

Abstract
(Cancer Sci 2010; 101: 743–750)
Although cytostatin analog protein phosphatase 2A (PP2A)‐specific inhibitors are promising candidates of a new type of anticancer drug, their development has been hindered because of their liability. To find new classes of PP2A‐specific inhibitors, we conducted a screening with microbial metabolites and found that rubratoxin A, a classical mycotoxin, is a highly specific and potent inhibitor of the enzyme. While rubratoxin A inhibits PP2A at Ki = 28.7 nm, it hardly inhibited any other phosphatases examined. Rubratoxin B, a close analog, also specifically but weakly inhibits PP2A at Ki = 3.1 μm. The inhibition of intracellular PP2A in cultured cells is obviously observed with 20 μm rubratoxin A treatment for 3 h, inducing the overphosphorylation in PP2A substrate proteins. Although rubratoxins and cytostatin differ in the apparent structures, these compounds share similarities in the structures in detail and PP2A‐binding manners. Rubratoxin A showed higher suppression of tumor metastasis and reduction of the primary tumor volume than cytostatin in mouse experiments. As a successor of cytostatin analogs, rubratoxin A should be a good compound leading to the development of antitumor drugs targeting PP2A.
The phosphorylation of intracellular protein plays a central role in the regulation of eukaryotic cell function and metabolism. Protein phosphatase (PP)2A is a ubiquitous serine/threonine phosphatase in eukaryotic cells, which regulates the phosphorylation state and function of a broad range of proteins.( 1 , 2 ) We previously isolated a PP2A‐specific inhibitor, cytostatin, from an actinomycete culture.( 3 , 4 , 5 ) Its analog, fostriecin, and other PP2A‐specific inhibitors have also been reported since mid 1990s.( 6 , 7 , 8 , 9 , 10 , 11 ) These new inhibitors have higher specificities to PP2A than okadaic acid, a previously identified inhibitor (PP1 IC5 0/PP2A IC50: fostriecin, 104–105; okadaic acid, 100),( 6 , 12 ) and have shown promising antitumor activities in animal experiments.( 6 , 9 , 13 ) A phase I clinical study of fostriecin was reported, but the result was ambiguous because of the instability of the compound and its short supply.( 14 ) Therefore, to further investigate the feasibility of PP2A‐specific inhibitors as an anticancer drug, we have been searching for new stable PP2A inhibitors from microbial metabolites.
In the present study, we found rubratoxin A as a new PP2A‐specific inhibitor. Rubratoxins are one of the oldest mycotoxins, whose existence as a hepatotoxic agent to livestock was first implied in 1957.( 15 ) Isolated from Penicillium rubrum, the structures of rubratoxins were determined in approximately the 1960s–1970s.( 16 , 17 ) Since then, numerous studies, usually using rubratoxin B, have reported the toxic effects of rubratoxins, including a case of rubratoxicosis in humans, and investigations are still being pursued.( 16 , 17 , 18 , 19 ) However, the toxicity mechanism remains ill‐defined because the target molecule of rubratoxins has not been elucidated. This is the first report to unveil the primary target molecule of rubratoxins, showing their specific and potent inhibition of PP2A activity and the convincing binding model. The antitumor and antimetastatic effects of rubratoxin A were also examined in several mouse models comparing the effects of cytostatin to evaluate the utility of PP2A as a novel target of cancer treatment.
Materials and Methods
Screening of the PP2A inhibitor.
The screening assay was performed on 96‐well plates using 100% acetone or 70% ethanol extract (24 mg/mL) libraries of microbial cultures consisting of fungi, actinomycetes, and other bacteria. Each extract was inoculated at 1 μL/well. The substrate, p‐nitrophenyl phosphate (pNPP), was dissolved at 200 mm in a reaction buffer composed of 50 mm Tris‐HCl (pH 8.5), 20 mm MgCl2, and 0.5 mm dithiothreitol (DTT) at 10 μL/well on the plate. A human blood cell PP2A (Millipore, Billerica, MA, USA) dissolved in a buffer composed of 20 mm MOPS (pH 7.5), 150 mm NaCl, 60 mm 2‐mercaptoethanol, 1 mm MgCl2, 1 mm EGTA, 0.1 mm MnCl2, 1 mm DTT, 10% glycerol, and 0.1 mg/mL bovine serum albumin at 0.5 units/9 mL was added at 90 μL/well to start the reaction. After incubation at 37°C for 30 min, the reaction product p‐nitrophenol was measured at 405 nm with a Multiskan MS microplate reader (Labsystems, Pitesti, Romania).
