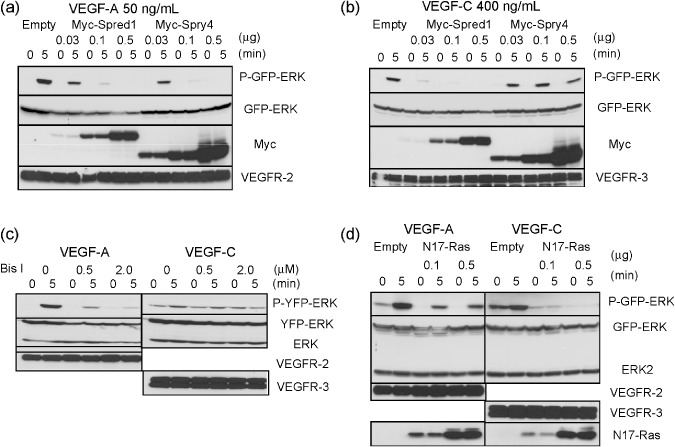
Effect of Sprouty/Spred overexpression on vascular endothelial growth factor (VEGF)‐A and VEGF‐C signaling. (a,b) Empty, Myc‐tagged Spred‐1 or Sprouty4 plasmids (0, 0.03, 0.1, 0.5 µg) were co‐transfected into 293T cells with VEGF receptor (VEGFR)‐2 (0.5 µg) (a) or VEGFR‐3 (0.5 µg) (b), and green fluorescent protein (GFP)‐ERK (0.5 µg). 293T cells were stimulated with 50 ng/mL VEGF‐A (a) or 400 ng/mL VEGF‐C (b). (c) 293T cells with VEGFR‐2 (0.5 µg) (left) or VEGFR‐3 (0.5 µg) (right), and yellow fluorescent protein (YFP)‐ERK (0.5 µg) were pre‐treated for 30 min with Bis I at the indicated concentration. 293T cells were stimulated with 50 ng/mL VEGF‐A (left) or 400 ng/mL VEGF‐C (right). (d) Empty or N17‐Ras plasmids (0, 0.1, 0.5 µg) were co‐transfected into 293T cells with VEGFR‐2 (0.5 µg) (left) or VEGFR‐3 (0.5 µg) (right), and GFP‐ERK (0.5 µg). 293T cells were stimulated with 50 ng/mL VEGF‐A (left) or 400 ng/mL VEGF‐C (right). (a–d) Cell extracts were immunoblotted with the indicated antibodies.
Next, we investigated whether N17‐Ras, a GDP‐bound dominant negative mutant, could inhibit VEGF‐A‐ or VEGF‐C‐induced ERK activation signaling in 293T cells which were expressing VEGFR‐2 or VEGFR‐3. While N17‐Ras did not inhibit VEGF‐A‐induced ERK activation, it did inhibit VEGF‐C‐induced ERK activation, which suggests that VEGF‐C signaling is Ras‐dependent (Fig. 4d). These results indicate that VEGF‐A/VEGFR‐2 signaling is PKC‐dependent and Ras‐independent, while VEGF‐C/VEGFR‐3 signaling is PKC‐independent and Ras‐dependent.
In previous studies, the function of Sprouty4 in various signaling pathways has been analyzed through overexpression experiments. Previously, we have shown that bFGF, which is an important angiogenic growth factor, induces stronger ERK activation in Sprouty4 KO MEFs than in WT MEFs.( 19 ) To determine the effect of the loss of Sprouty4 expression on VEGF‐A/VEGFR‐2 signaling, we stably expressed VEGFR‐2 in MEFs derived from WT and Sprouty4 KO embryos. VEGF‐A induced slightly stronger ERK and Akt activation in Sprouty4 KO MEFs than in WT MEFs (Fig. 5a). These data confirmed that Sprouty4 negatively regulated VEGF‐A mediated ERK activation, although the effect of the loss of Sprouty4 was not drastic in MEFs. In previous studies, we have shown functional redundancy between Sprouty2 and Sprouty4 in vitro as well as in vivo. ( 6 , 19 ) Thus, we hypothesized that such a slight difference between WT and Sprouty4 KO MEFs in VEGF‐A signaling was due to a compensation by Sprouty2. To examine this hypothesis, we used Sprouty2/Sprouty4 DKO MEFs expressing VEGFR‐2. As we expected, the differences in ERK and Akt activation were much more evident in Sprouty2/Sprouty4 DKO MEFs than in WT MEFs (Fig. 5b).
