

Abstract
Curcumin, the active component of turmeric, has been shown to protect against carcinogenesis and prevent tumor development in cancer. To enhance its potency, we tested the efficacy of synthetic curcumin analogues, known as FLLL11 and FLLL12, in cancer cells. We examined the impact of FLLL11 and FLLL12 on cell viability in eight different breast and prostate cancer cell lines. FLLL11 and FLLL12 (IC50 values 0.3–5.7 and 0.3–3.8 µmol/L, respectively) were substantially more potent than curcumin (IC50 values between 14.4–50 µmol/L). FLLL11 and FLLL12 were also found to inhibit AKT phosphorylation and downregulate the expression of HER2/neu. In addition, we demonstrate for the first time that FLLL11 and FLLL12 inhibit phosphorylation of signal transducer and activator of transcription (STAT) 3, an oncogene frequently found to be persistently active in many cancer types. The inhibition of STAT3 signaling was confirmed by the inhibition of STAT3 DNA binding and STAT3 transcriptional activity. Furthermore, FLLL11 and FLLL12 were more effective than curcumin in inhibiting cell migration and colony formation in soft agar as well as inducing apoptosis in cancer cells. These results indicate that FLLL11 and FLLL12 exhibit more potent activities than curcumin on the inhibition of STAT3, AKT, and HER‐2/neu, as well as inhibit cancer cell growth and migration, and may thus have translational potential as chemopreventive or therapeutic agents for breast and prostate cancers. (Cancer Sci 2009; 100: 1719–1727)
The cellular mechanisms contributing to breast and prostate cancer involve a multistep process that includes the inactivation of tumor suppressor genes and the dysregulation of several oncogenic pathways. Among the signaling pathways implicated are the AKT, HER2/neu, and signal transducers and activators of transcription (STAT) 3 pathways.( 1 , 2 , 3 , 4 , 5 ) Activated or phosphorylated AKT appears to play an important role in proliferation, chemoresistance, and resistance to hormone therapy of breast cancer cells.( 6 , 7 , 8 ) The AKT pathway has emerged as one of the key oncogenic pathways in breast cancer, carrying with it the potential for therapeutic intervention.( 9 ) Although AKT can be activated through a variety of mechanisms, its dysregulation in many breast cancers stems from overexpression of the HER2/neu protein.( 10 ) HER2/neu is a 185‐kDa surface membrane protein that is overexpressed in approximately 25–30% of breast cancers due to amplification of the HER2 gene.( 5 ) Patients with breast cancers that overexpress HER2/neu generally have a poor prognosis, experiencing shorter relapse times and low survival rates.( 11 ) Evidence suggests that cancer cells that overexpress HER2/neu may also be less sensitive to chemotherapy.( 12 , 13 )
STAT3 is a latent transcription factor and is one of the downstream signaling proteins for cytokine and growth factor receptors.( 14 , 15 ) Activation of these receptors induces the phosphorylation and subsequent dimerization of two STAT3 monomers through conserved SH2 domains.( 16 , 17 ) Persistently active STAT3 has been found with high frequency in a wide range of human cancer cell lines and tissues,( 18 ) where it has been implicated in stimulating cell proliferation, promoting angiogenesis, mediating immune evasion, and conferring increased resistance to apoptosis.( 19 , 20 , 21 , 22 , 23 ) Mora et al. reported constitutive activation of STAT3 in 37 out of 45 (82%) prostate tumor samples.( 3 ) Barton et al. also reported a similar observation except that all the prostate tumor samples they examined had constitutive activation of STAT3.( 4 ) The constitutive activation of STAT3 is also frequently detected in breast cancer specimens with advanced diseases.( 20 , 24 , 25 ) STAT3‐associated activities are apparently required for the continued survival of certain cancer cells, as interference of the STAT3 pathway has been found to result in growth inhibition and induction of apoptosis,( 26 , 27 , 28 ) furthering its interest as a potential chemotherapeutic target.( 29 , 30 , 31 , 32 )
Because the dysregulation of multiple oncogenic pathways is common among many cancers, an optimal therapeutic agent would need to be able to inhibit multiple pathways simultaneously while causing minimal deleterious side effects. One compound that may function in this capacity is curcumin, the bioactive component of the perennial herb Curcuma longa. Extensive research has revealed that the complex chemistry of curcumin allows it to influence multiple cell signaling pathways, giving it anti‐inflammatory, antioxidant, chemopreventive, and chemotherapeutic properties in addition to many others.( 33 ) Among curcumin's numerous molecular targets are components of the AKT, HER2/neu, and STAT3 pathways, whose inhibition requires exposure to relatively high concentrations of the agent.( 34 ) Although large dosages of curcumin tend to be well tolerated, testing in animal models and human clinical trials has revealed that the bioavailability of curcumin is low, owing to its poor absorption across the gut, limited tissue distribution, rapid metabolism, and subsequent elimination from the body.( 35 ) One potential method of circumventing this limitation has been the design and synthesis of curcumin structural analogues. Two such compounds, termed FLLL11 and FLLL12 (Fig. 1), were synthesized by our laboratories as part of a series of novel curcumin analogues. Initial screening revealed that these two compounds were particularly effective at inhibiting cancer cell viability, warranting a closer look at their anticarcinogenic properties. In our present study, we compared the inhibitory effects of FLLL11, FLLL12, and curcumin in breast and prostate cancer cells that express elevated levels of STAT3 and AKT phosphorylation as well as HER2/neu protein. Our results demonstrated that these two curcumin analogues can effectively inhibit STAT3, AKT, and HER2/neu pathways.
Figure 1.
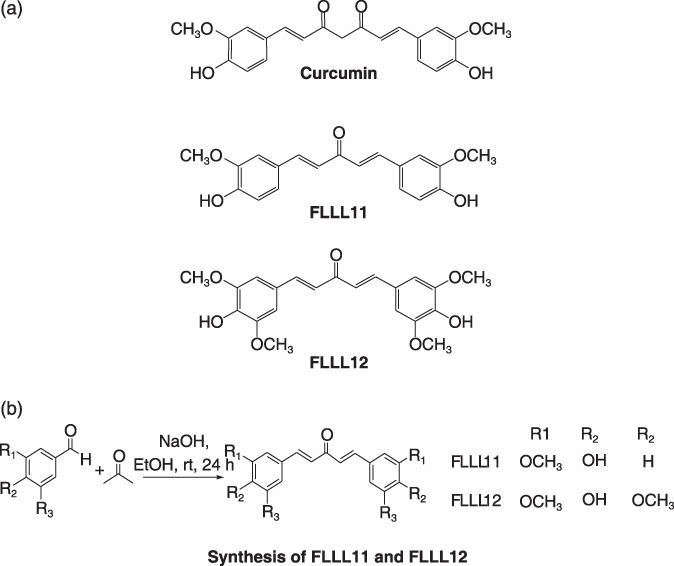
(a) The chemical structures of curcumin, FLLL11, and FLLL12. (b) The general synthesis scheme for FLLL11 and FLLL12.
