

Abstract
Hepatitis B virus (HBV) genotype C and the basic core promoter (BCP) mutations were reported to be associated with the development of hepatocellular carcinoma (HCC). In this study the full sequences of HBV genomes were analyzed in order to find the other predictors of HCC development. We determined the full sequences of HBV genomes in 24 genotype C carriers who developed HCC (HCC group) at the beginning of follow‐up and at the time of HCC diagnosis, and 20 patients who did not develop HCC (non‐HCC group) served as a control. The number of nucleotide and amino acid substitutions in most regions was higher in the HCC group than in the non‐HCC group, and the following substitutions and deletions were found more frequently in the HCC group than in the non‐HCC group: G1317A and T1341C/A/G in the X promoter region were detected in 13 and six of the HCC cases, four and none of the non‐HCC cases, respectively; and pre‐S2 deletion was detected in eight HCC and none of the non‐HCC cases. Compared with the wild type X promoter, the mutant type X promoters, M1 (G1317A), M2 (T1341C), and M4 (T1341G) showed increases in activity of 2.3, 3.8, and 1.4 times, respectively, in HepG2 cells. Substitutions and deletion of nucleotides of the HBV genome, especially the pre‐S2 deletion and G1317A and T1341C/A/G mutations may be useful markers for predicting the development of HCC. (Cancer Sci 2007; 98: 1921–1929)
Abbreviations
- aa
amino acid
- AP1
activator protein 1
- ASC
asymptomatic carrier
- ATF2
activating transcription factor 2
- BCP
basic core promoter
- C/EBP
CCAAT/enhancer‐binding proteins
- CLD
chronic liver diseases
- CP
core promoter
- CREB
cAMP response element‐binding protein
- DMEM
Dulbecco's modified minimal essential medium
- HBeAg
hepatitis B e antigen
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- LGE/mL
logarithm of genomic equivalent per milliliter
- MEGA
molecular evolutionary genetics analysis
- NRF1
nuclear respiratory factor 1
- nt
nucleotide
- ORFs
open reading frames
- PCR
polymerase chain reaction
- pHSA‐R
polymerized human serum albumin receptor
- Pol
polymerase
- RLU
relative light units
- RT
reverse transcriptase
- SD
standard deviation
- UPGMA
Unweighted Pair Group Method
- X‐PBP
X‐promoter‐binding protein.
Four hundred million people worldwide are chronically infected with HBV.( 1 ) Infection with HBV leads to a wide spectrum of clinical presentations ranging from an ASC state to chronic hepatitis with progression to HCC in some patients. HCC is one of the major malignant diseases in the world, ranking as the fifth most common cancer, and it is believed that chronic HBV infection is a major global cause of HCC.( 2 )
HBV consists of four ORFs: the X, precore/core, pre‐S/S, and Pol regions. Among these, the X and pre‐S/S regions have been reported to function as transcriptional transactivators.( 3 , 4 , 5 ) The domain between nt 221 and 573 in the pre‐S/S region was termed the transactivator region, and its 3′‐deleted form was shown to exert a transcriptional transactivator function.( 6 )
Although substitutions and deletions in the pre‐S1/pre‐S2/S region were reported in relation to the development of liver disease,( 7 , 8 ) it remains unclear whether differences in the full length sequence of HBV exist at different stages of infection, particularly between patients who develop HCC and those who do not. Kajiya et al. analyzed the full sequence of serial serum samples obtained from a patient who underwent long‐term follow‐up from the time prior to development of symptoms to the chronic active hepatitis stage, and suggested that pre‐S1 deletions and substitutions in the CP region may participate cooperatively in the progression of the disease.( 9 ) Takahashi et al. studied the full length nucleotide sequence of HBV genome in sera from 40 Japanese patients with HBsAg positive HCC, and reported frequent deletions and missense mutations in the preS2 region.( 10 )
In the present study, the full length sequence of the HBV genome were analyzed in the sera of patients who did not have HCC at the beginning of follow‐up and developed HCC thereafter, and in those of patients who did not develop HCC during follow‐up served as the control group, in order to find the predicators of HCC development.
Materials and Methods
Patients. HBV sequences were examined in the sera of 24 patients who developed HCC (HCC group) at the beginning of follow‐up (HCC 1B‐24B) and at the time of HCC diagnosis (HCC 1A‐24A). The control group (non‐HCC group) consisted of 20 patients matched with respect to age, sex, and follow‐up period who did not develop HCC, and were examined at the beginning of follow‐up (non‐HCC 1B‐20B) and at the end of follow‐up (non‐HCC 1A‐20A). The clinical and laboratory data for these patients are listed in Table 1.
Table 1.
Clinical and laboratory data of the 44 patients with and without HCC
| Gender | Age in year | HBeAg: anti‐HBe | HBV‐DNA (LGE/mL) | Follow‐up time (months) | WBC( × 103/µL) | RBC( × 106/µL) | PLT( × 103/µL) | AST(IU/L) | ALT(IU/L) | ALB(g/dL) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Non‐HCC group (B) | M:F = 14:6 | 41.35 ± 9.55 | 18:2 | 6.91 ± 2.90 | – | 5.09 ± 1.01 | 4.53 ± 0.45 | 182.55 ± 70.57 | 159.50 ± 415.58 | 214.55 ± 410.16 | 4.16 ± 0.34 |
| Non‐HCC group (A) | – | 48.55 ± 8.56 | 18:2 | 6.31 ± 1.74 | 87.45 ± 36.43 | 5.27 ± 1.47 | 4.36 ± 0.48 | 183.15 ± 70.95 | 147.40 ± 173.11 | 209.55 ± 240.01 | 4.02 ± 0.70 |
| HCC group (B) | M:F = 21:3 | 41.29 ± 10.51 | 14:10 | 6.21 ± 1.73 | – | 5.42 ± 1.62 | 4.54 ± 0.55 | 133.25 ± 63.93 | 89.96 ± 101.33 | 98.04 ± 92.79 | 4.17 ± 0.48 |
| HCC group (A) | – | 47.33 ± 9.14 | 15:9 | 5.48 ± 1.21 | 77.20 ± 41.86 | 4.68 ± 1.19 | 4.42 ± 0.55 | 118.33 ± 59.60 | 51.17 ± 36.86 | 49.50 ± 35.23 | 4.02 ± 0.57 |
| P (non‐HCC B vs HCC B) | 0.26 | 0.99 | 0.04 | 0.13 | – | 0.43 | 0.94 | 0.02 | 0.43 | 0.18 | 0.90 |
| P (non‐HCC A vs HCC A) | – | 0.65 | 0.04 | 0.39 | 0.40 | 0.15 | 0.74 | < 0.01 | 0.01 | < 0.01 | 1.00 |
The significance of differences (P‐value) in the indicated groups is shown. P < 0.05 is indicated by italic type. Non‐HCC group (B): at the beginning of follow‐up in patients who did not develop HCC. Non‐HCC group (A): at the end of follow‐up in patients who did not develop HCC. HCC group (B): at the beginning of follow‐up in patients who developed HCC. HCC group (A): at the diagnosis of HCC in patients who developed HCC. ALB, albumin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; HBeAg, hepatitis B e antigen; HCC, hepatocellular carcinoma; LGE/mL, logarithm of genomic equivalent per milliliter; PLT, platelet; RBC, red blood cells; WBC, white blood cells.