Purification of rubratoxins.
A rubratoxin‐producing Penicillium sp. strain was cultured in the autoclaved polished rice grain medium containing 2.5% soybean meal (Ajinomoto Takara, Tokyo, Japan) and 25% water at 26°C for 2 weeks. The 67% acetone extract of the culture was partitioned with BuOH/H2O. Rubratoxins were purified from the BuOH fraction by sequential chromatographies with an YMC*GEL ODS‐A column (6 nm, S‐150 μm; YMC, Kyoto, Japan), Inertsil ODS‐3 column (20 mm×250 mm; GL Sciences, Torrance, CA, USA), and Sephadex LH‐20 column (GE Healthcare, Chalfont St Giles, UK). For the octa decyl silyl (ODS) chromatography, isocratic 35% acetonitrile containing 0.1% trifluoroacetic acid was used as a solvent, while 35% acetonitrile was used for the Sephadex LH‐20 chromatography. Rubratoxins were identified by nuclear magnetic resonance (NMR) and mass spectrometry (MS) analyses with JEOL JNM‐A400 (JEOL, Tokyo, Japan), PE SCIEX API165 (Perkin Elmer, Waltham, MA, USA), and JEOL JMS‐T100LC (JEOL, Tokyo, Japan) spectrometers. The purity of rubratoxins A and B isolated and used in this study were estimated by HPLC (UV215nm) with a minimum of 96.6% and 93.2%, respectively.
Phosphatase inhibition assays.
Rabbit skeletal muscle PP1, recombinant PP2B with calmodulin, recombinant protein–tyrosine phosphatase‐1B (PTP‐1B) (Millipore, USA), and calf intestine alkaline phosphatase (CIP; New England Biolabs, Ipswich, MA, USA) were employed for the other phosphatase inhibition assays. The pNPP assays were performed as per PP2A in the buffers recommended by each enzyme’s manufacturers.
Immunoblotting.
The cells were cultured at 2 × 105 cells/mL as previously described( 5 ) for 24 h, and treated with the compounds for 3 h. The cell lysates were separated by sodium dodecylsulfate–polyacrylamide gel electrophoresis with 9% acrylamide gels, followed by the immunoblotting( 20 , 21 ) using Tris‐buffered saline (10 mm Tris‐HCl [pH 7.5] ad 150 mm NaCl). Horseradish peroxidase‐conjugated antirabbit immunoglobulin G (IgG) and antimouse IgG antibodies (GE Healthcare, UK) were used as the secondary antibody at 1/2000 and 1/5000, respectively. Antiphosphotyrosine mouse monoclonal antibody (Millipore, USA) was used at 1/1000. The other antibodies were rabbit polyclonal antibodies (Cell Signaling Technology, Beverly, MA, USA), which was used at 1/1000 or 1/2000.
Molecular modeling.
The rubratoxin A‐PP2A binding model, PP2A–RUB_AH, was created with Discovery Studio v2.1 (Accelrys Software, San Diego, CA, USA) based on the crystal structures of microcystin–LR and okadaic acid–PP2A (PDB code 2ie3 and 2ie4).( 22 )
The stereo structure of the rubratoxin A–dicarboxylic acid form, RUB_AH, was deduced from the structure of rubratoxin B( 23 , 24 ) converting the C26 carbonyl to the hydroxyl group and conjugating a water molecule to C23 and C24. RUB_AH was superimposed on microcystin–LR in the structure 2ie3 considering the direction of the inhibitors in the binding model of cytostatin analogs to PP2A.( 22 , 25 ) The carbonyl oxygens in the dicarboxylic acid (‐O‐CO‐C = C‐COO‐) of RUB_AH overlapped with the position of the oxygens of microcystin–LR (‐CO‐NH‐CH‐COO), thus it was fixed at the coordinate. The lactone ring positioned in the close vicinity of Cys269 was also fixed at the residue by Michael addition. The preliminary structure was energy minimized with the simulation module of Discovery Studio using the chemical force feedback force field, and the resultant model PP2A–RUB_AH was obtained.