Materials and Methods
Cell culture. MDA‐MB‐231, MCF‐7, MDA‐MB‐468, SK‐BR‐3, BT‐474, and MDA‐MB‐453 breast cancer cells, PC‐3 and DU145 prostate cancer cells, and WI‐38 normal human lung fibroblasts were acquired from the American Type Culture Collection (Manassas, VA, USA). Human mammary epithelial cells (HMEC) were purchased from Lonza Walkersville (Walkersville, MD, USA). These cancer and normal cells were maintained in DMEM, 1× with 4.5 g/L l‐glutamine and sodium pyruvate (Mediatech, Manassas, VA, USA), supplemented with 10% FBS (Invitrogen, Carlsbad, CA, USA) and 1% penicillin and streptomycin in cell culture incubators that were set at 37°C and aired with 5% CO2.
Western blot analysis. Breast and prostate cancer cells were treated with 5 or 10 µmol/L FLLL11, FLLL12, or curcumin (Sigma‐Aldrich, St. Louis, MO, USA) for 24 h. For Western blots, 50 µg of total protein from cancer cell lysates were subjected to SDS‐PAGE and transferred to PVDF membrane. Phospho‐specific STAT3 antibody (Tyr705), phospho‐independent STAT3 antibody, phospho‐specific AKT antibody (Ser473), Akt antibody, phospho‐specific ERK1/2 antibody (Thr202 and Tyr204), cleaved poly (ADP‐ribose) polymerase (PARP) antibody, cleaved caspase‐3, HER‐2/neu, and GAPDH antibody were purchased from Cell Signaling Technologies (Baverly, MA, USA). Membranes were blotted with these antibodies and scanned with the Storm PhosphorImager (Amersham Pharmacia Biotech, Piscataway, NJ, USA) using enhanced chemiluminescence plus reagents.
MTT cell viability assay. Breast and prostate cancer cells were seeded in 96‐well plates (3000 cells/well) in triplicate in 10% FBS. The following day, cells were treated with 0.5–5 µmol/L FLLL11 and FLLL12, or 0.5–30 µmol/L curcumin for 72 h. Then 25 µL MTT (M5655; Sigma‐Aldrich) was added to each sample. After 3.5 h, 100 µL N,N‐dimethylformamide (D4551; Sigma‐Aldrich) solubilization solution was added to each well. The absorbance was read at 590 nm the following day. The viability of the untreated cells was arbitrarily set at 100% and compared with the viability of curcumin‐, FLLL11‐, and FLLL12‐treated cells. IC50 were determined using Sigma Plot 9.0 software (SYSTAT Software, Chicago, IL, USA) using the four parameter logistic function standard curve analysis for dose response.
Determination of combinatorial effects. The ability of doxorubicin, FLLL11, and FLLL12 to act in a synergistic matter with regard to growth inhibition was determined as follows. Briefly, the log (fa/fu) was plotted against the concentration (D) for each compound alone or in combination, where fa is the fraction affected and fu is the unaffected fraction (1‐fa) of cells at each concentration. Calcusyn software (Biosoft, Great Shelford, Cambridge, UK) was used to determine the combinational index (CI) for each concentration of drug mixture used. A value of CI <1 represents a case where synergism of doxorubicin, FLLL11, and FLLL12 was present. CI values of 1 and >1 represent additive and antagonistic effects respectively.
STAT3 DNA binding activity and STAT3‐dependent transcriptional luciferase activity. MDA‐MB‐231, SK‐BR‐3, and DU145 breast and prostate cancer cells were treated with 10 µmol/L FLLL12, FLLL11, and curcumin, or DMSO for 24 h. The nuclear extracts were analyzed for STAT3 DNA binding activity using TransFactor Universal STAT3‐specific kits following the manufacturer's protocol (Clontech, Moutain View, CA, USA). To assess transcriptional activity, MDA‐MB‐231 cells were stably transfected with the pLucTKS3 luciferase reporter, a construct that contains seven copies of the STAT3 binding site in a thymidine kinase minimal reporter. Expression of luciferase is contingent upon activation of STAT3.( 36 ) These cells were grown to semiconfluency in six‐well plates and then treated with the listed concentrations of FLLL11, FLLL12, curcumin, or DMSO for 24 h in 5% FBS DMEM. The cells were washed with PBS and harvested in reporter lysis buffer. Twenty µL of lysate was used in the luciferase assay in accordance with the manufacturer's protocol (Promega, Madison, WI, USA). Samples were run in triplicate. The data were normalized via a bicinchoninic acid (BCA) protein assay (Thermo Scientific, Waltham, MA, USA) to determine the total amount of protein present in each lysate. Luciferase activity is expressed as counts per second per µg protein. Curcumin‐, FLLL11‐, and FLLL12‐treated STAT3 luciferase activity is presented relative to a pLucTKS3‐transfected sample treated with DMSO, arbitrarily set at 100%.
Soft agar colony formation assay. A 0.6% agar gel with 10% FBS in DMEM was prepared and added to the wells of a six‐well culture dish as a base agar. Five thousand cells per well were plated for anchorage‐independent growth analysis in 0.4% agar gel with 10% FBS in DMEM supplemented with the target treatment on top of the base agar. The MDA‐MB‐231 and SK‐BR‐3 breast cancer cells were allowed to grow at 37°C for 2 weeks. The effect of the drugs on anchorage‐independent growth was determined by colony growth. Colonies were stained with MTT dye (100 µL per well) and pictures of the colonies were taken with a Leica MZ 16FA inverted microscope (Leica Microsystems, Bannockburn, IL, USA) with a 7.4 Slider Camera (Diagnostic Instruments, Sterling Heights, MI, USA). Colonies were scored by counting with an inverted microscope. Numbers were normalized as a percentage of colonies formed in DMSO treatment.