All patients were followed at the Department of Medicine and Clinical Oncology, Chiba University. To compare differences in the same genotype, only cases with genotype C were examined since more than 80% of the patients were infected with this genotype.( 11 ) All of the patients were negative for second‐generation anti‐HCV antibody (Ortho Diagnostics, Tokyo, Japan). All serum samples were collected and stored at –20°C until analysis. Written informed consent was obtained from each patient included in the study.
Amplification and sequencing of the full HBV genome. To amplify the full sequence of the HBV genome, two pairs of synthetic oligonucleotide primers (p1, p2 and p3, p4) were used. PCR was carried out according to the methods described previously,( 12 ) with LA Taq (TaKaRa Bio Inc., Ohtsu, Japan) for 40 cycles in the first PCR and 35 cycles in the second PCR. The sequences between the joint points of the amplified PCR fragments were amplified using the precore/core primers (p5, p6 and p7, p8).( 11 )
For sequencing, 50 µL of the PCR product was purified with a MinElute PCR purification kit (QIAGEN, Hilden, Germany) and 50–100 ng of the products was used for sequencing with sequencing primers in both directions. The nucleotide sequences were determined directly with an ABI PRISM 3700 DNA Analyzer (Applied Biosystems, Foster City, CA, USA). Sequence alignments were carried out using the Auto Assembler of the GENETYX‐MAC program, version 10.1 (GENETYX Corp., Tokyo, Japan). The positions and sequences of the PCR primers and sequencing primers are listed in Table 2.
Table 2.
Positions and sequences of the primers used for polymerase chain reaction (PCR) amplification and sequencing
| Position † | Nucleotide sequence (5′–3′) | Polarity |
|---|---|---|
| PCR primer | ||
| p1 : 1821–1841 | TTTTTCACCTCTGCCTAATCA | Sense |
| p2 : 1825–1806 | AAAAAGTTGCATGGTGCTGG | Antisense |
| p3 : 1922–1940 | GAATTTGGAGCTTCTGTGG | Sense |
| p4 : 1788–1771 | GCCTACAGCCTCCTAGTA | Antisense |
| p5 : 1604–1623 | TCGCATGGAGACCACCGTGA | Sense |
| p6 : 2076–60 | ATAGCTTGCCTGAGTGC | Antisense |
| p7 : 1653–1672 | CATAAGAGGACTCTTGGACT | Sense |
| p8 : 1974–1957 | GGAAAGAAGTCAGAAGGC | Antisense |
| Sequencing primer | ||
| 242–258 | CAGAGTCTAGACTCGTGG | Sense |
| 687–668 | GGCACTAGTAAACTGAGCCA | Antisense |
| 456–475 | AAGGTATGTTGCCCGTTTGT | Sense |
| 771–752 | TACAGACTTGGCCCCCAATA | Antisense |
| 668–687 | TGGCTCAGTTTACTAGTGCC | Sense |
| 1103–1086 | GGCGAGAAAGTGAAAGCC | Antisense |
| 1054–1073 | ATGCCTTTATATGCATGTAT | Sense |
| 1436–1418 | GACGGGACGTAGACAAAGG | Antisense |
| 1267–1285 | CATACTGCGGAACTCCTAGC | Sense |
| 2470–2453 | TTATGAGTCCAAGGGATA | Antisense |
| 2301–2320 | CACCAAATGCCCCTATCTTA | Sense |
| 2656–2639 | GGATAGAACCTAGCAGGC | Antisense |
| 2637–2656 | ATGCCTGCTAGGTTCTATCC | Sense |
| 3155–3138 | CTTCCTGACTGCCGATTG | Antisense |
| 3082–3099 | CCTCAGGCTCAGGGCATA | Sense |
| 475–456 | ACAAACGGGCAACATACCTT | Antisense |
Nucleotide position based on the sequence of hepatitis B virus (HBV) genotype C (accession no. X01587). Primers p3 to p8 were also used for sequencing.
We selected the HBV genotype C X01587 (subtype adr4) strain as a prototype, in which the reverse transcriptase (RT) region (from nt 130–1161) in the Pol ORF is conserved.( 13 ) All of the sequences were compared with that of X01587. The number of nucleotide and amino acid substitutions was calculated. To confirm the results, we also compared all isolates with another HBV genotype C prototype, M12906 (subtype adr), in which the gene organization of HBV DNA was found to be well conserved irrespective of subtype,( 14 ) and it had 2.7% nucleotide (87 nt) divergence in the full genome compared with X01587. A difference was reported to exist when the results obtained using either of these two reference strains showed statistically significant differences from the sample sequences.
The numbers of synonymous and non‐synonymous substitutions in the ORF regions of HBV in the non‐HCC and the HCC groups were estimated using MEGA software (download from http://www.megasoftware.net) compared with X01587 and M12906.
Phylogenetic tree analysis. To examine the heterogeneity of the viral sequence, a phylogenetic tree was constructed by the UPGMA using the GENETYX‐MAC program version 10.1, based on the entire genomic sequences of the HBV isolates. The GenBank accession numbers of the representative genotype sequences C1‐C4 of HBV were AF473543 in C1, X01587 and M12906 in C2,( 15 ) (classified by Norder et al. as C2, C1 and C1, respectively( 16 )) X75656 in C3, and AB048704 in C4.( 16 , 17 )
Nucleotide sequences were multiple‐aligned using GENETYX‐MAC program version 10.1 and genetic distances were estimated using the Kimura 2‐parameter matrix.( 18 ) The percentages at nodes represent the levels of bootstrap support from 1000 resamplings of the data sets.
Preparation of plasmids with wild type and mutant type X promoters for transfection. pBluescript II SK+ plasmid containing full‐length HBV DNA was used to make four variants by substituting the G of nt 1317 of the X promoter with A (M1), and the T of nt 1341 with C (M2), A (M3), or G (M4) using the Quickchange site‐directed mutagenesis kit (Strategene). Mutations were verified by sequencing both strands of DNA in the regions of interest. Using the wild type and four mutant types of plasmid as the template, the region including enhancer I and X promoter was amplified by PCR using primers designed to introduce SacI and XhoI linker sequences (underlined) as follows: SacI CCGGAGCTCCCTGCGTTGATGCCTTTGTA and XhoI CCGCTCGAGGAAACGATGTATATTTGCGG. To investigate differences in the activation of wild type and mutant type X promoters, experiments were carried out using an in vivo reporter system. In brief, the PCR products were cut by SacI and XhoI, and cloned into the SacI and XhoI sites of vector pGL4.10 (Promega Corp, Madison, WI, USA), which contains the firefly luciferase reporter gene without promoter, and hence, expression of the firefly luciferase gene in the reporter plasmid was controlled by the inserted enhancer I and X promoter. Transfection efficiency was monitored through the cotransfection of pRL‐TK (Promega Corp, Madison, WI, USA), a control plasmid expressing Renilla reniformis (seapansy) luciferase driven by the herpes simplex virus thymidine kinase.
