

Abstract
Lipid metabolism is often elevated in cancer cells and plays an important role in their growth and malignancy. Acyl‐CoA synthetase (ACS), which converts long‐chain fatty acids to acyl‐CoA, is overexpressed in various types of cancer. However, the role of ACS in cancer remains unknown. Here, we found that ACS enzyme activity is required for cancer cell survival. Namely, the ACS inhibitor Triacsin c induced massive apoptosis in glioma cells while this cell death was completely suppressed by overexpression of ACSL5, the Triacsin c–resistant ACS isozyme, but not by overexpression of a catalytically inactive ACSL5 mutant. ACS inhibition by Triacsin c markedly potentiated the Bax‐induced intrinsic apoptotic pathway by promoting cytochrome c release and subsequent caspase activation. These effects were abrogated by ACSL5 overexpression. Correspondingly, ACS inhibition synergistically potentiated the glioma cell death induced by etoposide, a well‐known activator of apoptosis. Furthermore, in a nude mouse xenograft model, Triacsin c at a non‐toxic dose enhanced the antitumor efficacy of a low‐dose chemotherapy with etoposide. These results indicate that ACS is an apoptosis suppressor and that ACS inhibition could be a rational strategy to amplify the antitumor effect of etoposide. (Cancer Sci 2009)
Overexpression of lipogenic enzymes is a common characteristic of many cancers.( 1 ) In tumor cells, the supply of cellular fatty acids is highly dependent on de novo biosynthesis. FASN is a key enzyme that catalyzes the terminal step in the de novo synthesis of saturated fatty acids. Fatty acid synthase (FASN) is overexpressed in a wide variety of human epithelial cancer cells and plays a critical role in tumor growth and survival.( 2 ) In addition to FASN, several other enzymes involved in lipid metabolism have recently been shown to be involved in tumor growth and malignancy.( 3 , 4 ) These observations support the notion that elevated lipid metabolism could be a rational target for cancer treatment.
Acyl‐CoA synthetases (ACS) are enzymes that act downstream of FASN and convert long‐chain fatty acids to acyl‐CoA.( 5 ) This reaction is a crucial step in several lipid metabolism pathways, including phospholipid biosynthesis, lipid modification of cellular proteins, and β‐oxidation. In mammals, five ACS isozymes have been identified. Previous reports have indicated that several ACS isozymes, such as ACSL4 and ACSL5, are overexpressed in cancer cells.( 6 , 7 , 8 , 9 ) We recently identified Triacsin c, a potent inhibitor of ACS, as an agent that shows selective cytotoxicity to malignant cancer cells.( 10 ) These observations suggest that ACS could play a predominant role in cancer cell survival. Still, however, it remains unclear how ACS regulates cell death and whether the ACS inhibition could affect the chemosensitivity of cancer.
Activation of apoptotic pathways is a key mechanism by which anticancer agents kill tumor cells.( 11 ) Chemotherapeutic agents induce apoptosis through the intrinsic mitochondria‐dependent pathway that is activated mainly by the release of cytochrome c from the mitochondria. The released cytochrome c interacts with Apaf‐1 to form an Apaf‐1 multimer (apoptosome), which in turn activates caspase‐9 and the downstream caspases that participate in the execution phase of apoptosis.( 12 ) The activated caspases cleave many kinds of substrates, leading to cell death. Several factors, e.g. Bcl‐2 family members, suppress apoptosis on the mitochondria.( 13 ) Because these antiapoptotic factors are frequently overexpressed in cancer cells and are involved in chemotherapy resistance, trials have been undertaken to identify their specific inhibitors, some of which are now being tested clinically.( 14 , 15 , 16 )
In the present study, we examined the antiapoptotic role of ACS in cancer cells. We found that ACS enzyme activity was essential for glioma cell survival. Moreover, we demonstrate the combinational effect of ACS inhibition with etoposide.
Materials and Methods
Chemicals. Triacsin c was purchased from Sigma (St. Louis, MO, USA). Etoposide was purchased from Bristol‐Myers Squibb (New York, NY, USA). Caspase inhibitor Z‐VAD‐fmk and caspase substrate peptide DEVD‐MCA were purchased from Peptide Institute (Osaka, Japan).
Cell culture, detection of apoptotic cells, and measurement of cell growth. Human glioma SF268 and U251 cells were cultured in RPMI‐1640 supplemented with 10% heat‐inactivated fetal bovine serum and 100 µg/mL kanamycin in a humidified atmosphere of 5% CO2 and 95% air. Drug sensitivity was evaluated using the MTS method.( 17 ) In brief, we used a CellTiter 96AQueous One Solution Cell Proliferation Assay Kit (Promega, Tokyo, Japan). Twenty microliters of MTS and phenazine ethosulfate solution were added to the drug‐treated cells (100 µL/well in 96‐well plates) and the mixture was incubated at 37°C for 30–60 min. For quantitation of relative cell number, OD490 was measured using a microplate reader. To detect apoptotic cells, cell nuclei were stained with Hoechst 33342.( 18 ) Apoptotic cells were evaluated using such characteristic nuclear features as chromatin condensation and nuclear fragmentation.
Caspase and apoptosome assays. Cell lysates were prepared and caspase activity was measured using DEVD‐MCA as a substrate, as described previously.( 17 ) To estimate Bax‐induced caspase activation, we transfected cells with pCGBL‐HA‐Bax and pGVC, a luciferase‐expressing construct driven by a SV40 promoter and enhancer.( 18 ) After transfection and subsequent drug treatment, cell lysates were prepared and caspase activity was measured. To monitor the transfection efficiency, we also measured luciferase activities in cell lysates using the Luciferase Assay System (Promega). The caspase activity was normalized by the luciferase activity to estimate relative Bax‐dependent caspase activation. To measure apoptosome activity, cytosolic extracts were prepared as described previously( 17 ) and incubated with 10 µM cytochrome c and 1 mM dATP for 20–40 min. After the incubation, caspase activity was measured.