Cell migration assay. For cell migration assays, MDA‐MB‐231 breast cancer cells (3 × 105 cells per well) were plated in a six‐well plate. Approximately 24–48 h later, when the cells were 100% confluent, the monolayer was scratched using a 1‐mL pipette tip. Medium and non‐adherent cells were aspirated, the adherent cells were washed once, and new medium containing various concentrations of FLLL12 and FLLL11 (1–20 µmol/L) was added for 4 h. Cells were observed under the microscope after an additional 20 h. The inhibition of migration was assessed when the wound in the control was closed. ImageJ software, available from the NIH website (http://rsb.info.nih.gov/ij/), was used to quantify cell migration assay data. Three random pictures were taken for each treated well and the areas of the wounds measured using ImageJ software. The percentage of wound healed was then calculated using the formula: 100 – (final area/initial area × 100%).
Results
FLLL11 and FLLL12 are more potent inhibitors of the proliferation of breast cancer cells than curcumin. We determined the IC50 of a panel of 24 novel analogues of curcumin in MDA‐MB‐231 and MCF‐7 breast cancer cell lines.( 37 ) FLLL11 and FLLL12 (Fig. 1a) are among the most potent curcumin analogues with lowest IC50 values and we chose these two compounds for further testing in additional breast cancer cell lines. The synthesis of FLLL11 and FLLL12 is shown in Figure 1(b).
We next tested the inhibitory efficacy of FLLL11 and FLLL12 in four additional human breast cancer cell lines, MDA‐MB‐468, BT‐474, SKBR‐3, and MDA‐MB‐453. In total, six breast cancer cell lines were tested (Table 1) representing at least four distinct subtypes of breast cancer (based on molecular genetic profiling) that have prognostic significance:( 38 , 39 , 40 ) (1) MDA‐MB‐468 and MDA‐MB‐231 cells are basal‐like (estrogen receptor [ER] negative, progesterone receptor [PR] negative, and HER2 negative); (2) SKBR‐3 and MDA‐MB‐453 cells overexpress HER2 (ER−PR−HER2+); (3) BT‐474 cells are similar to luminal B (ER+PR+HER2+); and (4) MCF‐7 cells are similar to luminal A (ER+PR+HER2−). FLLL11 and FLLL12 were found to be more potent than curcumin at inhibiting cell viability in all six breast cancer cell lines (Table 1). The IC50 values for FLLL11 and FLLL12 ranged from 9‐ to 48‐fold lower than curcumin in these four subtypes of breast cancer cells (Table 1). FLLL11 and FLLL12 were also more potent than curcumin against the human prostate cancer cell lines PC‐3 and DU145. The IC50 values for FLLL11 and FLLL12 were 3.6–3.9 and 3.0–3.6 µmol/L, respectively, compared to IC50 values of 19.8–39.6 µmol/L observed with curcumin (Table 1). FLLL11 and FLLL12 were much more effective than curcumin against all of the cancer cell lines we initially evaluated. Importantly, these two compounds also maintained curcumin's reputed low toxicity against normal cell lines, as evidenced by their inability to decrease viability in the WI‐38 normal human lung fibroblasts (Table 1).
Table 1.
IC50 (µM) of FLLL12, FLLL11, and curcumin in human breast and prostate cancer cells and WI‐38 normal human fibroblasts
| Cell line | FLLL11 | FLLL12 | Curcumin |
|---|---|---|---|
| MDA‐MB‐468 | 0.3 | 0.3 | 14.4 |
| MDA‐MB‐231 | 2.8 | 2.7 | 25.6 |
| SK‐BR‐3 | 5.7 | 3.8 | 37.7 |
| BT‐474 | 5.6 | 1.8 | 41.9 |
| MDA‐MB‐453 | 4.7 | 1.3 | >50.0 |
| MCF‐7 | 2.4 | 1.7 | 21.5 |
| PC‐3 | 3.9 | 3.6 | 19.8 |
| DU145 | 3.6 | 3 | 39.6 |
| WI‐38 normal human lung fibroblasts | >1000.0 | >1000.0 | >1000.0 |
FLLL12 downregulates HER2/neu expression and AKT phosphorylation. The HER2/neu and AKT pathways play important roles in drug resistance, invasion, and angiogenesis, and influence a host of other oncogenic functions.( 6 , 7 , 41 , 42 , 43 ) Because HER2/neu overexpression is an important factor in the progression of many breast cancers, we tested whether FLLL11 and FLLL12 were capable of downregulating its expression. We observed that both FLLL11 and FLLL12 could downregulate protein levels of HER2/neu in BT‐474 and SK‐BR‐3 breast cancer cells; however, FLLL12 seemed to be slightly more potent (Fig. 2a,b). FLLL12 also downregulated HER2/neu expression in MDA‐MB‐453 breast cancer cells (Fig. 2c). In addition, FLLL12 but not FLLL11 inhibited AKT phosphorylation (serine 473) in MDA‐MB‐453 breast cancer cells (Fig. 2c) and PC‐3 prostate cancer cells (Fig. 2d). In MDA‐MB‐453 breast cancer cells, FLLL12 also decreased AKT expression (Fig. 2c). Both FLLL11 and FLLL12 induced apoptosis in these breast and prostate cancer cells, as evidenced by increased levels of cleaved PARP (Fig. 2a–d) and cleaved caspase‐3 (Fig. 2b–d). Similar treatment of these cell lines with equimolar concentrations of curcumin had little to no observable impact (Fig. 2a–d).
Figure 2.
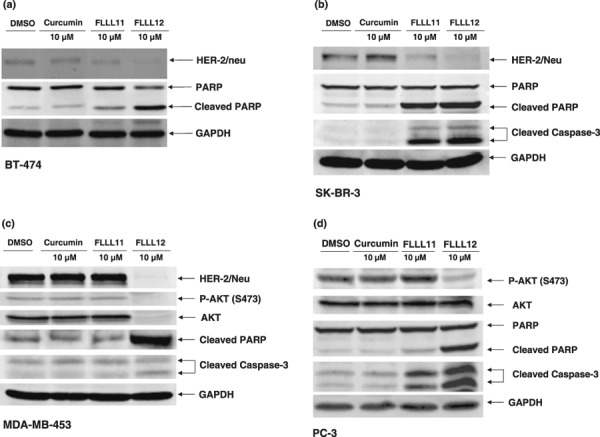
The inhibitory effects of AKT phosphorylation and HER‐2/neu protein expression by FLLL11, FLLL12, and curcumin in (a) BT‐474, (b) SK‐BR‐3, (c) MDA‐MB‐453, and (d) PC‐3 human cancer cell lines. Cancer cells were treated with 5–10 µmol/L FLLL11 and FLLL12, or 5–10 µmol/L curcumin for 24 h. Membranes were blotted with HER‐2/neu, phospho‐specific AKT (S473), cleaved poly (ADP‐ribose) polymerase (PARP), cleaved caspase‐3, and GAPDH antibodies.