Cell culture and transfection and luciferase assays. Huh7 and HepG2 cells were purchased from Health Science Research Resources Bank (Osaka, Japan). Cells were maintained in DMEM (Sigma‐Aldrich) supplemented with 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% fetal calf serum at 37°C in a 5% CO2 atmosphere.
Approximately 3 × 104 Huh7 and 2 × 104 HepG2 cells were plated in 24‐well tissue culture plates (Iwaki Glass, Tokyo, Japan) 24 h prior to transfection. The transfection complexes containing a total of 0.2 µg of plasmids (0.12 µg of pGL4.10 and 0.08 µg pRL‐TK) were added to each well using Effectene Transfection Reagent (Qiagen). Cells were harvested 24 h after transfection, and luciferase assays were carried out with the Dual–GloTM Assay system (Promega). Firefly and seapansy luciferase activities were measured as relative light units (RLU) with a luminometer (AB‐2200, ATTO. Tokyo, Japan).
Statistical analysis. All statistical analyses were carried out with Statview 5.0 software for Macintosh (SAS Institute Inc, Cary, NC). Statistical analyses were carried out by Student's t‐test, Fisher's exact probability test, and Mann–Whitney U‐test. Results were expressed as the mean ± standard deviation (SD). A two‐tailed P‐value of less than 0.05 was considered to be significant. Factors associated with the development of HCC were examined by multivariate Cox proportional regression analysis.
Results
Phylogenetic tree analysis of the full‐length HBV genomes. Phylogenetic tree analysis of the full sequence of the HBV genome of the 44 isolates showed that all were genotype C2, except for one case (Case No. HCC 24B) that had nucleotide sequence identity with AY206389 (genotype C, HCC strain) (data not shown).
Substitutions in nucleotide and amino acid sequences. Nucleotide and amino acid substitutions were examined in the 43 genotype C2 isolates. The number of nucleotide substitutions in the full genome was 68.35 ± 20.90 and 76.13 ± 11.59 at the beginning of follow‐up in the non‐HCC group and HCC group, respectively (P = 0.14). The number was 71.85 ± 21.64 and 80.26 ± 17.74 at the end of follow‐up in the non‐HCC group and HCC group, respectively (P = 0.20) (Table 3). The average nucleotide substitution rates in the full genome were 0.90 ± 0.96 × 10−3/nt per year and 1.60 ± 2.84 × 10−3/nt per year in the non‐HCC group and HCC group, respectively (P = 0.26). The number of amino acid substitutions in four ORFs was higher in the HCC group than in the non‐HCC group, but there were no significant differences between them either at the beginning or at the end of follow‐up (Table 4).
Table 3.
Number of nucleotide substitutions in the full genome and various regions of hepatitis B virus (HBV) in the non‐hepatocellular carcinoma (HCC) and HCC group
| Full genome | pre‐S1 | pre‐S2 | S | pre‐C/core | X | Pol | Enhancer I | Enhancer II | X promoter | core promoter | S1 promoter | S2 promoter | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (nt 0–3215) | (nt 2484–3204) | (nt 3205–154) | (nt 155–835) | (nt 1814–2452) | (nt 1376–1846) | (nt 2307–1620) | (nt 1060–1260) | (nt 1635–1714) | (nt1230–1376) | (nt 1519–1822) | (nt 2710–2800) | (nt 2960–3180) | |
| Non‐HCC group (B) | 68.35 ± 20.90 | 9.15 ± 3.73 | 4.40 ± 2.19 | 5.30 ± 2.16 | 10.30 ± 5.40 | 9.05 ± 3.25 | 54.55 ± 15.51 | 8.15 ± 3.05 | 1.70 ± 0.92 | 6.10 ± 1.99 | 5.30 ± 2.70 | 1.75 ± 1.12 | 7.95 ± 3.07 |
| Non‐HCC group (A) | 71.85 ± 21.64 | 9.75 ± 3.58 | 4.65 ± 2.28 | 6.05 ± 2.19 | 12.55 ± 5.67 | 8.90 ± 2.56 | 56.30 ± 16.70 | 7.65 ± 2.70 | 1.60 ± 0.88 | 6.05 ± 1.88 | 5.35 ± 1.93 | 1.85 ± 1.50 | 8.20 ± 2.89 |
| HCC group (B) | 76.13 ± 11.59 | 10.26 ± 2.14 | 3.78 ± 1.93 | 5.74 ± 1.60 | 13.00 ± 5.75 | 9.69 ± 1.87 | 59.04 ± 8.23 | 8.09 ± 2.15 | 1.83 ± 0.98 | 7.26 ± 1.66 | 5.48 ± 1.44 | 1.78 ± 0.85 | 9.04 ± 1.36 |
| HCC group (A) | 80.26 ± 17.74 | 10.61 ± 2.87 | 4.39 ± 2.43 | 6.52 ± 3.87 | 14.52 ± 6.49 | 9.91 ± 2.71 | 61.65 ± 13.14 | 7.96 ± 1.99 | 1.70 ± 0.82 | 7.04 ± 1.92 | 5.43 ± 1.56 | 2.26 ± 1.39 | 9.61 ± 2.02 |
| P (non‐HCC B vs HCC B) | 0.14 | 0.27 | 0.55 | 0.47 | 0.18 | 0.21 | 0.23 | 0.81 | 0.62 | 0.07 | 0.66 | 0.87 | 0.16 |
| P (non‐HCC A vs HCC A) | 0.20 | 0.57 | 0.43 | 0.61 | 0.25 | 0.39 | 0.31 | 0.75 | 0.95 | 0.06 | 0.88 | 0.35 | 0.12 |
The significance of differences (P‐value) in the indicated groups is shown. The number of nucleotide (nt) substitutions (mean±SD) in the full genome and the indicated regions of HBV are shown. Non‐HCC group (B): at the beginning of follow‐up in patients who did not develop HCC. Non‐HCC group (A): at the end of follow‐up in patients who did not develop HCC. HCC group (B): at the beginning of follow‐up in patients who developed HCC. HCC group (A): at the diagnosis of HCC in patients who developed HCC.
Table 4.