Mouse experiments.
The mouse experiments were conducted in accordance with a code of practice established by the ethical committee of the Microbial Chemistry Research Foundation (Shizuoka, Japan). The mice were purchased from Charles River (Yokohama, Japan) and maintained in a pathogen‐free barrier facility for 1 week before the experiments.
Results
Identification of rubratoxins as PP2A‐specific inhibitors.
To discover new PP2A‐specific inhibitors, we screened more than 20 000 microbial culture extracts and found a modest PP2A inhibitor, harzianic acid.( 10 ) Further investigations revealed a Penicillium sp. extract showing an extremely strong PP2A inhibition and containing a more potent PP2A inhibitor than harzianic acid. Two active compounds were purified and identified as rubratoxins A and B (Fig. 1a) by spectroscopic analyses, including various NMR and MS experiments (Appendix S1). Despite the absence of any record to date that rubratoxins are PP2A inhibitors, it is quite feasible that rubratoxins share some structural similarities to cytostatin and cantharidin (Fig. S1). In liquid chromatography–MS experiments using aqueous solvents, each rubratoxin showed an equilibrium between the molecules conjugated with (70–95% in 35% acetonitrile) or without a water molecule, indicating the majority of rubratoxins in aqueous solution existed as dicarboxylic acid forms opening a maleic anhydride moiety. The dicarboxylic acid form is considered to be the active form, like cantharidin and tautomycin.( 12 )
Figure 1.
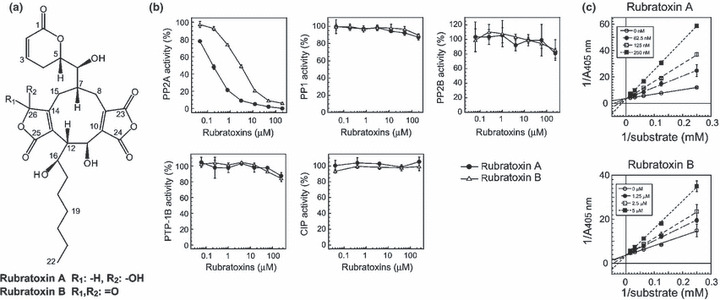
Protein phosphatase (PP)2A‐specific inhibition by rubratoxins. (a) Structures of rubratoxins A and B. (b) Effects of rubratoxins A and B on the activities of isolated PP2A, PP1, PP2B, recombinant protein–tyrosine phosphatase‐1B (PTP‐1B), and calf intestine alkaline phosphatase (CIP) in the p‐nitrophenyl phosphate dephosphorylation assays are shown. Mean ± SD. (n = 3). (c) Lineweaver–Burk plot of PP2A inhibition by rubratoxins A and B. PP2A inhibitory activity of rubratoxins A and B were assessed using varying substrate concentrations. Ki values of rubratoxins A and B to PP2A were calculated as 28.7 nm and 3.1 μm from the regression lines, respectively.
The inhibitory effects of rubratoxins A and B on isolated PP2A, PP1, PP2B, PTP‐1B, and CIP were examined by pNPP dephosphorylation assays. Rubratoxin A potently inhibited PP2A with an IC50 of 170 nm, while rubratoxin B modestly inhibited the enzyme with an IC50 of 2.95 μm (Fig. 1b). Both rubratoxins, up to 200 μm, failed to significantly inhibit any other phosphatases tested (Fig. 1b). The Ki values, as objective potency, of rubratoxins A and B against PP2A were 28.7 and 3.1 μm, respectively, and the plot patterns showed typical competitive inhibition (Fig. 1c).
Phosphorylation of the proteins in rubratoxin A‐treated cells.
The accumulation of phosphorylated proteins is typically observed in protein phosphatase inhibitor‐treated cells. Protein phosphorylation states in rubratoxin‐treated cells were monitored by immunoblotting using antibodies recognizing phosphorylated substrate peptides of kinases and phosphotyrosine. The protein phosphorylation was clearly increased in mouse B16‐BL6 melanoma, human HEK293 embryonic kidney, DU‐145 prostate cancer, and HepG2 hepatocellular carcinoma cells treated with 20 μm rubratoxin A for 3 h (Fig. 2). The overphosphorylation was obvious in AGC kinase substrates (phospho cyclic AMP‐dependent protien kinase (pPKA), phospho protein kinase C (pPKC),and pAkt substrates), while it was an ambiguous or cell‐ or substrate‐dependent manner in other kinase substrates (phospho cyclin‐dependent kinase (pCDK) and phospho ataxia telangiectasia mutated/ataxia telangiectasia and rad3‐related protein (pATM/ATR) substrates, and pTyrosine.