FLLL11 and FLLL12 are potent inhibitors of STAT3 phosphorylation and associated activities in breast and prostate cancer cells. We next examined whether FLLL11 and FLLL12 could inhibit STAT3 phosphorylation in the MDA‐MB‐231 and SK‐BR‐3 breast cancer cells and DU145 prostate cancer cells, all of which express elevated levels of phosphorylated and active STAT3.( 24 ) Both FLLL11 and FLLL12 inhibited STAT3 phosphorylation (Tyr705) in MDA‐MB‐231 (Fig. 3a), SK‐BR‐3 (Fig. 3b), and DU145 (Fig. 3c), but were ineffective at preventing phosphorylation of the ERK1/2 MAP kinase. With regard to STAT3 phosphorylation, FLLL12 was found to be a more potent inhibitor than FLLL11, if only to a slight extent. We also observed a correlation between reduction of phosphorylated STAT3 levels and the induction of apoptosis (determined by cleavage of PARP and caspase‐3), which again reflects the necessity of persistent activation of this transcription factor in many cancers.( 18 ) However, FLLL11 and FLLL12 (10 µmol/L) did not induce cleaved PARP and caspase‐3 in HMEC and WI‐38 normal human lung fibroblasts (Fig. 3d).
Figure 3.
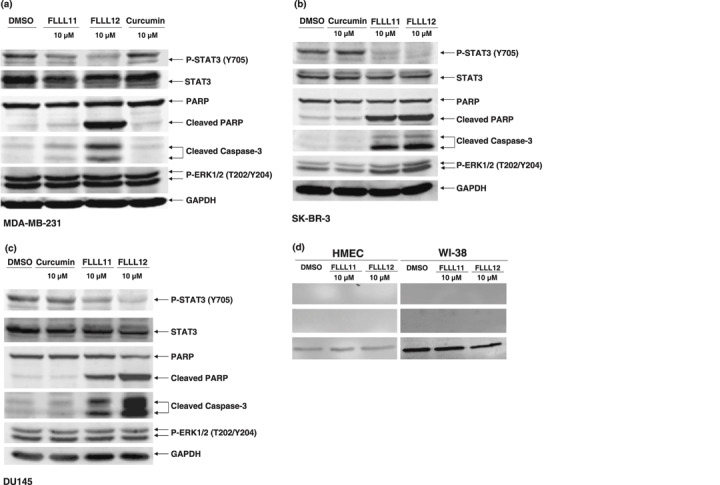
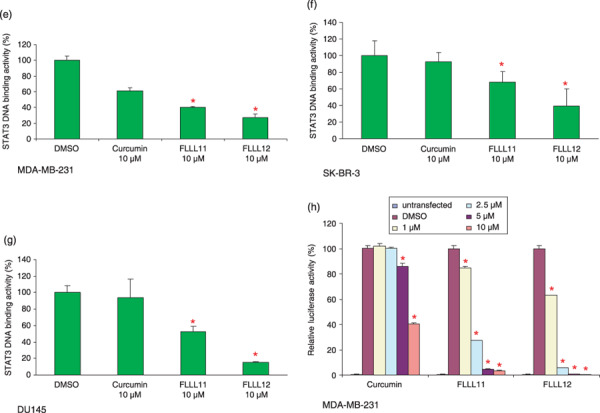
FLLL11 and FLLL12 inhibit STAT 3 phosphorylation in (a) MDA‐MB‐231, (b) SK‐BR‐3, and (c) DU145 breast and prostate cancer cells. Cells were treated for 24 h. Membranes were blotted with phospho‐specific STAT3, phospho‐independent STAT3, phospho‐specific ERK1/2, cleaved poly (ADP‐ribose) polymerase (PARP), cleaved caspase‐3, and GAPDH antibodies. (d) FLLL11 and FLLL12 (10 µmol/L) induce cleaved PARP and cleaved caspase‐3 in breast cancer cells but not in human mammary epithelial cells (HMEC) and WI‐38 normal human lung fibroblasts. The inhibitory effects of FLLL11 and FLLL12 on STAT3 DNA binding activity in (e) MDA‐MB‐231 (f), SK‐BR‐3, and (g) DU145 cancer cells. The nuclear extracts were analyzed for STAT3 DNA binding activity using STAT3‐specific TransFactor kit. Statistical significance (P < 0.01) relative to the DMSO vehicle control is designated by an asterisk. (h) The inhibitory effect of FLLL11 and FLLL12 on STAT3‐dependent transcriptional activity was analyzed in MDA‐MB‐231 breast cancer cells that stably integrate the STAT3‐dependent luciferase reporter construct. The data were subsequently normalized and presented here as percentages of the DMSO vehicle control. Asterisks denote statistically significant (P < 0.01) decreases in luciferase activity.
To further confirm the inhibition of STAT3 signaling by FLLL11 and FLLL12, we examined the impact these compounds had on STAT3 DNA binding and transcriptional activity in cancer cells. Treatment of MDA‐MB‐231, SK‐BR‐3, and DU145 with FLLL11 and FLLL12 resulted in significantly more inhibition of STAT3 DNA binding activity than that observed with curcumin (Fig. 3e–g). A luciferase assay was also used to gauge FLLL11‐ and FLLL12‐mediated repression of STAT3 transcriptional activity. Due to its high endogenous levels of phosphorylated STAT3 protein, MDA‐MB‐231 breast cancer cells were chosen to be stably transfected with pLucTKS3, a luciferase construct that features seven copies of the STAT3 binding site in a thymidine kinase minimal promoter. Expression of luciferase is thus contingent upon the phosphorylation and activation of STAT3.( 36 ) Each compound reduced STAT3‐mediated transcription of luciferase in a dose‐dependent manner, and FLLL11 and FLLL12 again showed much greater efficacy than curcumin (Fig. 3h). Dosage for dosage, FLLL12 displayed greater potency than FLLL11 in inhibiting STAT3‐dependent transcriptional activity, which was consistent with our earlier observations regarding the inhibition of STAT3 phosphorylation (Fig. 3a–c). Taken together, these results confirm that FLLL11 and FLLL12 are potent inhibitors of activated STAT3 in vitro.