Subcellular fractionation and western blot analysis. Subcellular fractions were obtained by using a ProteoExtract Subcellular Proteome Extraction kit (Calbiochem, San Diego, CA, USA). Each extract derived from the same number of the cells was subjected to SDS‐PAGE. To analyze apoptosis regulators’ expression, we washed cells in ice‐cold phosphate‐buffered saline (PBS) and lyzed them in TNE buffer (10 mM Tris‐HCl [pH 7.8], 1% Nonidet P‐40, 150 mM NaCl, 1 mM EDTA, and 10 µg/mL aprotinin) on ice for 30 min. After centrifugation at 12 000 g for 10 min at 4°C, the supernatant (TNE lysate) was subjected to SDS‐PAGE. For detection of cytochrome c release from the mitochondria, cytosolic extracts were prepared as described previously( 10 ) and were subjected to SDS‐PAGE. Western blot analysis was performed as described previously( 19 ) with the following primary antibodies: mouse anti‐FLAG (M2; Sigma), mouse anti‐PARP (BD Pharmingen, San Diego, CA, USA), rabbit anti‐EGFR (Cell Signaling Technology, Beverly, MA, USA), mouse anti‐voltage‐dependent anion channel (VDAC) (Ab‐4; Calbiochem), mouse anti‐cytochrome c (7H8.2C12; BD Pharmingen), rabbit anti‐HA (Y‐11; Santa Cruz Biotechnology, Santa Cruz, CA, USA) mouse anti‐α‐tubulin (B5‐1‐2; Sigma), mouse anti‐Bcl‐2 (BD Pharmingen), mouse anti‐Bcl‐XL (BD Pharmingen), or mouse anti‐Bax (BD Pharmingen).
Immunofluorescence staining. Cells were fixed with 2% paraformaldehyde/PBS and permeabilized with 0.5% NP40/PBS. The fixed cells were blocked in PBS containing 1% bovine serum albumin and incubated with rabbit anti‐FLAG (Sigma) and mouse anti‐cytochrome c (6H2.B4; BD Pharmingen) antibodies. These primary antibodies were detected with Rhodamine‐conjugated anti‐rabbit Ig and FITC‐conjugated anti‐mouse Ig, respectively. DNA was stained with 0.2 µg/mL of DAPI. Images were acquired using an Olympus IX‐71 microscope with a DP70 digital camera and Lumina Vision software (Mitani Corporation, Tokyo, Japan).
Vector construction. For the expression of human ACSL5 and its inactive mutant ACSL5‐MT, pHa‐ACSL5‐FLAG‐IRES‐DHFR and pHa‐ACSL5‐MT‐FLAG‐IRES‐DHFR were constructed as described previously.( 10 , 20 ) To construct an ACSL5 mutant, delta L, that lacked an amino‐terminus sequence (amino acid 2–41), we amplified, by polymerase chain reaction (PCR), the ACSL5 fragment that lacked amino acids 2–41 and subcloned it into the pHa vector to generate pHa‐ACSL5 (delta L)‐FLAG‐IRES‐DHFR. The full‐length cDNA for human Bax was amplified by PCR and subcloned into a pCGBL mammalian expression vector with an N‐terminal HA epitope tag to generate pCGBL‐HA‐Bax.
Transient transfection and retroviral infection. Transient transfection of pCGBL‐HA‐Bax was performed using Lipofectamine 2000 (Invitrogen, San Diego, CA, USA). For retroviral gene transfer, PA317 cells were transfected with pHa‐IRES‐DHFR (mock), pHa‐ACSL5‐FLAG‐IRES‐DHFR, or ACSL5 mutant constructs, selected with methotrexate (MTX), and culture supernatants of the MTX‐resistant PA317 cells were added to SF268 cells, as described previously.( 10 ) After retroviral infection and subsequent methotrexate selection (100 ng/mL), stably transduced cells were established.
Acyl‐CoA synthetase (ACS) enzyme assay. Total cell lysates were prepared by homogenizing cells in buffer A, and the ACS activity was measured, as described previously.( 10 ) In brief, the assay mixture contained 1.2 µM MgCl2, 5 µM ATP, 3 µM potassium fluoride, 0.1 µM coenzyme A, 3 µM 2‐mercaptoethanol, and 0.03 µM palmitic acid with 0.1 µCi of [14C]‐palmitic acid in a total volume of 20 µL. The reaction was initiated by adding 10 µL of cell lysates at 37°C and terminated after 1 h by adding 270 µL of isopropanol‐heptane‐aqueous 1 M H2SO4 (40:10:1 by volume). Then 180 µL of heptane and 120 µL of water were added and the upper layer was discarded. The lower layer was washed twice with 200 µL of heptane containing 15 mM palmitic acid and the radioactivity in 100 µL of the sample was counted in 1 mL of ACS II (Amersham, Tokyo, Japan).
Mouse xenograft therapeutic model. U251 cells (5 × 106 cells/mouse) were implanted subcutaneously in the right flanks of 6‐week‐old CAnN.Cg‐Foxn1nµ/CrlCrlj nude mice (Charles River Laboratories Japan, Kanagawa, Japan). Therapeutic experiments (five mice per group) were started approximately 20 days after implant when tumors reached 90–170 mm3, as measured with calipers (day 0). Etoposide (12 mg/kg/day) was administered i.v. on days 0, 1, and 2. Triacsin c (4 mg/kg/day) was administered by intratumoral injection in 40 µL of saline on days 0, 1, and 2. Control mice received the same volume of saline as the experimental mice. The length (L) and width (W) of the tumor mass were measured, and the tumor volume (TV) was calculated as: TV = (L × W2)/2. The difference in the growth rate between etoposide‐treated and the etoposide + Triacsin c–treated groups was tested by an autoregressive random errors model( 21 ) using statistical software R( 22 ) (version 2.7.1) with the agce package (version 1.2; http://www.R‐project.org). All animal procedures were performed in the animal experiment room of the Japanese Foundation for Cancer Research (JFCR) using protocols approved by the JFCR Animal Care and Use Committee.