Number of amino acid substitutions in various regions of hepatitis B virus (HBV) in the non‐hepatocellular carcinoma (HCC) and HCC groups
| Pre‐C/core(213 aa) | Pre‐S1(119 aa) | Pre‐S2(55 aa) | S(226 aa) | X(155 aa) | Pol(843 aa) | |
|---|---|---|---|---|---|---|
| Non‐HCC group (B) | 2.25 ± 2.81 | 3.70 ± 1.81 | 1.55 ± 1.28 | 3.45 ± 2.28 | 5.50 ± 2.24 | 24.35 ± 6.66 |
| Non‐HCC group (A) | 3.90 ± 3.48 | 4.05 ± 1.85 | 1.60 ± 1.05 | 3.75 ± 1.33 | 5.65 ± 1.84 | 24.30 ± 5.93 |
| HCC group (B) | 3.70 ± 3.39 | 3.96 ± 1.66 | 1.87 ± 1.42 | 4.26 ± 1.76 | 5.91 ± 1.53 | 24.61 ± 3.68 |
| HCC group (A) | 4.09 ± 3.91 | 4.22 ± 1.93 | 2.17 ± 1.80 | 4.61 ± 2.52 | 6.13 ± 1.91 | 25.91 ± 5.20 |
| P (non‐HCC B vs HCC B) | 0.17 | 0.87 | 0.43 | 0.19 | 0.47 | 0.97 |
| P (non‐HCC A vs HCC A) | 0.90 | 0.84 | 0.25 | 0.21 | 0.33 | 0.35 |
The significance of differences (P‐value) in the indicated groups is shown. The number of amino acid (aa) substitutions (mean±SD) in the indicated regions of HBV is shown. Non‐HCC group (B): at the beginning of follow‐up in patients who did not develop HCC. Non‐HCC group (A): at the end of follow‐up in patients who did not develop HCC. HCC group (B): at the beginning of follow‐up in patients who developed HCC. HCC group (A): at the diagnosis of HCC in patients who developed HCC. Pol, polymerase.
In the ORFs and regulatory elements, except for the pre‐S2 and enhancer I regions, the HCC group tended to have more nucleotide substitutions, both at the beginning and at the end of follow‐up, compared with the non‐HCC group (Table 3).
We also estimated synonymous and non‐synonymous substitutions in the ORF regions of HBV in the non‐HCC and HCC groups. In almost every region, there were more synonymous and non‐synonymous substitutions in the HCC group than in the non‐HCC group at both the beginning and end of follow‐up (5, 6).
Table 5.
Mean numbers of synonymous and non‐synonymous substitutions per nucleotide site in various regions of hepatitis B virus (HBV) in the non‐hepatocellular carcinoma (HCC) and HCC groups compared with X01587
| Pre‐C/core | Pre‐S1 | Pre‐S2 | S | X | Pol | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Syno | Non‐syno | Syno | Non‐syno | Syno | Non‐syno | Syno | Non‐syno | Syno | Non‐syno | Syno | Non‐syno | |
| Non‐HCC group (B) | 0.0465 | 0.0046 | 0.0640 | 0.0145 | 0.0592 | 0.0144 | 0.0090 | 0.0070 | 0.0567 | 0.0158 | 0.0527 | 0.0138 |
| Non‐HCC group (A) | 0.0510 | 0.0073 | 0.0692 | 0.0157 | 0.0636 | 0.0149 | 0.0084 | 0.0076 | 0.0538 | 0.0153 | 0.0542 | 0.0137 |
| HCC group (B) | 0.0570 | 0.0074 | 0.0927 | 0.0183 | 0.0096 | 0.0215 | 0.0052 | 0.0081 | 0.0579 | 0.0143 | 0.0617 | 0.0134 |
| HCC group (A) | 0.0615 | 0.0095 | 0.0841 | 0.0191 | 0.0118 | 0.0204 | 0.0070 | 0.0089 | 0.0563 | 0.0210 | 0.0630 | 0.0139 |
| P (non‐HCC B vs HCC B) | 0.05 | 0.21 | 0.001 | <0.05 | <0.001 | 0.10 | 0.01 | 0.39 | 0.77 | 0.31 | 0.03 | 0.72 |
| P (non‐HCC A vs HCC A) | 0.07 | 0.40 | 0.08 | 0.12 | < 0.001 | 0.19 | 0.61 | 0.31 | 0.56 | <0.01 | 0.07 | 0.87 |
The number of synonymous and non‐synonymous substitutions per nucleotide site in the indicated regions of HBV is shown. The significance of differences (P‐value) in the indicated groups is shown. P < 0.05 is indicated by italic type. Non‐HCC group (B): at the beginning of follow‐up in patients who did not develop HCC. Non‐HCC group (A): at the end of follow‐up in patients who did not develop HCC. HCC group (B): at the beginning of follow‐up in patients who developed HCC. HCC group (A): at the diagnosis of HCC in patients who developed HCC. Non‐syno, non‐synonymous; Pol, polymerase; syno, synonymous.
Table 6.
Mean numbers of synonymous and non‐synonymous substitutions per nucleotide site in various region of hepatitis B virus (HBV) in the non‐hepatocellular carcinoma (HCC) and HCC groups compared with M12906
| Pre‐C/core | Pre‐S1 | Pre‐S2 | S | X | Pol | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Syno | Non‐syno | Syno | Non‐syno | Syno | Non‐syno | Syno | Non‐syno | Syno | Non‐syno | Syno | Non‐syno | |
| Non‐HCC group (B) | 0.0320 | 0.0046 | 0.0579 | 0.0230 | 0.0279 | 0.0152 | 0.0150 | 0.0052 | 0.0397 | 0.0251 | 0.0450 | 0.0152 |
| Non‐HCC group (A) | 0.0355 | 0.0073 | 0.0628 | 0.0241 | 0.0326 | 0.0153 | 0.0144 | 0.0058 | 0.0401 | 0.0251 | 0.0459 | 0.0155 |
| HCC group (B) | 0.0379 | 0.0074 | 0.0713 | 0.0264 | 0.0094 | 0.0203 | 0.0112 | 0.0056 | 0.0361 | 0.0235 | 0.0457 | 0.0146 |
| HCC group (A) | 0.0427 | 0.0095 | 0.0693 | 0.0270 | 0.0119 | 0.0204 | 0.0130 | 0.0066 | 0.0374 | 0.0302 | 0.0501 | 0.0159 |
| P (non‐HCC B vs HCC B) | 0.21 | 0.21 | 0.08 | 0.06 | <0.01 | 0.17 | 0.01 | 0.78 | 0.29 | 0.36 | 0.85 | 0.48 |
| P (non‐HCC A vs HCC A) | 0.18 | 0.40 | 0.40 | 0.22 | <0.01 | 0.22 | 0.61 | 0.58 | 0.44 | <0.01 | 0.34 | 0.70 |
The significance of differences (P‐value) in the indicated groups is shown. P < 0.05 is indicated by italic type. The number of synonymous and non‐synonymous substitutions per nucleotide site in the indicated regions of HBV is shown. Non‐HCC group (B): at the beginning of follow‐up in patients who did not develop HCC. Non‐HCC group (A): at the end of follow‐up in patients who did not develop HCC. HCC group (B): at the beginning of follow‐up in patients who developed HCC. HCC group (A): at the diagnosis of HCC in patients who developed HCC. Non‐syno, non‐synonymous; Pol, polymerase; syno, synonymous.
The pre‐S2 region had more deletion mutants in the HCC group at both the beginning and end of follow‐up (Fig. 1), and the deletion mutants were not included in the nucleotide or amino acid substitutions in the HCC group, so the actual number of nucleotide and amino acid substitutions (including the synonymous and non‐synonymous substitutions) in the pre‐S2 region were underestimated in this study.
Figure 1.