FLLL11 and FLLL12 inhibit anchorage‐independent growth and cell migration in human breast cancer cells. The ability of transformed cells to grow and proliferate in the absence of substratum attachment is one of the hallmarks of malignancy and is vitally important in the formation of the tumor.( 44 ) As curcumin has been shown to downregulate STAT3( 45 ) and AKT( 46 ) phosphorylation, which may have an effect on anchorage independency,( 47 , 48 ) we hypothesized that curcumin would lead to a decrease in the number of colonies formed in soft agar and that FLLL11 and FLLL12 would show an even greater decrease. As expected, treatment with curcumin, FLLL11, and FLLL12 led to decreased colony formation in soft agar when compared to the untreated control in MDA‐MB‐231 and SK‐BR‐3 breast cancer cells (Fig. 4a,b). Compared to the control samples, a 5 µmol/L concentration of curcumin produced a decrease of nearly 60% in colony formation. Equal concentrations of FLLL11 and FLLL12, on the other hand, showed 95 and 80% reductions in colony numbers respectively.
Figure 4.
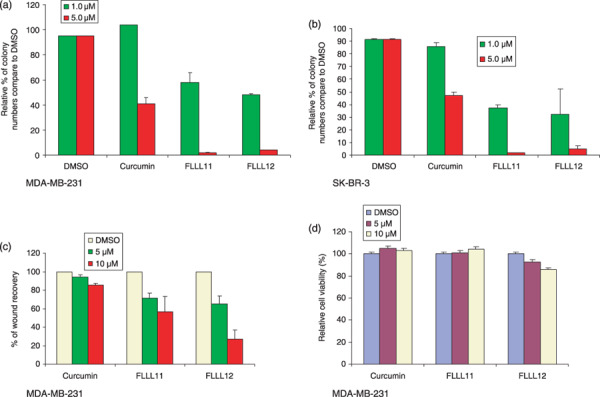
The ability of MDA‐MB‐231 breast cancer cells to form colonies in soft agar is decreased following treatment with 5 µmol/L curcumin or 1–5 µmol/L FLLL11 and FLLL12 in (a) MDA‐MB‐231 and (b) SK‐BR‐3 breast cancer cells. (c) Migration of MDA‐MB‐231 into a scratch wound is impeded after 4 h of treatment with curcumin, FLLL11, or FLLL12. (d) MTT assays of MDA‐MB‐231 cells reveal that the dosages of these agents used in the migration assay have minimal impact on viability over 4 h of drug treatment and an additional 20 h without treatment.
Cell migration is necessary in many physiological processes, such as wound healing and tumor metastasis. To assess whether FLLL11 and FLLL12 similarly affected cell migration, a wound healing assay was carried out with MDA‐MB‐231 breast cancer cells. Upon inflicting a scratch wound, these cells were treated with various concentrations of FLLL11 or FLLL12 for 4 h and returned to standard media in an attempt to minimize any cytotoxic effects that could potentially confound our observations. Following 20 h of further incubation, the areas of the wounds were measured using ImageJ software. Migration of the FLLL11‐ and FLLL12‐treated cells was impaired relative to the curcumin‐treated samples (Fig. 4c). FLLL12 has greater inhibitory effect than FLLL11 and curcumin (Fig. 4c). The ability of FLLL12 or FLLL11 to inhibit cell migration may not be due to their ability to inhibit cell proliferation (Fig. 4d).
Quantitative combinatorial effects between FLLL11, FLLL12, and doxorubicin. We next examined the possibility of synergistic effects of FLLL11 and FLLL12 with more established chemotherapeutic drugs, such as doxorubicin. Doxorubicin is a powerful inhibitor of DNA replication, and although it has proven effective against many types of cancer, it is associated with a host of acute and chronic side effects that increase in severity with cumulative dosing. Treatment of these cells with doxorubicin resulted in dose‐dependent decreases in their viability in a triple‐negative breast cancer cell line, MDA‐MB‐231. A representative combination figure is presented (Fig. 5a,b). As can be seen, all concentrations tested have a CI value smaller than 1, indicating synergism between doxorubicin and FLLL11 (Fig. 5a) or FLLL12 (Fig. 5b). These data clearly demonstrate a synergistic effect between doxorubicin and FLLL11 or FLLL12 that could potentially prove useful in therapy.
Figure 5.
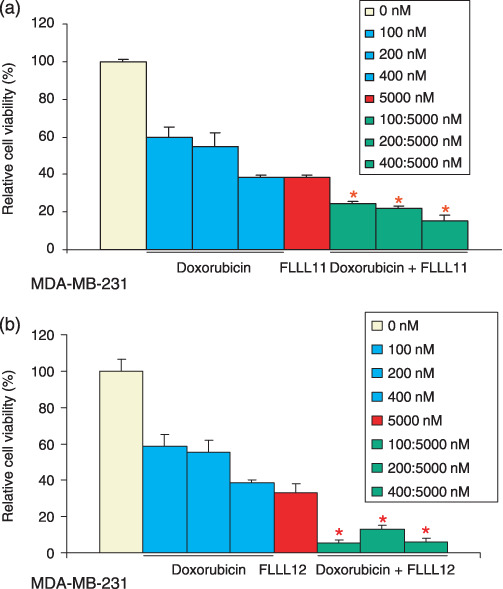
The inhibitory effects of cell viability by FLLL11 and FLLL12 in combination with doxorubicin in the MDA‐MB‐231 human breast cancer cell line. MDA‐MB‐231 breast cancer cells were treated with (a) FLLL11, (b) FLLL12, and doxorubicin for 72 h. The cell viability was determined by MTT assay. The untreated cells were set at 100% and the viability of doxorubicin‐, FLLL11‐, and FLLL12‐treated cells was determined relative to the untreated cells. Asterisks represent synergism (a combinational index less than 1).