Results
Acyl‐CoA synthetase (ACS) activity contributes to cancer cell survival. We have shown that Triacsin c, a specific inhibitor of ACS, induces cell death preferentially in tumor cells, but is less toxic to non‐cancerous cells.( 10 ) To confirm the requirement for ACS enzyme activity in cancer cell survival, we established cell lines that stably overexpressed either ACSL5 or its inactive mutant (ACSL5‐MT), since ACSL5 is a Triacsin c–resistant isozyme among five ACS members,( 23 ) and we postulated that the overexpression of ACSL5 could effectively reverse the inhibition of ACS activity by Triacsin c. We chose glioma cell lines for this experiment since ACSL5 is frequently overexpressed in malignant glioma.( 7 ) When retrovirally transduced in human glioma SF268 cells, wild‐type ACSL5 and the ACSL5‐MT proteins were stably expressed (Fig. 1a, inset). On the other hand, the ACS activity was exclusively increased in the wild‐type ACSL5‐expressed cells but not in ACSL5‐MT‐expressed cells under both Triacsin c–treated and –untreated conditions (Fig. 1a). Triacsin c inhibition of ACS activity was eliminated by wild‐type ACSL5 but not by ACSL5‐MT. These data indicate that ACSL5‐MT acts as an inactive mutant and that these systems work to determine the role of ACS activity in cancer cell survival. We examined the effect of wild‐type ACSL5 and ACSL5‐MT on Triacsin c–induced SF268 cell death. As shown in Fig. 1(b,c), Triacsin c (3 µM) strongly induced apoptosis and activated caspases in mock cells. Wild‐type ACSL5, which retained ACS activity in Triacsin c–treated cells, completely inhibited the caspase activation and apoptotic cell death, while ACSL5‐MT did not (Fig. 1b–d). Triacsin c–induced cell death would depend on caspase activation because a caspase inhibitor, Z‐VAD‐fmk, restored the viable cell number (Fig. 1d). These results indicate that ACS plays an essential role in SF268 cell survival, and that ACS inhibition induces apoptosis of the cells.
Figure 1.
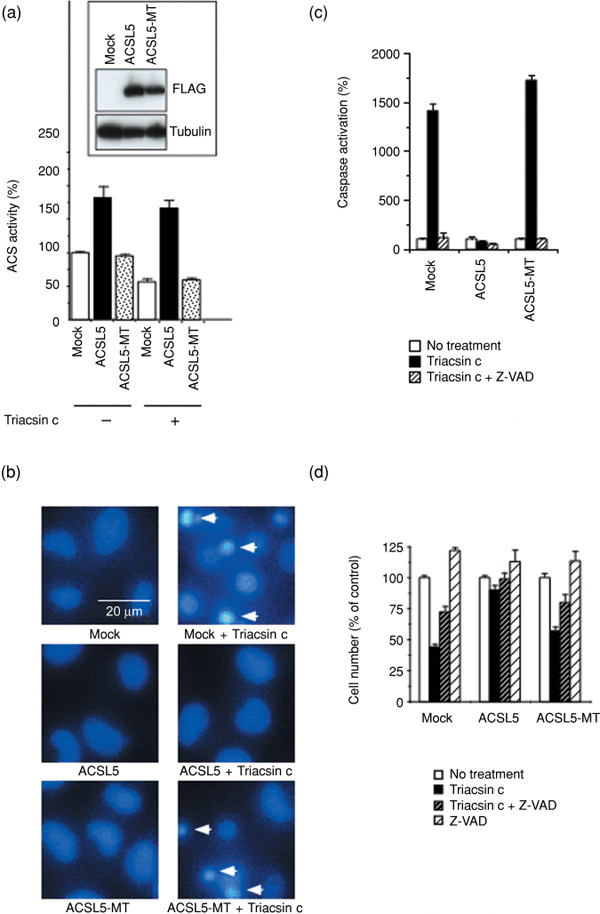
Acyl‐CoA synthetase (ACS) catalytic activity requirement for glioma cell survival. (a) Effect of 48‐h Triacsin c treatment (3 µM) on ACS activity in mock‐, ACSL5‐, and ACSL5‐MT–transduced SF268 cells. ACS activity was measured as described in ‘Materials and Methods’. Inset, The expressions of ACSL5 and ACSL5‐MT were examined by western blot with anti‐FLAG M2 antibody. The expression of α‐tubulin was analyzed as a loading control. (b) Apoptosis induction after Triacsin c treatment. Cells were treated as in (a). The cell nuclei were stained with Hoechst 33342. Induction of apoptosis in each cell was evaluated using the characteristic nuclear features of apoptosis, such as chromatin condensation and nuclear fragmentation. Arrows indicate apoptotic cells. (c) Caspase activation after Triacsin c treatment. Cells were left untreated or treated with 3‐µM Triacsin c in the absence or the presence of 50‐µM caspase inhibitor, Z‐Val‐Ala‐Asp(OMe)‐CH2F (Z‐VAD), for 30 h. Caspase activity was measured as described in ‘Materials and Methods’. (d) Cell number after Triacsin c treatment. Cells were treated as in (c) for 48 h and relative cell number was measured using the 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) method, as described in ‘Materials and Methods’. Data are mean values of three independent experiments. Error bars show standard deviations.