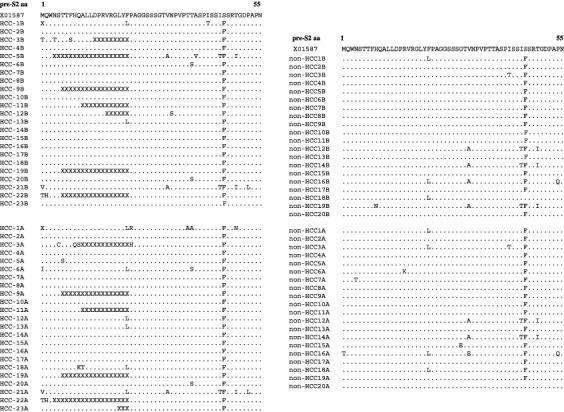
Deduced amino acid sequence encoded by the pre‐S2 region in the 44 hepatitis B virus (HBV) isolates. Dots indicate amino acids that are identical to the respective amino acid in the pre‐S2 region in X01587. X represents a deleted amino acid.
The number of synonymous and non‐synonymous substitution per nucleotide site was lower in the S region than in the other regions compared with both prototypes (5, 6); therefore, the S region appeared to be strongly conserved.( 19 )
Nucleotide and amino acid substitutions in the non‐HCC and HCC groups. The nucleotide and amino acid substitutions and positivity rates in amino acid sites of the four ORFs in the HCC and non‐HCC groups are listed in Table 7 and Fig. 2.
Table 7.
Nucleotide and amino acid substitutions and positive rates in several sites of the four open reading frames (ORFs) compared with X01587 and M12906
| Non‐HCC group (B) | Non‐HCC group (A) | HCC group (B) | HCC group (A) | P (non‐HCC B vs HCC B) | P (non‐HCC A vs HCC A) | |
|---|---|---|---|---|---|---|
| 1317 G‐A | 4 (20%) | 5 (25%) | 13 (56.5%) | 14 (60.9%) | 0.03 | 0.03 |
| 1341 T‐C/A/G | 0 (0%) | 0 (0%) | 3/2/1 (26.1%) | 3/3/1 (30.4%) | 0.02 | 0.01 |
| 1485 C‐T (Xaa38 P‐S) | 2 (10%) | 3 (15%) | 4 (17.4%) | 7 (30.4%) | 0.67 | 0.29 |
| 1653 C‐T (Xaa94H‐Y) | 5 (25%) | 6 (30%) | 6 (26.1%) | 6 (26.1%) | 1.00 | 1.00 |
| 1719T‐G (Xaa116L‐V) | 6 (30%) | 5 (25%) | 0 (0%) | 1 (4.3%) | <0.01 | 0.08 |
| 1753 T‐C/A/G (Xaa127I‐T/N/S) | 5/1/1 (35%) | 5/1/1 (35%) | 6/4/0 (43.5%) | 8/1/2 (47.8%) | 0.76 | 0.54 |
| 1762 A‐T (Xaa130K‐M) | 12 (60%) | 12 (60%) | 20 (87.0%) | 20 (87.0%) | 0.08 | 0.08 |
| 1764 G‐A (X131V‐I) | 11 (55%) | 15 (75%) | 21 (91.3%) | 21 (91.3%) | 0.01 | 0.22 |
| 1896 G‐A (preC28 W‐stop) | 4 (20%) | 7 (35%) | 9 (39.1%) | 9 (39.1%) | 0.20 | 1.00 |
| 1938 T‐C (core13 V‐A) | 0 (0%) | 0 (0%) | 2 (8.7%) | 4 (17.4%) | 0.49 | 0.11 |
| 2189 A‐C/T (core97 I‐L/F) | 3/0 (15%) | 5/1 (30%) | 5/0 (21.7%) | 4/1 (21.7%) | 0.70 | 0.73 |
| 2288 C‐A/G (core130 P‐T/A) | 2/0 (10%) | 4/1 (25%) | 8/1 (39.1%) | 6/2 (34.8%) | 0.04 | 0.53 |
| 3026 C‐A/T (pre‐S160 A‐V/E) | 0 (0%) | 0 (0%) | 4/1 (21.7%) | 5/1 (26.1%) | 0.05 | 0.02 |
| 53 T‐C (pre‐S222 F‐L) | 2 (10%) | 4 (20%) | 2 (8.7%) | 5 (21.7%) | 1.00 | 1.00 |
| Pre‐S2 deletion | 0 (0%) | 0 (0%) | 8 (34.8%) | 7 (30.4%) | <0.01 | 0.01 |
| 162 A‐G (S3 N‐S) | 8 (40%) | 8 (40%) | 12 (52.2%) | 12 (52.2%) | 0.54 | 0.54 |
| 357 T‐C (S68 I‐T) | 0 (0%) | 1 (5%) | 6 (26.1%) | 5 (21.7%) | 0.02 | 0.19 |
| 531 T‐C/G (S126 I‐T/S) | 3/2 (25%) | 3/3 (30%) | 7/2 (39.1%) | 7/2 (39.1%) | 0.35 | 0.75 |
| 706 A‐C (S184 V‐A) | 11 (55%) | 11 (55%) | 14 (60.9%) | 13 (56.5%) | 0.76 | 1.00 |
| 1386 G‐C/A (X5 V‐M/L) | 4/1 (25%) | 3/1 (20%) | 4/4 (34.8%) | 3/6 (39.1%) | 0.33 | 0.20 |
| 1320 A‐C (Pol743 K‐N) | 1 (5%) | 1 (5%) | 7 (30.4%) | 7 (30.4%) | 0.05 | 0.05 |
P‐values for comparisons between the indicated groups are shown. P < 0.05 is indicated by italic type. Non‐hepatocellular carcinoma (HCC) group (B): at the beginning of follow‐up in patients who did not develop HCC. Non‐HCC group (A): at the end of follow‐up in patients who did not develop HCC. HCC group (B): at the beginning of follow‐up in patients who developed HCC. HCC group (A): at the diagnosis of HCC in patients who developed HCC.
Figure 2.
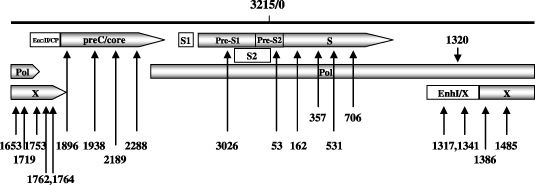
Schematic diagram of the point mutations shown in Table 7. The number on the top of the full hepatitis B virus (HBV) genome refer to the EcoRI site (3215/0). Position of the open reading frames (ORFs) encoding precore/core (preC/core), pre‐S1/pre‐S2/S, polymerase (Pol), and X protein are shown by the gray rectangle and the arrow indicates the transcription direction. Enhancer I/X promoter (EnhI/X), enhancer II/core promoter (EnhII/CP), S1 promoter (S1) and S2 promoter (S2) are indicated by the open rectangle.
At the end of follow‐up, a comparison of the mutation frequency in the HCC group and the non‐HCC group showed statistically significant substitution from adenine to guanine at 1317 (G1317A, hereafter) (14/23 vs 5/20, P = 0.03), T1341C/A/G (7/23 vs 0/20, P = 0.01), and C3026A/T (6/23 vs 0/20, P = 0.02) (Table 7). The frequency of these changes was also different at the beginning of follow‐up between the HCC and the non‐HCC groups (Table 7).