Figure 5.
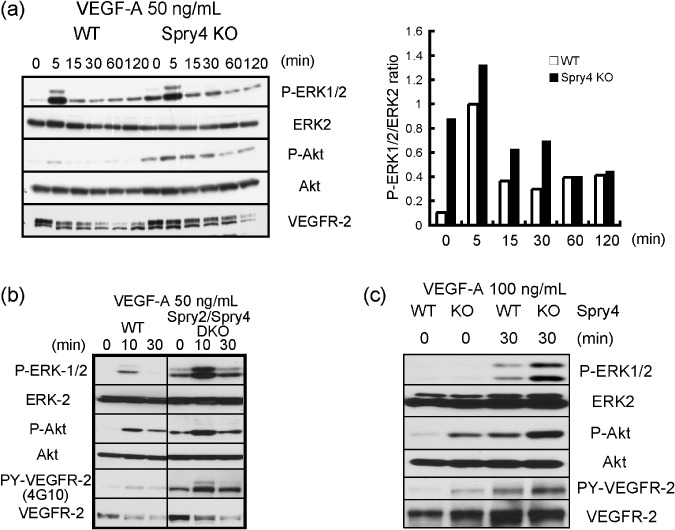
Influence of Sprouty4 deficiency on vascular endothelial growth factor (VEGF)‐A signaling. (a,b) Wild‐type (WT) and Sprouty4 knockout (KO) mouse embryonic fibroblasts (MEFs) (a) or WT and Sprouty2/Sprouty4 double KO (DKO) MEFs (b), which stably expressed VEGFR‐2, were stimulated with 50 ng/mL VEGF‐A. The relative ratio of the band intensity of phosphorylated ERK1/2 versus that of total ERK2 is shown in the right (a). (c) Inferior venae cavae of WT and Sprouty4 KO were stimulated with 100 ng/mL VEGF‐A. (a–c) Cell extracts were immunoblotted with the indicated antibodies.
To confirm the effect of the loss of Sprouty4 expression on VEGF‐A in blood vessels, we stimulated blood vessels derived from WT and Sprouty4 KO mice with VEGF‐A. VEGF‐A induced stronger ERK and Akt activation in Sprouty4 KO blood vessels than in WT blood vessels (Fig. 5c). This suggests that Sprouty4 is an important negative regulator of VEGF‐A signaling in blood vessels.
Inhibition of S1P signaling by Sprouty4. Recently, S1P has emerged as an important mediator of angiogenesis by way of the endothelial differentiation gene (Edg) receptor, a member of the GPCR family (S1PR 1–5).( 18 ) S1P binds to S1PR and activates ERK and Akt.( 18 ) Since Elk‐1 is phosphorylated and activated by ERK, it has been shown that ERK activation can be monitored by measuring the Elk‐1‐dependent transcription levels.( 7 ) Interestingly, our Elk‐1 reporter assay indicated that the overexpression of Sprouty4 has suppressive effects on S1P‐induced ERK activation in MEFs (Fig. 6a). Our western blot analysis indicated that S1P induced not only ERK and Akt activation but also protein kinase D (PKD, also known as protein kinase Cµ) activation (Fig. 6b,c), which has been shown to be activated via the VEGFR‐2/PLC‐γ/PKC pathway( 27 , 28 ) and inhibited by Sprouty412. The overexpression of Sprouty4 efficiently suppressed S1P‐induced PKD, ERK, and Akt activation in MEFs (Fig. 6b). Conversely, S1P induced stronger PKD and ERK activation in Sprouty4 KO MEFs than in WT MEFs, although Akt activation was almost similar in WT MEFs and Sprouty4 KO MEFs (Fig. 6c).
Figure 6.