Figure 2.
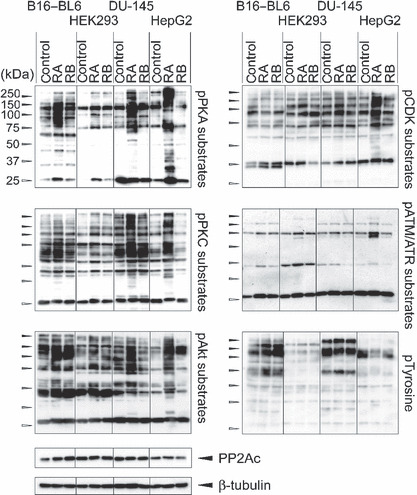
Overphosphorylation of proteins in rubratoxin A‐treated cells. Logarithmically‐growing B16–BL6, HEK293, DU‐145, and HepG2 cells were treated with 20 μm rubratoxins (rubratoxin A [RA] or rubratoxin B [RB]) or vehicle (control; 0.07% MeCN) for 3 h. Cell lysates containing 10 μg protein were separated by sodium dodecylsulfate–polyacrylamide gel electrophoresis, and the phosphorylation states of proteins were analyzed by immunoblotting using antibodies recognizing the phosphorylated substrates of protein kinase A (pPKA), protein kinase (pPKC), Akt, cyclin‐dependent kinase (pCDK), ataxia telangiectasia mutated ataxia telangiectasia mutated and rad3‐related protein (pATM/pATR), and phosphorylated tyrosine (pTyrosine). Expression level of the protein phosphatase (PP)2A catalytic subunit and β‐tubulin were detected as the controls.
We then investigated the phosphorylation states of individual PP2A‐substrate proteins. Rubratoxin A generally and obviously augmented the phosphorylation of PP2A‐specific substrate cyclic AMP response element binding protein (CREB),( 26 ) its related protein activating transcription factor‐1 (ATF‐1), and c‐Myc,( 27 ) although the phosphorylated c‐Myc signal was too weak to be detected in B16–BL6 and HEK293 (Fig. 3a). Rubratoxin A did not generally induced the increase of phosphorylation in a PP1 substrate cdc25C( 28 ) and pleckstrin homology domain leucine‐rich repeat protein phosphatase (PHLPP) substrate Akt( 29 ) (Fig. 3b). Mitogen‐activated protein (MAP) kinases extracellular signal‐regulated kinase (ERK)1/2, Jun‐N‐terminal kinase (JNK), and p38 are known to be dephosphorylated by PP2A and MAP kinase phosphatases.( 30 , 31 ) While the phosphorylation of JNK and p38 specifically increased by rubratoxin A treatment (Fig. 3a), such effects of ERK1/2 were only observed in HepG2 cells (Fig. 3b). This could reflect the ratio of the expression levels of PP2A and MAP kinase phosphatases, or the preference of each MAP kinase for the certain phosphatase.
Figure 3.
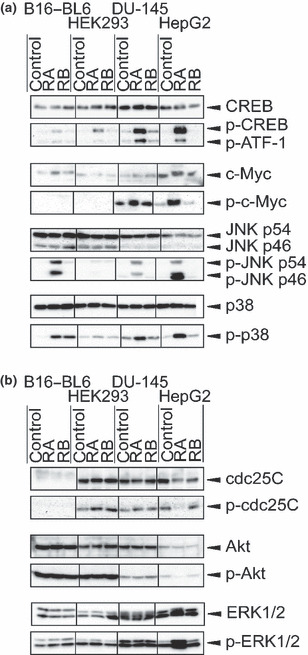
Overphosphorylation of protein phosphatase (PP)2A substrate proteins by rubratoxin (RA) treatment. Intracellular proteins specifically overphosphorylated by rubratoxin A treatment (a) and those independent of the treatment (b) are shown. Immunoblotting were performed using the protein‐ or phosphorylated protein‐specific antibodies. Recognition sites of phosphorylated proteins are cyclic AMP response element binding protein (CREB) Ser133, c‐Myc Thr58/Ser62, Jun‐N‐terminal kinase (JNK) Thr183/Tyr185, p38 Thr180/Tyr182, cdc25C Ser216, Akt Ser473, and ERK1/2 extracellular signal‐regulated kinase (ERK)1/2 Thr180/Tyr204. Phosphorylated activating transcription factor‐1 (ATF‐1) was concomitantly detected by the phosphorylated CREB antibody. Phosphorylated cdc25C antibody recognizes only the human protein, while the other antibodies recognize both human and mouse proteins.