Furthermore, comparison of the frequency of mutations in the HCC group with that in the non‐HCC group at the beginning of follow‐up showed the following changes more frequent in the HCC group besides the above mentioned G1317A, T1341C/A/G (Table 7); G1764A with change of V to I at codon 131 in the X region (21/23 vs 11/20, P = 0.01), C2288A/G with changes of P to T/A at codon 130 in the core region (9/23 vs 2/20, P = 0.04), and T357C with a change of I to T at codon 68 in the S region (6/23 vs 0/20, P = 0.02) (Table 7).
In the pre‐S2 region, deletions between nt 3205–54 were detected in eight (34.8%) and seven (30.4%) of 23 cases in the HCC group at the beginning and the end of follow‐up, respectively, but were not detected in the 20 non‐HCC cases (P = 0.0042 and P = 0.01, respectively). The ranges of the deleted amino acids are listed in Fig. 1. All deletions ended at codon 22 in the pre‐S2 region.
To further evaluate the effect of the above changes at the beginning of follow‐up on the development of HCC, multivariate analysis (proportional hazards model) was carried out using the variables that were significantly associated with the presence of HCC at the beginning of follow‐up as determined by Student's t‐test and Fisher's exact probability test (1, 7), showing only the G1317A mutation and HBeAg negativity were related with the development of HCC (Table 8).
Table 8.
Factors associated with the development of hepatocellular carcinoma (HCC): multivariate cox proportional analysis
| Factor | Multivariate analysis | P‐value | |
|---|---|---|---|
| Risk ratio | 95% CI | ||
| HBeAg | |||
| Presence | 1 | ||
| Absence | 9.60 | 2.01–45.78 | 0.005 |
| PLT(×103/µL) | |||
| <100 | 1 | ||
| ≥100 | 0.41 | 0.12–1.39 | 0.15 |
| T357C change | |||
| Absence | 1 | ||
| Presence | 1.85 | 0.25–13.97 | 0.55 |
| Pre‐S2 deletion | |||
| Absence | 1 | ||
| Presence | 0.79 | 0.18–3.47 | 0.75 |
| G1317A change | |||
| Absence | 1 | ||
| Presence | 3.63 | 1.26–10.45 | 0.02 |
| T1341C/A/G change | |||
| Absence | 1 | ||
| Presence | 0.68 | 0.18–2.52 | 0.56 |
| G1764A change | |||
| Absence | 1 | ||
| Presence | 3.90 | 0.73–20.92 | 0.11 |
| C2288A/G change | |||
| Absence | 1 | ||
| Presence | 0.71 | 0.24–2.10 | 0.53 |
CI, confidence interval. P < 0.05 is indicated by italic type.
Comparison of the activity of wild type and mutant type X promoters. The relative luciferase activity of firefly luciferase was compared among the wild type and mutant type X promoters. M1 (G1317A), M2 (T1341C), and M4 (T1341G) increased the activity of the X promoter 2.3, 3.8, and 1.4 times, respectively, compared with the wild type X promoter in HepG2 cells, but there were no significant changes in Huh7 cells (Fig. 3).
Figure 3.
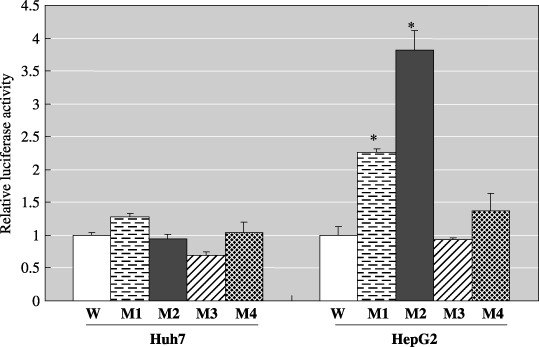
Activation of the wild type and mutant type X promoters, M1 (G1317A), M2 (T1314C), M3 (T1314A), and M4 (T1314G) in Huh7 and HepG2 cells. The observed firefly luciferase activities (mean ± SD) were normalized to the mean Renilla reniformis luciferase activities. *Indicates mean values in mutant type X promoters that are significantly different from that of the wild type X promoter (P < 0.05).
Discussion
This study showed that the number of nucleotide and deduced amino acid substitutions in the full genome and in each ORF region were higher in the HCC group than in the non‐HCC group. The immune response to HBV‐encoded antigens is responsible for both viral clearance and disease pathogenesis during HBV infection. The dominant cause of viral persistence during chronic HBV infection is the development of a weak antiviral immune response to the viral antigen.( 20 ) The nucleotide and deduced amino acid substitutions in the HBV genome will change the target epitopes of some HBV proteins, may help HBV to evade host‐immune surveillance, and thus may contribute to the progression to HCC. The X region in particular had significantly different non‐synonymous substitutions between the non‐HCC group and the HCC group at the end of follow‐up (5, 6), so these non‐synonymous substitutions in HBx may abolish the antiproliferative and apoptotic effect of HBx, thereby causing uncontrolled growth and multiple‐step hepatocarcinogenesis.( 21 )
Furthermore, we found several mutations and deletions in the HBV genome more frequently in the HCC group than in the non‐HCC group at the beginning of follow‐up. These included G1317A and T1341C/A/G mutation in the X promoter and pre‐S2 deletion. Nt 1317 and 1341 were overlapped with the Pol region but these two mutations did not change the amino acid sequence of Pol. The results of the reporter assay showed that the mutant promoters with G1317A and T1341C/G retained higher X promoter activity in HepG2 cells. We therefore speculate that these point mutations might increase the synthesis of HBx mRNA and could be associated with the development of HCC, since HBx has been shown to function as a transcriptional transactivator of a variety of viral and cellular promoter and/or enhancer elements,( 22 ) and the expression of HBx was also reported to be closely related to the pathogenesis of HCC.( 23 )
X‐promoter‐binding protein (X‐PBP) specially interacts with a 16‐bp sequence (nt 1228–1243 in our study) which has promoter activity in X gene transcription. This target sequence is located in the 58‐bp sequence (nt 1211–1268 in our study), which could be enhanced by the HBV enhancer.( 24 ) Recently, NRF1 was reported to specially bind the 21‐bp (including the 16‐bp sequence above) minimal promoter and to positively contribute to transcription of the X gene.( 25 ) In addition to the X‐PBP and NRF‐1 binding sites, other binding sites interact with the transcription factors C/EBP, CREB/ATF2, AP1 located about 130 bp upstream of the X protein‐encoding region.( 26 ) In our study, nt 1317 and nt 1341 were not located in the aforementioned binding sites that interact with X‐PBP and other transcription factors, but changes at nt 1317 and nt 1341 influenced the activity of the X promoter in HepG2 cells, indicating that changes at these two sites might influence X gene transcription to some degree. However, the mutant promoter with T1341A did not change X promoter activity, which was also detected in 8.7% (2/23) of HCC group patients at the beginning of follow‐up, and X promoter activity did not differ between wild type and mutant type X promoters in Huh7 cells; effects of these mutant types of X promoters warrant further study.