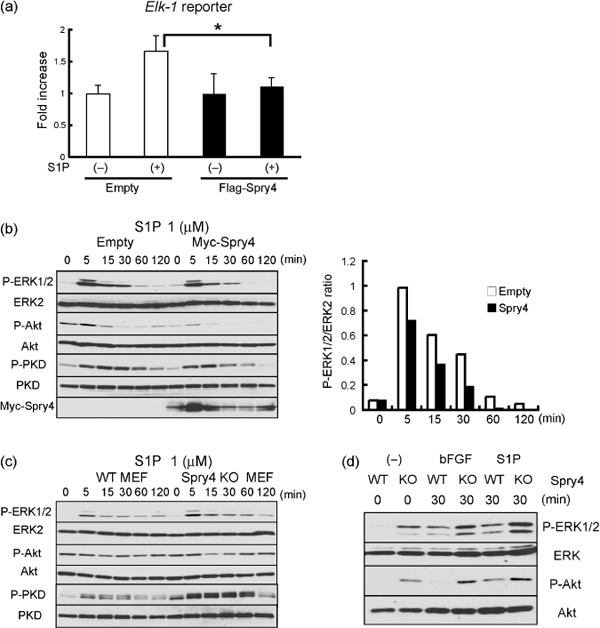
Negative regulation of sphingosine‐1‐phosphate (S1P) signaling by Sprouty4. (a) Flag‐tagged control or Sprouty plasmid (0.5 µg) was transfected into mouse embryonic fibroblasts (MEFs) with the Elk‐1 reporter. Cells were treated with (+) or without (–) 1 µM S1P for 6 h and then analyzed with the luciferase assay. The results are expressed as fold‐increase in luciferase activity with S1P relative to that without S1P. Data shown are means ± SEM. *P < 0.05. (b) MEFs stably expressing empty or Myc‐tagged Sprouty4 plasmid were stimulated with 1 µM S1P. The relative ratio of the band intensity of phosphorylated ERK1/2 versus that of total ERK2 is shown in the right. (c) Wild‐type (WT) and Sprouty4 knockout (KO) MEFs were stimulated with 1 µM S1P. (d) Inferior venae cavae of WT and Sprouty4 KO were stimulated with 50 ng/mL basic fibroblast growth factor (bFGF) or 1 µM S1P. (b–d) Cell extracts were immunoblotted with the indicated antibodies.
Finally, to confirm the effect of the loss of Sprouty4 expression on bFGF or S1P in blood vessels, we stimulated blood vessels derived from WT and Sprouty4 KO mice with bFGF or S1P. Both bFGF and S1P induced stronger ERK and Akt activation in Sprouty4 KO blood vessels than in WT blood vessels (Fig. 6d).
These results suggest that Sprouty4 deficiency accelerates a wide variety of angiogenic factor signaling pathways, including not only VEGF‐A and bFGF signals but also S1P signals.
Discussion
In this study we investigated the physiological function of Sprouty4 in angiogenesis by performing a knockout analysis of Sprouty4. Sprouty4 deficiency enhanced tumor growth and angiogenesis. We found that Sprouty4 suppressed bFGF‐ and S1P‐induced angiogenesis as well as VEGF‐A‐induced angiogenesis. Moreover, we showed that Sprouty4 is a negative regulator for VEGF‐A but not for VEGF‐C signaling in vitro, which is mostly because Sprouty4 does not suppress Ras‐dependent signals. We also found that Sprouty4 inhibits not only VEGF‐A and bFGF signals but also S1P signals. These findings suggest that Sprouty4 is an important negative regulator of various signals in angiogenesis, but not for lymphangiogenesis, and that it plays a key function in tumor environment.
The expression of Sprouty/Spred‐family genes is suppressed in breast, prostate, and liver cancers; accordingly, they are considered to be tumor suppressor genes.( 13 , 14 ) Recently, we reported that germline loss‐of‐function mutations in SPRED1 cause a neurofibromatosis 1‐like phenotype.( 29 ) This is the first report that mutations in the Sprouty/Spred family of genes are involved in human disease. Although there is no doubt that Sproutys play important roles in cancer cells themselves, it was unclear whether Sproutys are also important factors in the tumor microenvironment. The current study revealed that the loss of Sprouty4 enhances tumor angiogenesis and tumor growth, and that Sprouty4 plays an important role in tumor microenvironment.