Discussion
Breast cancer remains the second most common cancer after skin cancer in women and is second in mortality only to lung cancer. Likewise, prostate cancer is the second most common type of cancer found in American men, other than skin cancer. Curcumin has been shown to protect against carcinogenesis or prevent tumor formation and development in several cancer types and also to suppress angiogenesis and metastasis in a variety of animal tumor models.( 34 , 49 , 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 ) From the published literature, curcumin has been shown to exhibit inhibitory effects in breast cancer cells( 51 , 58 ) and in prostate cancer cells.( 59 , 60 , 61 ) Curcumin has been shown to inhibit multiple pathways including the STAT3, HER2/neu, and AKT pathways, albeit at high concentrations.( 34 , 45 , 62 ) These results suggest that curcumin might be an ideal agent to target multiple oncogenic pathways in breast and prostate cancer. Although the growth suppressive activity and bioavailability of curcumin in humans may be effective as a preventive agent, it may not be sufficient as an effective therapeutic agent in cancer. Therefore, more potent curcumin analogues that can inhibit STAT3, AKT, and HER2/neu pathways with lower doses are needed as more effective therapeutic agents for cancer treatments.
We demonstrated that two new analogues, FLLL11 and FLLL12, are more potent than curcumin at inhibiting cell viability, cell migration, and colony formation in soft agar, and they induced apoptosis in human breast and prostate cancer cells. In addition, we demonstrated that FLLL11 and FLLL12 can synergize with doxorubicin to suppress the growth of MDA‐MB‐231 breast cancer cells. Besides FLLL11 and FLLL12, additional curcumin analogues such as dimethoxycurcumin, EF‐24, GO‐Y030, and others have been reported.( 63 , 64 , 65 , 66 ) However, none of the current curcumin analogues have been reported to be able to inhibit the STAT3, HER2/neu, and AKT pathways. Our results demonstrate that two new curcumin analogues, FLLL11 and FLLL12, can effectively inhibit all three of these pathways. Numerous reports have shown that inhibiting STAT3 activation by a dominant‐negative mutant, antisense oligonucleotides, small interfering RNA, and other methods in human cancer cells suppresses proliferation and induces apoptosis in vitro and tumorigenicity in vivo. ( 4 , 67 ) These results demonstrate that STAT3 is crucial to the survival and growth of tumor cells. Our data show that FLLL11 and FLLL12 are more potent than curcumin at inhibiting STAT3 phosphorylation, STAT3 DNA binding, and STAT3‐dependent transcriptional activity in breast and/or prostate cancer cells. Blocking signaling to STAT3 by FLLL11 and FLLL12 may represent one of the more potent therapeutic approaches to targeting the STAT3 pathway in breast and prostate cancers that express constitutive STAT3 signaling. In addition, constitutive AKT signaling appears to play an important role in proliferation, chemoresistance, and resistance to hormone therapy of prostate( 2 , 68 , 69 ) and breast cancer cells.( 7 , 8 ) Furthermore, patients with breast cancer that overexpress HER2/neu have a poor prognosis, shorter relapse time, and low survival rate.( 11 ) Our results showed that FLLL12, but not FLLL11, is more potent than curcumin in downregulating AKT phosphorylation in breast and prostate cancer cells. Our results also showed that FLLL11 and FLLL12 are more potent than curcumin in downregulating the expression of HER2/neu. In summary, our findings demonstrated that the curcumin analogues FLLL11 and FLLL12 could have translational potential as novel cancer therapeutic agents. For Absorption, Distribution, Meabolism, Excretion, and Toxicity (ADMET) pharmacokinetic and pharmacodynamic assessments, fifty parameters have been calculated and it appears that FLLL11 and FLLL12 pass the drug‐likeness test. Of particular note are the cell permeability indexes of FLLL11, which although acceptable, are not as good as FLLL12. This may explain, at least in part, the lower efficacy of FLLL11 observed in our studies. Thus far, FLLL12 is the best choice, and has 86% similarity to propafenone and trimethobenzamide. Therefore, FLLL12 may be a better agent than FLLL11 and curcumin for the future development of chemotherapeutics to target breast and prostate cancers.
Acknowledgments
This work was supported in part by a Susan Komen Breast Cancer Research grant to Jiayuh Lin and a Congressionally Directed Medical Research Programs of Prostate Cancer Research grant to Pui‐Kai Li.
References
- 1. Ni Z, Lou W, Leman E, Gao A. Inhibition of constitutively activated Stat3 signaling pathway suppresses growth of prostate cancer cells. Cancer Res 2000; 60: 1225–8. [PubMed] [Google Scholar]
- 2. Davies M, Koul D, Dhesi H et al . Regulation of Akt/PKB activity, cellular growth, and apoptosis in prostate carcinoma cells by MMAC/PTEN. Cancer Res 1999; 59: 2551–6. [PubMed] [Google Scholar]
- 3. Mora LB, Buettner R, Seigne J et al . Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res 2002; 62: 6659–66. [PubMed] [Google Scholar]
- 4. Barton BE, Karras JG, Murphy TF, Barton A, Huang HF. Signal transducer and activator of transcription 3 (STAT3) activation in prostate cancer: Direct STAT3 inhibition induces apoptosis in prostate cancer lines. Mol Cancer Ther 2004; 3: 11–20. [PubMed] [Google Scholar]
- 5. Slamon D, Godolphin W, Jones L et al . Studies of the HER‐2/neu proto‐oncogene in human breast and ovarian cancer. Science 1989; 244: 707–12. [DOI] [PubMed] [Google Scholar]
- 6. Tokunaga E, Kimura Y, Mashino K et al . Activation of PI3K/Akt signaling and hormone resistance in breast cancer. Breast Cancer 2006; 13: 137–44. [DOI] [PubMed] [Google Scholar]
- 7. Knuefermann C, Lu Y, Liu B et al . HER2/PI‐3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene 2003; 22: 3205–12. [DOI] [PubMed] [Google Scholar]
- 8. Clark A, West K, Streicher S, Dennis P. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther 2002; 1: 707–17. [PubMed] [Google Scholar]