Role of subcellular ACS in cancer cell survival. As shown above, ACSL5 overexpression compensated for the decrease of ACS activity in Triacsin c–treated cells and suppressed Triacsin c–induced cell death. Since rat ACSL5 is expressed predominantly on the mitochondrial membrane,( 24 ) we speculated that human ACSL5 could also be expressed on the organelle and would compensate for Triacsin c–induced loss of the ACS activity, leading to cell death suppression. To test this hypothesis, we first examined the subcellular localization of human ACSL5 by immunofluorescence staining (Fig. 2a). Consistent with the previous report of rat ACSL5, human ACSL5 was colocalized with cytochrome c, a marker protein of mitochondria. Furthermore, the patterns of immunofluorescence staining suggested that the human ACSL5 was localized not only to the mitochondria but could also exist at the perinuclear region and, to a lesser extent, in the nucleoplasm (Fig. 2a). To determine the localization of human ACSL5 further, we performed subcellular fractionation of cellular proteins. As shown in Fig. 2(b), ACSL5 was not only present in the organelle/membrane fraction (O/M), which contained mitochondria, but also in the nuclear fraction (N). These data confirmed that ACSL5 was localized in the nuclei of cancer cells as well as to the mitochondria. To clarify the importance of subcellular ACS in SF268 cell survival, we generated deletion mutants of ACSL5 and examined their subcellular localizations. We found that the mutant delta L (Fig. 2b) exclusively lost its nuclear localization, although the amino‐terminus sequence that the mutant lacked did not contain any nuclear localization sequence (data not shown, searched by the PSORT II prediction algorithm at http://psort.ims.u‐tokyo.ac.jp/form2.html). On the other hand, this mutant still retained the organelle localization and ACS activity. When retrovirally transduced in SF268 cells, both wild‐type ACSL5 and the delta L proteins were stably expressed, and, as in the wild‐type ACSL5‐expressed cells, ACS activity clearly increased in the delta L–expressed cells (Suppl. Fig. 1). These results indicated that the delta L was catalytically active. While the wild‐type ACSL5 was detected in both the organelle/membrane and the nuclear fractions, the delta L mutant lost its nuclear localization and instead was included in the organelle/membrane and the cytosol (C) fractions (Fig. 2b). Immunofluorescence staining further revealed that the delta L was still localized to the mitochondria (Suppl. Fig. 2). These data indicate that the N‐terminal region deleted in the delta L mutant is required for ACSL5 targeting to the nuclei but not to the mitochondria. We compared the effect of ACSL5 and the delta L on Triacsin c–induced cell death and found that, as well as wild‐type ACSL5, the delta L mutant efficiently suppressed caspase activation and cell death caused by Triacsin c (Fig. 2c,d). These results indicate that the nuclear ACS could not be required for survival of SF268 cells.
Figure 2.
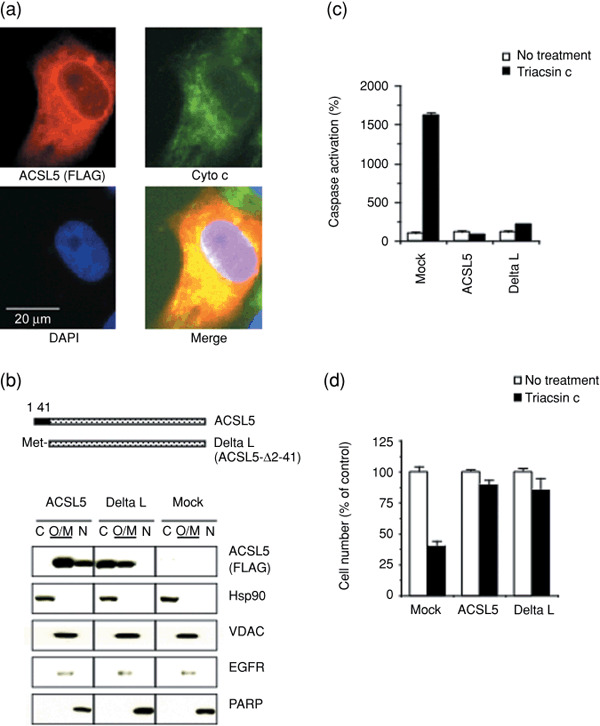
Organelle‐localized acyl‐CoA synthetase (ACS) is critical for cancer cell survival. (a) Localization of human ACSL5 in SF268 cells. ACSL5 (FLAG) and the mitochondria marker cytochrome c (Cyto c) were detected by indirect immunofluorescence stain of ACSL5‐transduced SF268 cells with anti‐FLAG M2 (red) and anti‐cytochrome c (green) antibodies, respectively. DAPI staining of DNA is shown in blue. (b) Subcellular fractionation of ACSL5 and its N‐terminal deletion mutant (delta L) in SF268 cells. Cytoplasmic (C), organelle/membrane (O/M), and nuclear (N) fractions were prepared, and subjected to western blot analysis with the indicated primary antibodies. Blots with anti‐FLAG M2 antibody (FLAG) indicate the expression of ACLS5. Blots with anti‐Heat shock protein (Hsp)‐90, voltage‐dependent anion channel (VDAC), epidermal growth factor receptor (EGFR), and poly(ADP‐ribose) polymerase (PARP) antibodies demonstrate the purity of their respective fractions. (c) Caspase activation in SF268/ACSL5 and SF268/delta L cells after Triacsin c treatment. Mock‐, ACSL5‐, and delta L–transduced SF268 cells were left untreated or treated with 3‐µM Triacsin c for 30 h. Caspase activity was measured as described in ‘Materials and Methods’. (d) Cell number after Triacsin c treatment. Cells were treated as in (c) for 48 h and relative cell number was measured using the 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) method. In (c) and (d), data are mean values of three independent experiments, and error bars show standard deviations.
Enhancement of the intrinsic apoptosis pathway by ACS inhibition. To determine the role of ACS in glioma cell survival, we examined whether ACS could act as a repressor against mitochondria‐dependent apoptosis. Mitochondrial apoptosis inducers, such as Bax protein, can directly activate this intrinsic apoptosis pathway.( 25 , 26 ) Therefore, we tested the effect of ACS inhibition on Bax‐induced apoptosis. As shown in Figure 3(a), Bax activated caspases in a dose‐dependent manner. Sublethal dose of Triacsin c potentiated this Bax‐induced caspase activation. Moreover, this effect was strongly suppressed by compensation of ACS activity by ACSL5 overexpression. These results indicate the antagonistic role of ACS in the mitochondrial apoptosis pathway, and ACS inhibition potentiates this pathway. To clarify the molecular mechanisms further, we examined the effect of ACS inhibition on Bax‐induced cytochrome c release from the mitochondria to cytoplasm. When cells were expressed with Bax alone, cytochrome c release to the cytosol was below detection levels in western blot analysis. On the other hand, we found that the cytochrome c release was clearly enhanced by Triacsin c, which was suppressed by ACSL5 expression (Fig. 3b). By contrast, Triacsin c treatment or ACSL5 overexpression did not affect the in vitro apoptosome‐dependent caspase activation that was initiated when exogenous cytochrome c was added to the cytosolic extracts (Fig. 3c). These results indicate that ACS antagonizes the intrinsic apoptosis pathway at the point of cytochrome c release and upstream of subsequent apoptosome activation. On the mitochondria, the cytochrome c release is regulated by Bcl‐2 family members.( 13 , 15 ) So next, we examined the effect of ACS inhibition on the protein expression of the apoptosis regulators in mock and ACSL5‐overexpressed cells. We found that ACS inhibition did not affect protein levels of the mitochondrial Bcl‐2 family members, Bcl‐2, Bcl‐XL, and Bax (Suppl. Fig. 3), suggesting the involvement of other factors in the apoptosis enhancement.