The roles of Sprouty and Spred proteins during gastrulation in Xenopus tropicalis have been compared elsewhere.( 30 ) Spred proteins preferentially inhibit the Ras/ERK cascade that directs mesoderm formation, whereas Sprouty proteins block the Ca2+ and PKCδ signals required for morphogenetic movements during gastrulation. Thus, the expression of Sprouty or Spred genes at specific times during gastrulation might redirect FGF signals toward mesoderm formation or morphogenesis, respectively.( 30 ) VEGF‐A/VEGFR‐2 signaling is Ras‐independent and PLC‐γ/PKC‐dependent, while VEGF‐C/VEGFR‐3 signaling is Ras‐dependent and PKC‐independent (Fig. 4c,d and Ref. 26). In mammalian development, Sprouty2 and Sprouty4 are expressed mainly in blood endothelial cells, while Spred1 and Spred2 are expressed mainly in lymphatic endothelial cells.( 8 ) In addition to this difference in expression, Sproutys and Spreds may have different functions in endothelial cells. The effect of Sprouty deletion is more specific to VEGF‐A signaling than to VEGF‐C signaling, while the effect of Spred deletion is more specific to VEGF‐C signaling than to VEGF‐A signaling (Koji Taniguchi, unpublished data, 2009; Fig. 5a,b and Ref. 8). Indeed, Sprouty2 and Sprouty4 single‐deficient mice exhibit defects of blood vessels rather than lymphatic vessels, while Spred1/Spred2 DKO mice exhibit abnormal lymphatic vessel development and nearly normal blood vessel development (Koji Taniguchi, unpublished data, 2009; and Ref. 8). Drawing an analogy from the functional differences between Sproutys and Spreds that have been observed in Xenopus tropicalis, we propose that Sproutys inhibit PLC‐γ/PKC‐dependent VEGF‐A signaling and angiogenesis, while Spreds inhibit Ras‐dependent VEGF‐C signaling and lymphangiogenesis.
Angiogenesis is the more complex of these two processes, as it is triggered not only by VEGF‐A but also by bFGF, S1P, angiopoietins, and others.( 16 , 17 , 18 ) It is already known that Sproutys can inhibit various RTK signals,( 1 , 2 ) and we have previously shown that the loss of Sprouty expression results in hyperactivation of bFGF signaling.( 19 ) In addition, we have shown that Sprouty4 can inhibit not only RTK signals but also GPCR signals such as S1P and LPA (Fig. 6a–d and Ref. 12). The possibility that Sproutys regulate RTK and GPCR signals other than the VEGF‐A signal in angiogenesis in vivo needs to be investigated further.
In conclusion, Sprouty4 is a physiologically important negative regulator of angiogenic signals in tumors and may be useful as a new therapeutic target for cancer therapy.
Acknowledgments
We thank T. Yoshioka, K. Fukuse, M. Asakawa, and N. Shiino for technical assistance, and N. Soma for manuscript preparation. We also thank Dr H. Nishinakamura and Dr K. Hamada for technical advice. This study was supported by Grants‐in‐Aid for Scientific Research (S) and for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO); the Naito Foundation; and the Astellas Foundation for Research on Metabolic Disorders.
References
- 1. Kim HJ, Bar‐Sagi D. Modulation of signalling by Sprouty: a developing story. Nat Rev Mol Cell Biol 2004; 5: 441–50. [DOI] [PubMed] [Google Scholar]
- 2. Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative‐feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol 2006; 16: 45–54. [DOI] [PubMed] [Google Scholar]
- 3. Cabrita MA, Christofori G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis 2008; 11: 53–62. [DOI] [PubMed] [Google Scholar]
- 4. Wakioka T, Sasaki A, Kato R et al . Spred is a Sprouty‐related suppressor of Ras signalling. Nature 2001; 412: 647–51. [DOI] [PubMed] [Google Scholar]
- 5. Kato R, Nonami A, Taketomi T et al . Molecular cloning of mammalian Spred‐3 which suppresses tyrosine kinase‐mediated Erk activation. Biochem Biophys Res Commun 2003; 302: 767–72. [DOI] [PubMed] [Google Scholar]
- 6. Sasaki A, Taketomi T, Wakioka T, Kato R, Yoshimura A. Identification of a dominant negative mutant of Sprouty that potentiates fibroblast growth factor‐ but not epidermal growth factor‐induced ERK activation. J Biol Chem 2001; 276: 36804–8. [DOI] [PubMed] [Google Scholar]
- 7. Sasaki A, Taketomi T, Kato R et al . Mammalian Sprouty4 suppresses Ras‐independent ERK activation by binding to Raf1. Nat Cell Biol 2003; 5: 427–32. [DOI] [PubMed] [Google Scholar]