Since more pre‐S2 deletion isolates were detected in the pre‐follow up sera of patients who developed HCC, it is likely that the deletion mutant in pre‐S2 is predictive of HCC development. Naturally occurring pre‐S2 deletion has been found in serum samples from HBV carriers.( 27 , 28 ) Huy et al. studied 387 HBV DNA positive serum samples of individuals from 12 countries, and pre‐S2 deletion was observed in 33 samples (8.5%), and was found exclusively at the 5′‐terminus of pre‐S2 region, and pre‐S2 mutants were detected at an even higher rate in HCC cases (34.7%).( 29 ) Takahashi et al. examined sera from 40 cases of HBV‐related HCC and found that 10 of 40 (25%) isolates had pre‐S2 deletions, and mutations of codon 22 were detected in 30% of cases.( 10 ) These results are in accordance with the present study, which showed the detection of pre‐S2 deletions in seven of 23 (30.4%) isolates in the HCC group. A noteworthy finding was that all deletions detected in the pre‐S2 region ended at amino acid 22 of the pre‐S2 region in the current study (Fig. 1).
In the pre‐S2 region, pHSA‐R (aa 3–16),( 30 ) has been identified, against which neutralizing antibodies develop.( 31 ) Previous studies have shown that HLA‐restricted B‐ and T‐cell epitopes of HBV exist in the middle envelope proteins, and middle envelope proteins with a pre‐S2 internal deletion were not recognized in vitro by a putative neutralizing antiserum, suggesting that these variants can evade immune recognition in vivo,( 32 ) and change the course of HBV infection.( 33 ) The deleted region in this study mainly included the pHSA‐R and B‐cell epitope. Chen et al. reported that the detection of pre‐S deletions overlapping with the pHSA‐R region were detected in 57.1% of HCC patients with genotype C, and most of the deletions ended at nt 60.( 34 ) The pre‐S2‐deleted mutant has been reported to display tumor‐promoting phenotypes in Huh7 cells, for example, enhanced proliferation and clonal expansion abilities,( 35 ) and to cause strong oxidative stress and overall genomic instability,( 36 ) the induced genomic instability surely enhances HCC development,( 36 ) and this may be related with the oncogenic properties of this mutant virus.( 31 ) Furthermore, integration of the truncated pre‐S/S gene has been proposed to enhance the development of HCC by expressing a transactivating capacity.( 37 )
It has been suggested that older age, liver cirrhosis and positive HBeAg are important risk factors for HCC.( 38 , 39 , 40 ) Recently, BCP mutations (A1762T/G1764A) were reported to be associated with the development of HCC.( 41 , 42 ) However, other studies have reported that BCP mutations do not predict HCC development and that genotype C HBV infection is an independent risk factor for HCC development.( 43 , 44 ) In this study, all patients were infected with HBV genotype C, and A1762T/G1764A mutations were more common for the HBV genotype C than genotype B,( 45 ) therefore A1762T/G1764A mutations have higher positive rates in both the non‐HCC and HCC groups. Although the G1764A mutation differed significantly only at the beginning of the follow‐up between the non‐HCC and the HCC group, A1762T/G1764A mutations showed a higher positive rate in the HCC group than in the non‐HCC group at both the beginning of follow‐up and the end of follow‐up.
Although several factors differed significantly, according to univariate analysis, between the HCC and non‐HCC groups at the beginning of follow‐up (Table 7), multivariate analysis showed that only the G1317A mutation and HBeAg negativity were associated with the development of HCC. Since pre‐S2 deletions and T1341C/A/G overlapped with each other or with other variables such as A1762T and G1764A mutants, they may be not independent predictors of the development of HCC.
In conclusion, some substitution and deletion mutations may be predictive factors for the development of HCC. G1317A, T1341C/A/G mutations in the X promoter region and pre‐S2 deletion in chronic HBV carriers may be useful markers for predicting the risk of developing HCC.
Acknowledgments
We thank Dr Kato N. for providing the plasmid containing full‐length HBV DNA, and Professor Mizokami M. for providing information on the MEGA software.
References
- 1. Lok AS, McMahon BJ. Chronic hepatitis B: update of recommendations. Hepatology 2004; 39: 857–61. [DOI] [PubMed] [Google Scholar]
- 2. Waris G, Siddiqui A. Regulatory mechanisms of viral hepatitis B and C. J Biosci 2003; 28: 311–21. [DOI] [PubMed] [Google Scholar]
- 3. Koike K. Hepatitis B virus HBx gene and hepatocarcinogenesis. Intervirology 1995; 38: 134–42. [DOI] [PubMed] [Google Scholar]
- 4. Levrero M, Balsano C, Avantaggiati ML et al . Hepatitis B virus and hepatocellular carcinoma: a possible role for the viral transactivators. Ital J Gastroenterol 1991; 23: 576–83. [PubMed] [Google Scholar]
- 5. Kekule AS, Lauer U, Meyer M et al . The preS2/S region of integrated hepatitis B virus DNA encodes a transcriptional transactivator. Nature 1990; 343: 457–61. [DOI] [PubMed] [Google Scholar]
- 6. Lauer U, Weiss L, Hofschneider PH et al . The hepatitis B virus pre‐S/S (t) transactivator is generated by 3′ truncations within a defined region of the S gene. J Virol 1992; 66: 5284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pollicino T, Campo S, Raimondo G. PreS and core gene heterogeneity in hepatitis B virus (HBV) genomes isolated from patients with long‐lasting HBV chronic infection. Virology 1995; 208: 672–7. [DOI] [PubMed] [Google Scholar]
- 8. Pollicino T, Zanetti AR, Cacciola I et al . Pre‐S2 defective hepatitis B virus infection in patients with fulminant hepatitis. Hepatology 1997; 26: 495–9. [DOI] [PubMed] [Google Scholar]
- 9. Kajiya Y, Hamasaki K, Nakata K et al . Full‐length sequence and functional analysis of hepatitis B virus genome in a virus carrier: a case report suggesting the impact of pre‐S and core promoter mutations on the progression of the disease. J Viral Hepat 2002; 9: 149–56. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi K, Akahane Y, Hino K et al . Hepatitis B virus genomic sequence in the circulation of hepatocellular carcinoma patients: comparative analysis of 40 full‐length isolates. Arch Virol 1998; 143: 2313–26. [DOI] [PubMed] [Google Scholar]
- 11. Sumi H, Yokosuka O, Seki N et al . Influence of hepatitis B virus genotypes on the progression of chronic type B liver disease. Hepatology 2003; 37: 19–26. [DOI] [PubMed] [Google Scholar]
- 12. Gunther S, Li BC, Miska S et al . A novel method for efficient amplification of whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J Virol 1995; 69: 5437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stuyver LJ, Locarnini SA, Lok A et al . Nomenclature for antiviral‐resistant human hepatitis B virus mutations in the polymerase region. Hepatology 2001; 33: 751–7. [DOI] [PubMed] [Google Scholar]