Figure 3.
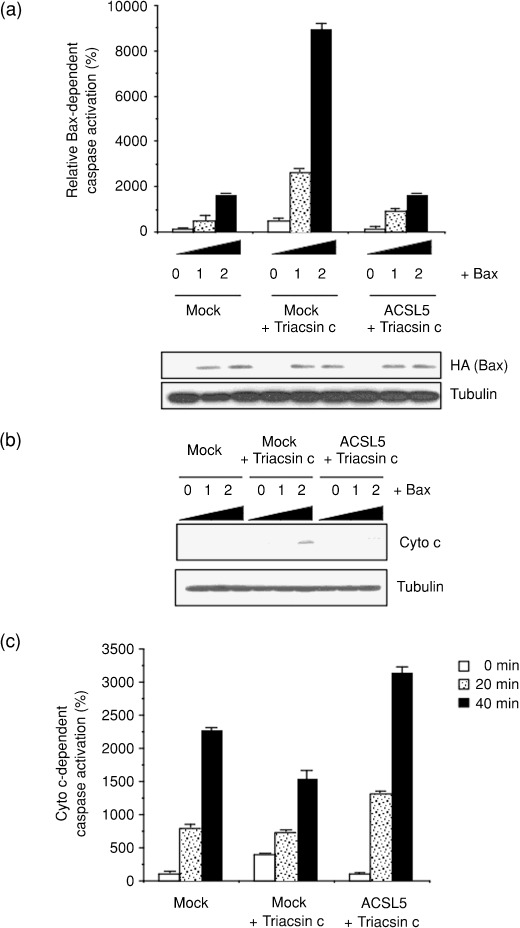
Potentiating the mitochondrial apoptosis pathway by acyl‐CoA synthetase (ACS) inhibition. (a) To estimate Bax‐induced caspase activation, mock‐ and ACSL5‐transduced SF268 cells were seeded in six‐well plates and transiently transfected with pCGBL‐HA‐Bax (0, 0.1, and 0.2 µg/well) and pGVC, a luciferase‐expressing construct (0.4 µg/well). At 6 h after transfection, cells were left untreated or were treated with 1 µM Triacsin c for an additional 24 h. Each cell lysate was prepared and caspase activity measured as described in ‘Materials and Methods’. The expression of Bax (HA) was examined by western blot. The expression of α‐tubulin was analyzed as a loading control. (b) Cells were transiently transfected with the Bax plasmid vector and then were treated with Triacsin c as in (a). Cytochrome c release from the mitochondria to cytoplasm was monitored by western blot analysis. (c) Mock‐ and ACSL5‐transduced SF268 cells were left untreated or treated with 1 µM Triacsin c for 24 h. Cytosolic extracts were prepared and incubated with 10 µM cytochrome c and 1 mM dATP for 0–40 min. After the incubation, caspase activity was measured, as described in ‘Materials and Methods’. In (a) and (c), data are mean values of three independent experiments. Error bars show standard deviations.
Potentiation of etoposide‐induced cell death by ACS inhibition. Because ACS inhibition potentiated activation of the intrinsic apoptosis pathway, we examined whether ACS inhibition also enhances chemotherapeutic agent–induced death of cancer cells. We initially examined combinations of several chemotherapeutic drugs with Triacsin c, and determined the optical conditions for maximal efficacy. As a result, we found the synergistic effect of ACS inhibition with etoposide. As a result, we found the combinational effect of Triacsin c with etoposide, which induces apoptosis through the mitochondria‐mediated pathway.( 27 , 28 ) When SF268/mock cells were treated with either etoposide (0.3 µg/mL, 48 h) or a sublethal dose of Triacsin c (1 µM, 48 h), caspases were activated 3.5‐fold or 13‐fold, respectively, compared to non‐treated cells (Fig. 4a). Co‐treatment with etopiside and Triacsin c significantly enhanced the caspase activation (33‐fold). This synergism in caspase activation by etoposide and Triacsin c was quenched by ACSL5 overexpression. Correspondingly, the sublethal dose of Triacsin c (1 µM) significantly potentiated etoposide‐induced apoptosis (Fig. 4b) and the loss of cell numbers (Fig. 4c). Again, these effects of Triacsin c were canceled by ACSL5 overexpression. These results indicate that inhibiting ACS activity would be a rational strategy to potentiate etoposide‐induced cytotoxicity.
Figure 4.