- 9. LoPiccolo J, Granville CA, Gills JJ, Dennis PA. Targeting Akt in cancer therapy. Anticancer Drugs 2007; 18: 861–74. [DOI] [PubMed] [Google Scholar]
- 10. She QB, Chandarlapaty S, Ye Q et al . Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling PLoS ONE 2008; 3: e3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang SC, Hung MC. HER2 overexpression and cancer targeting. Semin Oncol 2001; 28(5 Suppl. 16): 115–24. [DOI] [PubMed] [Google Scholar]
- 12. Pegram MD, Lipton A, Hayes DF et al . Phase II study of receptor‐enhanced chemosensitivity using recombinant humanized anti‐p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu‐overexpressing metastatic breast cancer refractory to chemotherapy treatment. J Clin Oncol 1998; 16: 2659–71. [DOI] [PubMed] [Google Scholar]
- 13. Bitran JD, Samuels B, Trujillo Y, Klein L, Schroeder L, Martinec J. Her2/neu overexpression is associated with treatment failure in women with high‐risk stage II and stage IIIA breast cancer (>10 involved lymph nodes) treated with high‐dose chemotherapy and autologous hematopoietic progenitor cell support following standard‐dose adjuvant chemotherapy. Clin Cancer Res 1996; 2: 1509–13. [PubMed] [Google Scholar]
- 14. Zhong Z, Wen Z, Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin‐6. Science 1994; 264: 95–8. [DOI] [PubMed] [Google Scholar]
- 15. Horvath CM, Darnell JE. The state of the STATs: recent developments in the study of signal transduction to the nucleus. Curr Opin Cell Biol 1997; 9: 233–9. [DOI] [PubMed] [Google Scholar]
- 16. Sasse J, Hemmann U, Schwartz C et al . Mutational analysis of acute‐phase response factor/Stat3 activation and dimerization. Mol Cell Biol 1997; 17: 4677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shuai K, Horvath CM, Huang LH, Qureshi SA, Cowburn D, Darnell JE Jr. Interferon activation of the transcription factor Stat91 involves dimerization through SH2‐phosphotyrosyl peptide interactions. Cell 1994; 76: 821–8. [DOI] [PubMed] [Google Scholar]
- 18. Yu H, Jove R. The STATs of cancer – new molecular targets come of age. Nat Rev Cancer 2004; 4: 97–105. [DOI] [PubMed] [Google Scholar]
- 19. Alas S, Bonavida B. Inhibition of constitutive STAT3 activity sensitizes resistant non‐Hodgkin's lymphoma and multiple myeloma to chemotherapeutic drug‐mediated apoptosis. Clin Cancer Res 2003; 9: 316–26. [PubMed] [Google Scholar]
- 20. Buettner R, Mora L, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res 2002; 8: 945–54. [PubMed] [Google Scholar]
- 21. Shen Y, Devgan G, Darnell JJ, Bromberg J. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV‐induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc Natl Acad Sci USA 2001; 98: 1543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Real P, Sierra A, De Juan A, Segovia J, Lopez‐Vega J, Fernandez‐Luna J. Resistance to chemotherapy via Stat3‐dependent overexpression of Bcl‐2 in metastatic breast cancer cells. Oncogene 2002; 21: 7611–18. [DOI] [PubMed] [Google Scholar]
- 23. Wang T, Niu G, Kortylewski M et al . Regulation of the innate and adaptive immune responses by Stat‐3 signaling in tumor cells. Nat Med 2004; 10: 48–54. [DOI] [PubMed] [Google Scholar]
- 24. Garcia R, Bowman T, Niu G et al . Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001; 20: 2499–513. [DOI] [PubMed] [Google Scholar]
- 25. Watson C, Miller W. Elevated levels of members of the STAT family of transcription factors in breast carcinoma nuclear extracts. Br J Cancer 1995; 71: 840–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aoki Y, Feldman G, Tosato G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003; 101: 1535–42. [DOI] [PubMed] [Google Scholar]
- 27. Burke W, Jin X, Liu R, Huang M, Reynolds RK, Lin J. Inhibition of constitutively active Stat3 pathway in ovarian and breast cancer cells. Oncogene 2001; 20: 7925–34. [DOI] [PubMed] [Google Scholar]
- 28. Calvin D, Nam S, Buettner R, Sekharam M, Torres‐Roca J, Jove R. Inhibition of STAT3 activity with STAT3 antisense oligonucleotide (STAT3‐ASO) enhances radiation‐induced apoptosis in DU145 prostate cancer cells. Int J Radiat Oncol Biol Phys 2003; 57: S297. [Google Scholar]
- 29. Takeda K, Noguchi K, Shi W et al . Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA 1997; 94: 3801–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akira S. Roles of STAT3 defined by tissue‐specific gene targeting. Oncogene 2000; 19: 2607–11. [DOI] [PubMed] [Google Scholar]
- 31. Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res 2007; 13: 5665–9. [DOI] [PubMed] [Google Scholar]
- 32. Aggarwal BB, Sethi G, Ahn KS et al . Targeting signal‐transducer‐and‐activator‐of‐transcription‐3 for prevention and therapy of cancer: modern target but ancient solution. Ann NY Acad Sci 2006; 1091: 151–69. [DOI] [PubMed] [Google Scholar]
- 33. Hatcher H, Planalp R, Cho J, Torti FM, Torti SV. Curcumin: from ancient medicine to current clinical trials. Cell Mol Life Sci 2008; 65 (11): 1631–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aggarwal B, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol 2006; 71: 1397–421. [DOI] [PubMed] [Google Scholar]
- 35. Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm 2007; 4: 807–18. [DOI] [PubMed] [Google Scholar]
- 36. Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol 1998; 18: 2545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fuchs J, Pandit B, Bhasin D et al . Structure–activity relationship studies of curcumin analogues. Bioorg Med Chem Lett 2009; 19: 2065–9. [DOI] [PubMed] [Google Scholar]
- 38. Sorlie T, Perou CM, Tibshirani R et al . Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001; 98: 10 869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brenton JD, Carey LA, Ahmed AA, Caldas C. Molecular classification and molecular forecasting of breast cancer: ready for clinical application? J Clin Oncol 2005; 23: 7350–60. [DOI] [PubMed] [Google Scholar]
- 40. Carey LA, Perou CM, Livasy CA et al . Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006; 295: 2492–502. [DOI] [PubMed] [Google Scholar]
- 41. Harari DY, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene 2000; 19: 6102–14. [DOI] [PubMed] [Google Scholar]