The protein phosphorylation patterns in 20 μm rubratoxin A‐treated cells were similar to those in 20–50 nm okadaic acid‐treated cells (Fig. S2a), suggesting that rubratoxin A inhibits PP2A in cells with at least the same specificity of okadaic acid, and its effect on the other proteins, including other phosphatases, was negligible. Despite the failure at 20 μm (2, 3), a higher concentration of rubratoxin B induced a similar overphosphorylation of proteins (Fig. S2b). The effects of rubratoxins were enhanced by increasing the concentration of the vehicle (Fig. S2b).
Binding simulation of rubratoxin A and PP2A.
To consolidate the PP2A‐specific inhibition by rubratoxin A, binding simulation was conducted. Since okadaic acid and microcystin–LR bind to the same catalytic site interacting with surrounding amino acids in a highly overlapping manner( 22 ) and the fact that rubratoxins are also considered to bind to the site (Fig. 1c), we constructed the rubratoxin A‐PP2A binding model based on the crystal structures of the inhibitor–PP2A complex (Fig. 4a–c).
Figure 4.
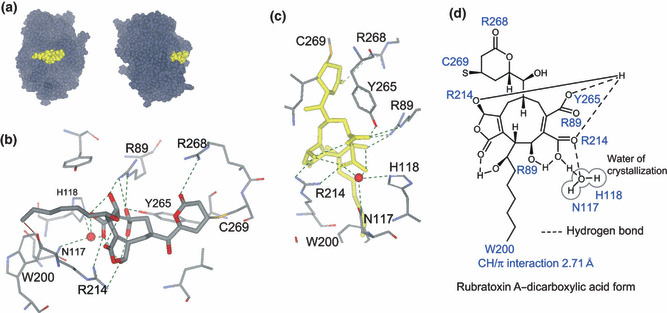
Binding model of rubratoxin A to protein phosphatase (PP)2A. (a) Binding model of rubratoxin A–dicarboxylic acid form (yellow) and PP2A catalytic subunit α (dark gray), PP2A–RUB_AH, are shown from two angles. (b) Binding site of PP2A–RUB_AH is magnified. Rubratoxin A and amino acid residues of PP2A involved in the interactions are depicted with thick and thinner lines, respectively. Atoms in the structure C, O, N, and S are gray, red, blue, and yellow, respectively. Red sphere represents oxygen atom of the crystallization water. Hydrogen or ionic bonds are shown with green dashed lines, and the CH/π interaction is shown with a brown line. (c) Binding site of PP2A–RUB_AH is shown from a different angle. To clarify the hydrogen bonds, rubratoxin A is highlighted in yellow. (d) Interactions in the binding complex are illustrated. Amino acid residues forming covalent, hydrogen, and ionic bonds, and CH/π interaction with rubratoxin A, are shown beside the interacting parts.
There are three characteristics in the binding model. The first is the interactions around the α,β‐unsaturated δ‐lactone ring. Besides the covalent bond of Cys269 to C3, Arg268 and the carbonyl oxygen on the ring form a hydrogen bond (Fig. 4b–d). These interactions are also indicated in the binding of cytostatin analogs to PP2A,( 25 , 32 ) and a similar covalent bond was reported in the binding of N‐methyldehydroalanine of microcystin–LR to Cys269.( 22 ) Ceramidastin, a close analog of rubratoxin B( 33 ) whose lactone ring is opened and reduced, hardly inhibited PP2A (IC50 > 200 μm; Hiroyuki Inoue, personal communication, 2009). Cys269 and Arg268 are conserved only in PP2A and its related proteins PP4 and PP6, whereas they are absent from other protein phosphatases, such as PP1, PP2B, PP5, and PP7. Those interactions are regarded as a significant determinant for the high specificity of rubratoxins and cytostatin analogs to inhibit PP2A.