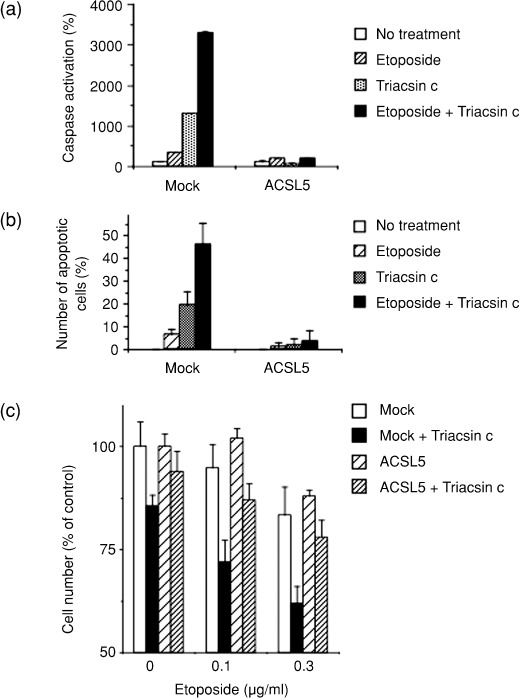
Potentiating etoposide‐induced cell death by acyl‐CoA synthetase (ACS) inhibition. (a) Enhanced activation of caspase by etoposide in combination with Triacsin c. Mock‐ and ACSL5‐transduced SF268 cells were left untreated or were treated with 0.3 µg/mL of etoposide, 1 µM Triacsin c or 0.3 µg/mL of etoposide and 1 µM Triacsin c for 48 h. Caspase activity was measured as described in ‘Materials and Methods’. (b) Potentiation of apoptosis by etoposide in combination with Triacsin c. Mock‐ and ACSL5‐transduced SF268 cells were treated as in (a), and apoptotic cells were evaluated and counted. (c) Effect of ACS inhibition on etoposide‐induced cytotoxicity. Mock‐ and ACSL5‐transduced SF268 cells were left untreated or were treated with the indicated concentrations of etoposide in the absence or presence of 1 µM Triacsin c for 48 h. Cell viability was measured, using the 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) method. Data are mean values of three independent experiments. Error bars show standard deviations.
To test whether ACS inhibition could increase the antitumor efficacy of etoposide, we developed a tumor xenograft model in nude mice. Because SF268 cells could not form stable tumors in nude mice (our unpublished observation), we chose another implantable glioma cell line, U251, for this in vivo study. Mice were treated with saline (control), etoposide, Triacsin c, or etoposide in combination with Triacsin c. Under the limited dose conditions in Figure 5(a), etoposide or Triacsin c alone did not show apparent antitumor effects. Strikingly, however, cotreatment with etoposide and Triacsin c significantly retarded tumor growth. During the treatment, no toxic death or significant body weight change was observed (Fig. 5b). These results indicate that the ACS inhibition potentiates the antitumor effect of etoposide in vivo with minimal side effects in the mice.
Figure 5.
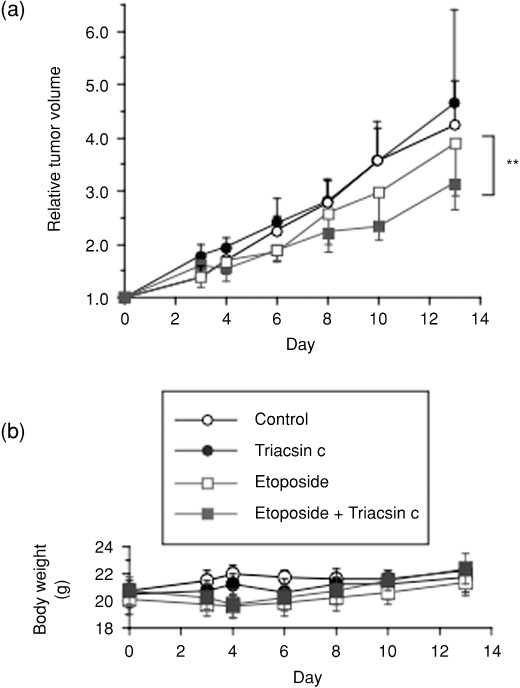
Triacsin c enhances the efficacy of etoposide in vivo. Therapeutic experiments (five mice per group) were started (day 0) when U251 tumors reached 90–170 mm3. Etoposide (12 mg/kg/day) was administrated i.v. on days 0, 1, and 2. Triacsin c (4 mg/kg/day) was administrated by intratumoral injection in 40 µL of saline on days 0, 1, and 2. Control mice received the same volume of saline. Relative tumor volumes and body weight changes of the mice are shown in (a) and (b), respectively. Data are mean values for five mice, and error bars show standard deviations. Statistical evaluations were performed as described in ‘Materials and Methods’. **P < 0.01.
Discussion
ACS catalyzes a critical step in both the anabolic and catabolic pathways of fatty acid metabolism. In our present study, we showed that ACS enzyme activity is a critical factor for cancer cell survival and apoptosis inhibition. When cancer develops, excessive mitogenic signals, due to oncogenic activation or uncontrolled cell cycle progression, are coupled with constitutive activation of the intrinsic apoptosis machinery.( 29 ) Under these conditions, expressions of antiapoptotic factors are requisite for cancer cell survival, and these factors could be the cells’ Achilles heel. In fact, agents or strategies that suppress antiapoptotic proteins or directly activate the mitochondria‐dependent apoptosis pathway (apoptosome pathway) selectively induce tumor cell death or potentiate the chemosensitivity of tumor cells.( 13 , 14 , 16 , 30 , 31 ) Our present results suggest that ACS could be one such factor essential for cancer cell survival and whose inhibition induces tumor‐selective cell death.
ACS inhibition potentiated Bax‐ and etoposide‐induced apoptosis (3, 4). Consistently, the overexpression of ACSL5 suppressed apoptosis induced by etoposide (Fig. 4a,b). These data indicate that ACS could act as an apoptosis inhibitor in tumor cells.
We have shown that ACSL5 nearly completely suppressed Triacsin c–induced cell death. Since small molecules could have off‐targets, we cannot exclude the possibility of an ACS‐unrelated, off‐target effect of Triacsin c. However, such effect would be minor, if any, in our experimental settings, because the catalytically inactive ACSL5 mutant did not suppress the Triacsin c–induced cell death. Thus, Triacsin c could induce apoptosis mainly through ACS inhibition. Meanwhile, because ACS have five isoforms, it is difficult to perform gene‐knockdown experiments or to examine the effect of dominant‐negative mutants.