- 42. Perez‐Tenorio G, Stal O. Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br J Cancer 2002; 86: 540–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arboleda M, Lyons J, Kabbinavar F et al . Overexpression of AKT2/protein kinase Bβ leads to up‐regulation of β1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res 2003; 63: 196–206. [PubMed] [Google Scholar]
- 44. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 45. Bharti A, Donato N, Aggarwal B. Curcumin (diferuloylmethane) inhibits constitutive and IL‐6‐inducible STAT3 phosphorylation in human multiple myeloma cells. J Immunol 2003; 171: 3863–71. [DOI] [PubMed] [Google Scholar]
- 46. Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal‐regulated kinase signaling pathways. Mol Pharmacol 2007; 72: 29–39. [DOI] [PubMed] [Google Scholar]
- 47. Diehl KM, Grewal N, Ethier SP, Woods‐Ignatoski KM. p38MAPK‐activated AKT in HER‐2 overexpressing human breast cancer cells acts as an EGF‐independent survival signal. J Surg Res 2007; 142: 162–9. [DOI] [PubMed] [Google Scholar]
- 48. Schlessinger K, Levy DE. Malignant transformation but not normal cell growth depends on signal transducer and activator of transcription 3. Cancer Res 2005; 65: 5828–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hanif R, Qiao L, Shiff SJ, Rigas B. Curcumin, a natural plant phenolic food additive, inhibits cell proliferation and induces cell cycle changes in colon adenocarcinoma cell lines by a prostaglandin‐independent pathway. J Lab Clin Med 1997; 130: 576–84. [DOI] [PubMed] [Google Scholar]
- 50. Kawamori T, Lubet R, Steele VE et al . Chemopreventive effect of curcumin, a naturally occurring anti‐inflammatory agent, during the promotion/progression stages of colon cancer. Cancer Res 1999; 59: 597–601. [PubMed] [Google Scholar]
- 51. Aggarwal BB, Shishodia S, Takada Y et al . Curcumin suppresses the paclitaxel‐induced nuclear factor‐κB pathway in breast cancer cells and inhibits lung metastasis of human breast cancer in nude mice. Clin Cancer Res 2005; 11: 7490–8. [DOI] [PubMed] [Google Scholar]
- 52. Bachmeier B, Nerlich AG, Iancu CM et al . The chemopreventive polyphenol Curcumin prevents hematogenous breast cancer metastases in immunodeficient mice. Cell Physiol Biochem 2007; 19: 137–52. [DOI] [PubMed] [Google Scholar]
- 53. Azuine MA, Bhide SV. Chemopreventive effect of turmeric against stomach and skin tumors induced by chemical carcinogens in Swiss mice. Nutr Cancer 1992; 17: 77–83. [DOI] [PubMed] [Google Scholar]
- 54. Azuine MA, Bhide SV. Protective single/combined treatment with betel leaf and turmeric against methyl (acetoxymethyl) nitrosamine‐induced hamster oral carcinogenesis. Int J Cancer 1992; 51: 412–15. [DOI] [PubMed] [Google Scholar]
- 55. Ikezaki S, Nishikawa A, Furukawa F et al . Chemopreventive effects of curcumin on glandular stomach carcinogenesis induced by N‐methyl‐N′‐nitro‐N‐nitrosoguanidine and sodium chloride in rats. Anticancer Res 2001; 21: 3407–11. [PubMed] [Google Scholar]
- 56. Frank N, Knauft J, Amelung F, Nair J, Wesch H, Bartsch H. No prevention of liver and kidney tumors in Long–Evans Cinnamon rats by dietary curcumin, but inhibition at other sites and of metastases. Mutat Res 2003; 523–524: 127–35. [DOI] [PubMed] [Google Scholar]
- 57. Gururaj AE, Belakavadi M, Venkatesh DA, Marme D, Salimath BP. Molecular mechanisms of anti‐angiogenic effect of curcumin. Biochem Biophys Res Commun 2002; 297: 934–42. [DOI] [PubMed] [Google Scholar]
- 58. Ramachandran C, Fonseca HB, Jhabvala P, Escalon EA, Melnick SJ. Curcumin inhibits telomerase activity through human telomerase reverse transcritpase in MCF‐7 breast cancer cell line. Cancer Lett 2002; 184: 1–6. [DOI] [PubMed] [Google Scholar]
- 59. Dorai T, Cao YC, Dorai B, Buttyan R, Katz AE. Therapeutic potential of curcumin in human prostate cancer. III. Curcumin inhibits proliferation, induces apoptosis, and inhibits angiogenesis of LNCaP prostate cancer cells in vivo . Prostate 2001; 47: 293–303. [DOI] [PubMed] [Google Scholar]
- 60. Mukhopadhyay A, Bueso‐Ramos C, Chatterjee D, Pantazis P, Aggarwal BB. Curcumin downregulates cell survival mechanisms in human prostate cancer cell lines. Oncogene 2001; 20: 7597–609. [DOI] [PubMed] [Google Scholar]
- 61. Chendil D, Ranga RS, Meigooni D, Sathishkumar S, Ahmed MM. Curcumin confers radiosensitizing effect in prostate cancer cell line PC‐3. Oncogene 2004; 23: 1599–607. [DOI] [PubMed] [Google Scholar]
- 62. Chaudhary LR, Hruska KA. Inhibition of cell survival signal protein kinase B/Akt by curcumin in human prostate cancer cells. J Cell Biochem 2003; 89: 1–5. [DOI] [PubMed] [Google Scholar]
- 63. Tamvakopoulos C, Dimas K, Sofianos Z et al . Metabolism and anticancer activity of the curcumin analogue, dimethoxycurcumin. Clin Cancer Res 2007; 13: 1269–77. [DOI] [PubMed] [Google Scholar]
- 64. Ohtsu H, Xiao Z, Ishida J et al . Antitumor agents. 217. Curcumin analogues as novel androgen receptor antagonists with potential as anti‐prostate cancer agents. J Med Chem 2002; 45: 5037–42. [DOI] [PubMed] [Google Scholar]
- 65. Mosley C, Liotta D, Snyder J. Highly active anticancer curcumin analogues. Adv Exp Med Biol 2007; 595: 77–103. [DOI] [PubMed] [Google Scholar]
- 66. Youssef D, Nichols C, Cameron T, Balzarini J, De Clercq E, Jha A. Design, synthesis, and cytostatic activity of novel cyclic curcumin analogues. Bioorg Med Chem Lett 2007; 17: 5624–9. [DOI] [PubMed] [Google Scholar]
- 67. Gao L, Zhang L, Hu J et al . Down‐regulation of signal transducer and activator of transcription 3 expression using vector‐based small interfering RNAs suppresses growth of human prostate tumor in vivo . Clin Cancer Res 2005; 11: 6333–41. [DOI] [PubMed] [Google Scholar]
- 68. Lin HK, Yeh S, Kang HY, Chang C. Akt suppresses androgen‐induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc Natl Acad Sci USA 2001; 98: 7200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Graff JR, Konicek BW, McNulty AM et al . Increased AKT activity contributes to prostate cancer progression by dramatically accelerating prostate tumor growth and diminishing p27Kip1 expression. J Biol Chem 2000; 275: 24 500–5. [DOI] [PubMed] [Google Scholar]