The second characteristic is the insertion of the hydrophobic chain into the hydrophobic pocket around Trp200, as seen in the crystal structures or binding models of other compound–PP2A binding complexes (Fig. 4b–d). CH/π interaction( 34 ) was implied between the methyl terminal of rubratoxin A and Trp200.
The third is the intermolecular and intramolecular hydrogen and ionic bond networks at the center of the molecule, as shown in Figure 4(c,d). When rubratoxin A was simply replaced with rubratoxin B in this model, the distances of C23–C26, C24‐C26, and C16–C25 were enlarged (Fig. S3). Therefore, to tighten the structure of rubratoxin A to adapt for the appropriate interaction with the surrounding amino acid residues, the intramolecular hydrogen bonds could play a major role in the difference of the inhibitory potency of rubratoxins A and B. Similar interactions with the surrounding amino acids were observed in our binding model of cytostatin–PP2A, although the ligand was slightly separated from Arg214 (Fig. S4), confirming that rubratoxins and cytostatin analogs inhibit the enzyme in a similar manner and that dicarboxylic acid in rubratoxin A could be a substitute for phosphate in cytostatin analogs.
Antitumor effect of rubratoxin A.
The antitumor and antimetastatic effects of rubratoxin A were examined in several mouse experiments. In an experimental metastatic model using C57BL/6N mice intravenously inoculated with B16–BL6 cells (Table 1), only 3 days of rubratoxin A pre‐administration before cellular inoculation clearly suppressed the formation of lung metastatic foci. Although cytostatin also suppressed metastasis in this model, the same dose of rubratoxin A showed 15% increased inhibitory activity.
Table 1.
Antimetastatic effect of rubratoxin A and cytostatin
| Mouse | No. lung foci | ||||
|---|---|---|---|---|---|
| Control | Rubratoxin A | Cytostatin (1.6 mg/kg) | |||
| 0.1 mg/kg | 0.4 mg/kg | 1.6 mg/kg | |||
| 1 | 81 | 79 | 43 | 23 | 62 |
| 2 | 71 | 60 | 26 | 11 | 22 |
| 3 | 70 | 56 | 22 | 7 | 21 |
| 4 | 67 | 54 | 8 | 6 | 7 |
| 5 | 64 | 34 | 7 | 5 | 6 |
| 6 | 62 | 21 | 6 | 5 | 2 |
| 7 | 59 | 10 | 0 | 4 | 0 |
| 8 | 51 | — | — | — | — |
| 9 | 50 | — | — | — | — |
| 10 | 48 | — | — | — | — |
| 11 | 45 | — | — | — | — |
| 12 | 45 | — | — | — | — |
| 13 | 43 | — | — | — | — |
| 14 | 35 | — | — | — | — |
| Mean | 56.5 | 44.9 | 16 | 8.7 | 17.1 |
| SD | 13.2 | 24.2 | 15.1 | 6.7 | 21.6 |
| Inhibition(%) | — | 20.61 | 71.68 | 84.58 | 69.66 |
| P‐value | — | NS | <0.001 | <0.001 | <0.005 |
Six‐week‐old female C57BL/6N mice were divided into five groups (n = 7 or 14) and intraperitoneally injected with 0.1, 0.4, or 1.6 mg/kg rubratoxin A, 1.6 mg/kg cytostatin, or vehicle (10% dimethylsulfoxide and 0.5% Tween‐80 in saline) every 24 h for 3 days. Mice were intravenously inoculated with 2.5 × 104 cells/100 μL of B16–BL6 melanoma cells at 24 h after the last injection of the agent. Fourteen days after the cellular inoculation, the mice were killed and the number of lung metastatic foci was counted under a microscope. NS, not significant.