ACS shows various subcellular localizations.( 24 ) The ACS inhibitor–induced apoptosis was suppressed by ACSL5, which localizes on the mitochondria and in the nuclei. Moreover, the ACSL5 mutant lacking nuclear localization still inhibited the ACS inhibitor–induced cell death. These data indicate that the nuclear ACS is not required for cancer cell survival and suggest that the organelle ACS could be a critical factor for the survival. To be sure of the role of the organelle ACS, however, further studies with an ACSL5 mutant that lacks the organelle localization could be required. Given the fact that ACSL5 is the only known ACS isozyme that localizes to the mitochondria and is frequently overexpressed in human cancers, it is suggested that ACSL5 could play an important role in tumorigenesis or malignant transformation by means of inhibiting mitochondrial apoptosis pathway. Meanwhile, we should take into account additional unknown mitochondrial ACS enzymes that are involved in this mechanism, since a Triacsin c–sensitive ACS activity is known to exist on rat mitochondria in spite of the fact that ACSL5 is Triacsin c–resistant.( 23 )
How could the organelle ACS function as an antagonist of apoptosis? Mitochondrial ACS was thought to be involved in β‐oxidation of fatty acid leading to energy production.( 5 ) Recently, it was shown that ACSL5 partitions exogenous fatty acids toward triacylglycerol synthesis and storage.( 32 ) These pathways could play a role in apoptosis inhibition. We have reported that Triacsin c reduces the level of cardiolipin, a phospholipid that is localized on the mitochondria and involved in cytochrome c anchorage on the mitochondria membrane.( 10 ) However, it was recently reported that down‐regulation of cardiolipin alone is not sufficient to induce cytochrome c release.( 33 ) Here, we also showed that the expressions of the Bcl‐2 family members were not significantly changed after ACS inhibition. These data suggest that other mechanisms could also be involved in the potentiation of cytochrome c release by ACS inhibition.
In the present study, we demonstrated that ACS inhibition enhances the etoposide‐induced cell death of human glioma cells. We examined combinations of several agents with Triacsin c and also found a synergistic effect of the ACS inhibitor with SN‐38 (data not shown). Lipid metabolism is selectively activated in a wide variety of cancers, and ACS is overexpressed in such cancers as glioma, colon cancer, and hepatocellular cancer.( 7 , 8 , 9 , 34 ) Our data suggest that ACS inhibition would be a rational strategy to potentiate chemosensitivity of cancer. Additional studies, including the combinational effect of ACS inhibition with other antitumor agents, could further clarify the importance of ACS as a new therapeutic target for cancer.
Abbreviations
| ACS | Acyl‐CoA synthetase |
| DAPI | 4,6‐diamino‐2‐phenylindole |
| DEVD‐MCA | Acetyl‐Asp‐Glu‐Val‐Asp‐(4‐methyl‐coumaryl‐7‐amide) |
| EGFR | Epidermal growth factor receptor |
| FASN | Fatty acid synthase |
| Hsp | Heat shock protein |
| MTS | 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐ 2‐(4‐sulfophenyl)‐2H‐tetrazolium |
| PARP | Poly(ADP‐ribose) polymerase |
| Z‐VAD‐fmk | Z‐Val‐Ala‐Asp(OMe)‐CH2F |
Supporting information
Fig. S1. Acyl‐CoA synthetase (ACS) enzyme activity of the delta L mutant. ACS activity in mock‐, ACSL5‐, and delta L–transduced SF268 cells was measured as described in ‘Materials and Methods’.
Fig. S2. Localization of the ACSL5 delta L mutant in SF268 cells. The delta L mutant of ACSL5 (FLAG) and a mitochondria marker, cytochrome c (Cyto c), were detected by indirect immunofluorescence staining of delta L–transduced SF268 cells with anti‐FLAG M2 (red) and anticytochrome c (green) antibodies, respectively. DAPI staining of DNA is shown in blue.
Fig. S3. Effect of acyl‐CoA synthetase (ACS) inhibition on mitochondria‐dependent apoptosis pathway regulators. Mock‐ and ACSL5‐transduced SF268 cells were left untreated or were treated with 1‐µM Triacsin c in the absence or presence of a 50 µM caspase inhibitor, Z‐Val‐Ala‐Asp(OMe)‐CH2F (Z‐VAD), for 36 h. Expression of proteins that regulate cytochrome c release from mitochondria (Bcl‐2, Bcl‐XL, and Bax) was analyzed by western blot.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We thank the National Cancer Institute as well as Takao Yamori for providing us human glioma cell lines. We also thank members in our laboratory for helpful discussions. This work was supported by grants‐in‐aid for Cancer Research on Priority Areas (T. Mashima, T. Tsuruo, H. Seimiya) and Young Scientists (T. Mashima) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
- 1. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007; 7: 763–77. [DOI] [PubMed] [Google Scholar]
- 2. Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res 2006; 66: 5977–80. [DOI] [PubMed] [Google Scholar]
- 3. Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA interference‐mediated silencing of the acetyl‐CoA‐carboxylase‐alpha gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res 2005; 65: 6719–25. [DOI] [PubMed] [Google Scholar]
- 4. Mashima T, Seimiya H, Tsuruo T. De novo fatty acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer 2009; 100: 1369–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coleman RA, Lewin TM, Van Horn CG, Gonzalez‐Baro MR. Do long‐chain acyl‐CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? J Nutr 2002; 132: 2123–6. [DOI] [PubMed] [Google Scholar]
- 6. Cao Y, Pearman AT, Zimmerman GA, McIntyre TM, Prescott SM. Intracellular unesterified arachidonic acid signals apoptosis. Proc Natl Acad Sci U S A 2000; 97: 11280–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamashita Y, Kumabe T, Cho YY et al . Fatty acid induced glioma cell growth is mediated by the acyl‐CoA synthetase 5 gene located on chromosome 10q25.1‐q25.2, a region frequently deleted in malignant gliomas. Oncogene 2000; 19: 5919–25. [DOI] [PubMed] [Google Scholar]