- 8. Taniguchi K, Kohno R, Ayada T et al . Spreds are essential for embryonic lymphangiogenesis by regulating vascular endothelial growth factor receptor 3 signaling. Mol Cell Biol 2007; 27: 4541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gross I, Bassit B, Benezra M, Licht JD. Mammalian sprouty proteins inhibit cell growth and differentiation by preventing ras activation. J Biol Chem 2001; 276: 46460–8. [DOI] [PubMed] [Google Scholar]
- 10. Hanafusa H, Torii S, Yasunaga T, Nishida E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol 2002; 4: 850–8. [DOI] [PubMed] [Google Scholar]
- 11. Yusoff P, Lao DH, Ong SH et al . Sprouty2 inhibits the Ras/MAP kinase pathway by inhibiting the activation of Raf. J Biol Chem 2002; 277: 3195–201. [DOI] [PubMed] [Google Scholar]
- 12. Ayada T, Taniguchi K, Okamoto F et al . Sprouty4 negatively regulates protein kinase C activation by inhibiting phosphatidylinositol 4,5‐biphosphate hydrolysis. Oncogene 2009; 28: 1076–88. [DOI] [PubMed] [Google Scholar]
- 13. Lo TL, Fong CW, Yusoff P et al . Sprouty and cancer: the first terms report. Cancer Lett 2006; 242: 141–50. [DOI] [PubMed] [Google Scholar]
- 14. Yoshida T, Hisamoto T, Akiba J et al . Spreds, inhibitors of the Ras/ERK signal transduction, are dysregulated in human hepatocellular carcinoma and linked to the malignant phenotype of tumors. Oncogene 2006; 25: 6056–66. [DOI] [PubMed] [Google Scholar]
- 15. Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer 2002; 2: 795–803. [DOI] [PubMed] [Google Scholar]
- 16. Hanahan D. Signaling vascular morphogenesis and maintenance. Science 1997; 277: 48–50. [DOI] [PubMed] [Google Scholar]
- 17. Shibuya M. Vascular endothelial growth factor‐dependent and – independent regulation of angiogenesis. BMB Rep 2008; 41: 278–86. [DOI] [PubMed] [Google Scholar]
- 18. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 2008; 9: 139–50. [DOI] [PubMed] [Google Scholar]
- 19. Taniguchi K, Ayada T, Ichiyama K et al . Sprouty2 and Sprouty4 are essential for embryonic morphogenesis and regulation of FGF signaling. Biochem Biophys Res Commun 2007; 352: 896–902. [DOI] [PubMed] [Google Scholar]
- 20. Nishinakamura H, Minoda Y, Saeki K et al . An RNA‐binding protein alphaCP‐1 is involved in the STAT3‐mediated suppression of NF‐kappaB transcriptional activity. Int Immunol 2007; 19: 609–19. [DOI] [PubMed] [Google Scholar]
- 21. Hamada K, Sasaki T, Koni PA et al . The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev 2005; 19: 2054–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tammela T, Zarkada G, Wallgard E et al . Blocking VEGFR‐3 suppresses angiogenic sprouting and vascular network formation. Nature 2008; 454: 656–60. [DOI] [PubMed] [Google Scholar]
- 23. Mamluk R, Klagsbrun M, Detmar M, Bielenberg DR. Soluble neuropilin targeted to the skin inhibits vascular permeability. Angiogenesis 2005; 8: 217–27. [DOI] [PubMed] [Google Scholar]
- 24. Reynolds LE, Wyder L, Lively JC et al . Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nature Med 2002; 8: 27–34. [DOI] [PubMed] [Google Scholar]
- 25. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003; 9: 669–76. [DOI] [PubMed] [Google Scholar]
- 26. Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk‐1 is essential for VEGF‐A‐dependent activation of PLC‐gamma and DNA synthesis in vascular endothelial cells. EMBO J 2001; 20: 2768–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wong C, Jin ZG. Protein kinase C‐dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. J Biol Chem 2005; 280: 33262–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qin L, Zeng H, Zhao D. Requirement of protein kinase D tyrosine phosphorylation for VEGF‐A165‐induced angiogenesis through its interaction and regulation of phospholipase Cgamma phosphorylation. J Biol Chem 2006; 281: 32550–8. [DOI] [PubMed] [Google Scholar]
- 29. Brems H, Chmara M, Sahbatou M et al . Germline loss‐of‐function mutations in SPRED1 cause a neurofibromatosis 1‐like phenotype. Nat Genet 2007; 39: 1120–6. [DOI] [PubMed] [Google Scholar]
- 30. Sivak JM, Petersen LF, Amaya E. FGF signal interpretation is directed by Sprouty and Spred proteins during mesoderm formation. Dev Cell 2005; 8: 689–701. [DOI] [PubMed] [Google Scholar]