In a spontaneous metastatic model using C57BL/6N mice subcutaneously inoculated with Lewis lung carcinoma (3LL) cells, the intraperitoneal administration of 1.6 mg/kg/day rubratoxin A significantly reduced the size of the primary tumor (Fig. 5a,b). The spontaneous lung metastasis showed a distinct decrease in the number and size of foci in the rubratoxin A‐treated mice (Fig. 5c,d). Toxic effects, including body weight loss (Fig. S5a) and hepatotoxicity with a partially whitened liver (3/5 mice, <3% of liver surface), were observed with the administration of 1.6 mg/kg/day rubratoxin A. Although cytostatin at 3.2 mg/kg/2 days apparently induced a similar growth suppression of the primary tumor size (Fig. 5a), the final tumor weights and lung metastasis at day 21 showed no significant reduction (Fig. 5b,c). The toxic effect of 3.2 mg/kg/2 days cytostatin resulted in partially whitened liver (3/5 mice) and was comparable with 1.6 mg/kg/day rubratoxin A, although no body weight loss was observed in this treatment (Fig. S5a). In a follow‐up experiment with almost the same condition, similar effects of rubratoxin A were reproduced (Fig. S5b–e). The antitumor and antimetastatic effects of rubratoxin A were better than or comparable to those of doxorubicin and lentinan, while the weight loss by rubratoxin A was milder than that by doxorubicin in the treatments. Considering the sustained use of doxorubicin for cancer treatments, the toxicity of rubratoxin A appears manageable with further investigation and administration protocols.
Figure 5.
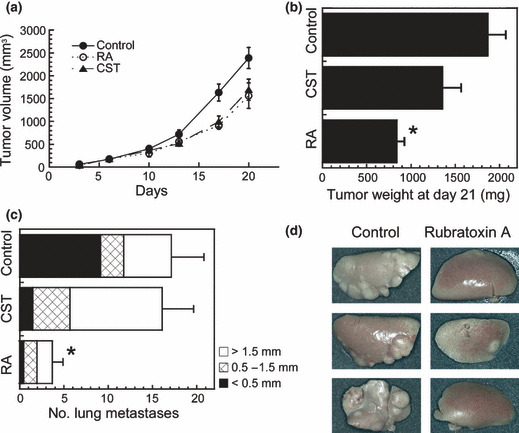
Antitumor and antimetastatic effects of rubratoxin A (RA). C57BL/6N mice were subcutaneously inoculated with 1 × 106 cells/100 μL saline of Lewis lung carcinoma cells in the left flank (day 0) and intraperitoneally injected with 1.6 mg/kg/day RA (n = 5), 3.2 mg/kg/2 days cytostatin (CST, n = 5), or vehicle (control; 250 μL of 5% dimethylsulfoxide, 0.5% Tween‐80, n = 7). Primary tumor sizes were measured with a caliper twice per week (a). Mice were killed at day 21, and the weights of excised primary tumors (b) and number of each size of lung metastatic foci (c) were measured. Some of the representative lung lobes from RA‐treated or non‐treated mice are shown in (d). (*) Difference between control and RA treatment was significant (P < 0.01). Graph data are mean ± SE. For clarification, only the results of the representative three groups are shown among the total 13 groups of mice that were treated with various conditions of RA or CST at the same time.
We also conducted an experiment using immunodeficient SCID (CB17/Icr–Prkdcscid/CrlCrlj) and NOD–SCID (non‐obese diabetic [NOD].CB17‐Prkdcscid/J) mice bearing 3LL cells (Fig. S6). Although the primary tumor weight in SCID mice was not significantly reduced, the antimetastatic effects of rubratoxin A were observed in both mouse models. The numbers of lung metastatic foci decreased to 13–22% of the control in SCID and C57BL/6N mice and to 35% in NOD–SCID mice by rubratoxin A treatment.
Discussion
In the present study, we found that rubratoxin A specifically and potently inhibited PP2A. The enzyme inhibition assays and phosphorylation patterns of cellular proteins ensured its high specificity to PP2A, and the existence of any other target molecules was not implied. Although it is still difficult to exclude the possibility that rubratoxin A has other target molecules, as in the case of other inhibitors in general, one can consider it as a highly specific and potent PP2A inhibitor comparable with cytostatin analogs. Rubratoxin B also specifically inhibited PP2A in our assays, albeit weaker than rubratoxin A. Considering the ubiquity of PP2A, the inhibition of the enzyme would be the main cause for most of the previously reported biological effects of rubratoxin B.