- 8. Cao Y, Dave KB, Doan TP, Prescott SM. Fatty acid CoA ligase 4 is up‐regulated in colon adenocarcinoma. Cancer Res 2001; 61: 8429–34. [PubMed] [Google Scholar]
- 9. Gassler N, Herr I, Schneider A et al . Impaired expression of acyl‐CoA synthetase 5 in sporadic colorectal adenocarcinomas. J Pathol 2005; 207: 295–300. [DOI] [PubMed] [Google Scholar]
- 10. Mashima T, Oh‐hara T, Sato S et al . p53‐defective tumors with a functional apoptosome‐mediated pathway: a new therapeutic target. J Natl Cancer Inst 2005; 97: 765–77. [DOI] [PubMed] [Google Scholar]
- 11. Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer 2005; 5: 876–85. [DOI] [PubMed] [Google Scholar]
- 12. Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol 2007; 8: 405–13. [DOI] [PubMed] [Google Scholar]
- 13. Mashima T, Tsuruo T. Defects of the apoptotic pathway as therapeutic target against cancer. Drug Resist Updat 2005; 8: 339–43. [DOI] [PubMed] [Google Scholar]
- 14. Reed JC, Pellecchia M. Apoptosis‐based therapies for hematologic malignancies. Blood 2005; 106: 408–18. [DOI] [PubMed] [Google Scholar]
- 15. Meng XW, Lee SH, Kaufmann SH. Apoptosis in the treatment of cancer: a promise kept? Curr Opin Cell Biol 2006; 18: 668–76. [DOI] [PubMed] [Google Scholar]
- 16. Letai AG. Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat Rev Cancer 2008; 8: 121–32. [DOI] [PubMed] [Google Scholar]
- 17. Yang L, Mashima T, Sato S et al . Predominant suppression of apoptosome by inhibitor of apoptosis protein in non‐small cell lung cancer H460 cells: therapeutic effect of a novel polyarginine‐conjugated Smac peptide. Cancer Res 2003; 63: 831–7. [PubMed] [Google Scholar]
- 18. Mashima T, Udagawa S, Tsuruo T. Involvement of transcriptional repressor ATF3 in acceleration of caspase protease activation during DNA damaging agent‐induced apoptosis. J Cell Physiol 2001; 188: 352–8. [DOI] [PubMed] [Google Scholar]
- 19. Mashima T, Naito M, Tsuruo T. Caspase‐mediated cleavage of cytoskeletal actin plays a positive role in the process of morphological apoptosis. Oncogene 1999; 18: 2423–30. [DOI] [PubMed] [Google Scholar]
- 20. Mashima T, Sato S, Sugimoto Y et al . Promotion of glioma cell survival by acyl‐CoA synthetase 5 under extracellular acidosis conditions. Oncogene 2009; 28: 9–19. [DOI] [PubMed] [Google Scholar]
- 21. Heitjan DF, Manni A, Santen RJ. Statistical analysis of in vivo tumor growth experiments. Cancer Res 1993; 53: 6042–50. [PubMed] [Google Scholar]
- 22. R development core team . R: a language and environment for statistical computing. R foundation for statistical computing. Vienna: 2007. (URL: http://www.R‐project.org).
- 23. Kim JH, Lewin TM, Coleman RA. Expression and characterization of recombinant rat Acyl‐CoA synthetases 1, 4, and 5. Selective inhibition by triacsin C and thiazolidinediones. J Biol Chem 2001; 276: 24667–73. [DOI] [PubMed] [Google Scholar]
- 24. Lewin TM, Kim JH, Granger DA, Vance JE, Coleman RA. Acyl‐CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J Biol Chem 2001; 276: 24674–9. [DOI] [PubMed] [Google Scholar]
- 25. Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci U S A 1998; 95: 4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martin AG, Fearnhead HO. Apocytochrome c blocks caspase‐9 activation and Bax‐induced apoptosis. J Biol Chem 2002; 277: 50834–41. [DOI] [PubMed] [Google Scholar]
- 27. Yoshida A, Pourquier P, Pommier Y. Purification and characterization of a Mg2+‐dependent endonuclease (AN34) from etoposide‐treated human leukemia HL‐60 cells undergoing apoptosis. Cancer Res 1998; 58: 2576–82. [PubMed] [Google Scholar]
- 28. Perkins CL, Fang G, Kim CN, Bhalla KN. The role of Apaf‐1, caspase‐9, and bid proteins in etoposide‐ or paclitaxel‐induced mitochondrial events during apoptosis. Cancer Res 2000; 60: 1645–53. [PubMed] [Google Scholar]
- 29. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004; 432: 307–15. [DOI] [PubMed] [Google Scholar]
- 30. Tsuruo T, Naito M, Tomida A et al . Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci 2003; 94: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006; 25: 4798–811. [DOI] [PubMed] [Google Scholar]
- 32. Mashek DG, McKenzie MA, Van Horn CG, Coleman RA. Rat long chain acyl‐CoA synthetase 5 increases fatty acid uptake and partitioning to cellular triacylglycerol in McArdle‐RH7777 cells. J Biol Chem 2006; 281: 945–50. [DOI] [PubMed] [Google Scholar]
- 33. Ott M, Zhivotovsky B, Orrenius S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ 2007; 14: 1243–7. [DOI] [PubMed] [Google Scholar]
- 34. Sung YK, Hwang SY, Park MK et al . Fatty acid‐CoA ligase 4 is overexpressed in human hepatocellular carcinoma. Cancer Sci 2003; 94: 421–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Acyl‐CoA synthetase (ACS) enzyme activity of the delta L mutant. ACS activity in mock‐, ACSL5‐, and delta L–transduced SF268 cells was measured as described in ‘Materials and Methods’.
Fig. S2. Localization of the ACSL5 delta L mutant in SF268 cells. The delta L mutant of ACSL5 (FLAG) and a mitochondria marker, cytochrome c (Cyto c), were detected by indirect immunofluorescence staining of delta L–transduced SF268 cells with anti‐FLAG M2 (red) and anticytochrome c (green) antibodies, respectively. DAPI staining of DNA is shown in blue.
Fig. S3. Effect of acyl‐CoA synthetase (ACS) inhibition on mitochondria‐dependent apoptosis pathway regulators. Mock‐ and ACSL5‐transduced SF268 cells were left untreated or were treated with 1‐µM Triacsin c in the absence or presence of a 50 µM caspase inhibitor, Z‐Val‐Ala‐Asp(OMe)‐CH2F (Z‐VAD), for 36 h. Expression of proteins that regulate cytochrome c release from mitochondria (Bcl‐2, Bcl‐XL, and Bax) was analyzed by western blot.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item