- 14. Kobayashi M, Koike K. Complete nucleotide sequence of hepatitis B virus DNA of subtype adr and its conserved gene organization. Gene 1984; 30: 227–32. [DOI] [PubMed] [Google Scholar]
- 15. Huy TT, Ushijima H, Quang VX et al . Genotype C of hepatitis B virus can be classified into at least two subgroups. J Gen Virol 2004; 85: 283–92. [DOI] [PubMed] [Google Scholar]
- 16. Norder H, Courouce AM, Coursaget P et al . Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004; 47: 289–309. [DOI] [PubMed] [Google Scholar]
- 17. Tanaka Y, Orito E, Yuen MF et al . Two subtypes (subgenotypes) of hepatitis B virus genotype C: a novel subtyping assay based on restriction fragment length polymorphism. Hepatol Res 2005; 33: 216–24. [DOI] [PubMed] [Google Scholar]
- 18. Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 1980; 16: 111–20. [DOI] [PubMed] [Google Scholar]
- 19. Mizokami M, Orito E, Ohba K et al . Constrained evolution with respect to gene overlap of hepatitis B virus. J Mol Evol 1997; 44: S83–90. [DOI] [PubMed] [Google Scholar]
- 20. Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol 1995; 13: 29–60. [DOI] [PubMed] [Google Scholar]
- 21. Sirma H, Giannini C, Poussin K et al . Hepatitis B virus X mutants, present in hepatocellular carcinoma tissue abrogate both the antiproliferative and transactivation effects of HBx. Oncogene 1999; 18: 48 448–59. [DOI] [PubMed] [Google Scholar]
- 22. Yen TS. Hepadnaviral X protein: Review of recent progress. J Biomed Sci 1996; 3: 20–30. [DOI] [PubMed] [Google Scholar]
- 23. Urashima T, Saigo K, Kobayashi S et al . Identification of hepatitis B virus integration in hepatitis C virus‐infected hepatocellular carcinoma tissues. J Hepatol 1997; 26: 771–8. [DOI] [PubMed] [Google Scholar]
- 24. Nakamura I, Koike K. Identification of a binding protein to the X gene promoter region of hepatitis B virus. Virology 1992; 191: 533–40. [DOI] [PubMed] [Google Scholar]
- 25. Tokusumi Y, Zhou S, Takada S. Nuclear respiratory factor 1 plays an essential role in transcriptional initiation from the hepatitis B virus gene promoter. J Virol 2004; 78: 10 856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo WT, Bell KD, Ou JH. Characterization of the hepatitis B virus Enh1 Enhancer and X promoter complex. J Virol 1991; 65: 6686–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Raimondo G, Campo S, Smedile V et al . Hepatitis B virus variant, with a deletion in the preS2 and two translational stop codons in the precore regions, in a patient with hepatocellular carcinoma. J Hepatol 1991; 13: S74–7. [DOI] [PubMed] [Google Scholar]
- 28. Santantonio T, Jung MC, Schneider R et al . Hepatitis B virus genomes that cannot synthesize pre‐S2 proteins occur frequently and as dominant virus populations in chronic carriers in Italy. Virology 1992; 188: 948–52. [DOI] [PubMed] [Google Scholar]
- 29. Huy TT, Ushijima H, Win KM et al . High prevalence of hepatitis B virus pre‐s mutant in countries where it is endemic and its relationship with genotype and chronicity. J Clin Microbiol 2003; 41: 5449–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pontisso P, Schiavon E, Fraiese A et al . Antibody to the hepatitis B virus receptor for polymerized albumin in acute infection and in hepatitis B vaccine recipients. J Hepatol 1986; 3: 393–8. [DOI] [PubMed] [Google Scholar]
- 31. Fan YF, Lu CC, Chen WC et al . Prevalence and significance of hepatitis B virus (HBV) pre‐S mutants in serum and liver at different replicative stages of chronic HBV infection. Hepatology 2001; 33: 277–86. [DOI] [PubMed] [Google Scholar]
- 32. Tai PC, Suk FM, Gerlich WH et al . Hypermodification and immune escape of an internally deleted middle‐envelope (M) protein of frequent and predominant hepatitis B virus variants. Virology 2002; 292: 44–58. [DOI] [PubMed] [Google Scholar]
- 33. Gerken G, Paterlini P, Manns M et al . Assay of hepatitis B virus DNA by polymerase chain reaction and its relationship to pre‐S‐ and S‐encoded viral surface antigens. Hepatology 1991; 13: 158–66. [PubMed] [Google Scholar]
- 34. Chen BF, Liu CJ, Jow GM et al . High prevalence and mapping of pre‐S deletion in hepatitis B virus carriers with progressive liver diseases. Gastroenterology 2006; 130: 1153–68. [DOI] [PubMed] [Google Scholar]
- 35. Wang HC, Wu HC, Chen CF et al . Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre‐S mutants that may induce endoplasmic reticulum stress. Am J Pathol 2003; 163: 2441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hsieh YH, Su IJ, Wang HC et al . Pre‐S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004; 25: 2023–32. [DOI] [PubMed] [Google Scholar]
- 37. Caselmann WH, Meyer M, Kekule AS et al . A trans‐activator function is generated by integration of hepatitis B virus preS/S sequences in human hepatocellular carcinoma DNA. Proc Natl Acad Sci USA 1990; 87: 2970–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McMahon BJ, Holck P, Bulkow L et al . Serologic and clinical outcomes of 1536 Alaska Natives chronically infected with hepatitis B virus. Ann Intern Med 2001; 135: 759–68. [DOI] [PubMed] [Google Scholar]
- 39. Fattovich G, Giustina G, Schalm SW et al . Occurrence of hepatocellular carcinoma and decompensation in western European patients with cirrhosis type B. The EUROHEP Study Group on Hepatitis B Virus and Cirrhosis. Hepatology 1995; 21: 77–82. [DOI] [PubMed] [Google Scholar]
- 40. Yang HI, Lu SN, Liaw YF et al . Hepatitis B e antigen and the risk of hepatocellular carcinoma. N Engl J Med 2002; 347: 168–74. [DOI] [PubMed] [Google Scholar]
- 41. Kao JH. Hepatitis B virus genotypes and hepatocellular carcinoma in Taiwan. Intervirology 2003; 46: 400–7. [DOI] [PubMed] [Google Scholar]
- 42. Baptista M, Kramvis A, Kew MC. High prevalence of 1762(T) 1764(A) mutations in the basic core promoter of hepatitis B virus isolated from black Africans with hepatocellular carcinoma compared with asymptomatic carriers. Hepatology 1999; 29: 946–53. [DOI] [PubMed] [Google Scholar]
- 43. Chan HL, Tsang SW, Wong ML et al . Genotype B hepatitis B virus is associated with severe icteric flare‐up of chronic hepatitis B virus infection in Hong Kong. Am J Gastroenterol 2002; 97: 2629–33. [DOI] [PubMed] [Google Scholar]
- 44. Chan HL, Hui AY, Wong ML et al . Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut 2004; 53: 1494–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kao JH, Chen PJ, Lai MY et al . Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology 2003; 124: 327–34. [DOI] [PubMed] [Google Scholar]