Taking advantage of their high specificity to PP2A, cytostatin analog compounds were expected to function as good molecular probes. However, because of their liability, their applications have been limited, while less specific PP2A inhibitors, such as okadaic acid and calyculin A, have been broadly used in biochemical and cell biological protocols. Rubratoxin A is stable, as no degradation was observed in normal stocks for 2 years and in 0.2 N HCl treatment at 60°C for 30 min. With the higher stability and comparable high specificity, rubratoxin A will be a successor of cytostatin analogs as a PP2A‐specific molecular probe and a lead compound of clinical drugs for cancer treatment.
Less specific PP2A inhibitors, such as okadaic acid and microcystin–LR, are known as tumor promoters, whereas it is unclear whether rubratoxin A has such a function. Rubratoxin A, as well as okadaic acid, induces the increase of c‐Myc oncogene phosphorylation and accumulation in cultured cells, which might lead to some tumor‐promotion effects. Side‐by‐side experiments for these compounds using appropriate mouse models might be required. However, we observed promising anticancer effects of rubratoxin A instead of any tumor promotion in mouse experiments, and the tumor‐promoting effects of okadaic acid class compounds are experimentally induced under the presence of initiator mutagens.( 35 ) Even if rubratoxin A has some tumor‐promoting effects, they should be negligible or manageable. Although another problem, hepatotoxicity, also warrants a solution, the dramatic antimetastatic effects of rubratoxin A and cytostatin are regarded as a common effect of PP2A‐specific inhibitors, which should be considered in cancer treatments. Clarifying the difference in the molecular mechanisms in hepatotoxicity and the anticancer activity of rubratoxin A will lead to less toxic and potent anticancer drugs.
The activation of natural killer (NK) cells is considered to be a cause of the antimetastatic effects of cytostatin,( 9 ) and rubratoxin A was also found to activate NK cells in some experiments (Fig. S7). The phosphorylation of CREB, as shown in this study, might be involved in NK cell activation via the stimulation of interleukin (IL)‐2 release from T cells.( 36 ) However, weaker but still obvious antimetastatic effects of rubratoxin A have been observed in NK cell‐deficient NOD–SCID mice. The activity of other leukocytes existing in NOD–SCID mice, the impairment of cell adhesion via focal adhesion kinase phosphorylation and paxillin phosphorylation, and the secretion of tissue inhibitor of metalloproteinases‐1 (TIMP‐1) could also contribute to the antimetastatic effects.( 5 , 37 )
The systemic administration of rubratoxins and other PP2A inhibitors with higher hydrophilicity, such as microcystins and cytostatin analogs, induces hepatic disorder in mice, whereas okadaic acid and calyculin A, which are hydrophobic inhibitors, have not been reported to induce such toxicity.( 12 , 35 )Hepatotoxicity is a common effect of hydrophilic PP2A inhibitors. Since rubratoxin B enhances the secretion of several cytokines, such as IL‐8, tumor necrosis factor‐α, macrophage colony stimulating factor (M‐CSF), and granulocyte macrophage colony stimulating factor (GM‐CSF) from liver‐resident cells, it was suggested that macrophages and neutrophils are recruited and activated in the liver, which attack liver tissue and cause the hepatic disorder.( 38 , 39 ) Microcystins also induce IL‐8 secretion in neutrophils, and the infiltration of neutrophils into the liver tissue is regarded as an important factor for microcystin‐induced liver injury.( 40 ) Although the detailed mechanism of the hepatotoxicity is still obscure, some common principles, including the inhibition of the enzyme, increment of cytokine secretion, and infiltration of leukocytes into liver, can contribute to the hydrophilic PP2A inhibitor‐induced hepatotoxicity.
In conclusion, in the present study, we report that a classical mycotoxin, rubratoxin A, discovered approximately 50 years ago without an identity of its initial target molecule, is a highly specific and potent PP2A inhibitor. Rubratoxin A has broad and efficient potential for use in biochemical and cell biological areas as an excellent molecular probe. With its dramatic antimetastatic effects, rubratoxin A should become a lead compound among a new class of anticancer drugs. Further studies to elucidate the molecular mechanism of how PP2A inhibition leads to antimetastatic effects and hepatotoxicity are in progress.
Supporting information
Fig. S1. Chemical structures of rubratoxins A and B, cantharidin and its dicarboxylic acid form cantharidic acid, and cytostatin analogs.
Fig. S2. Effects of okadaic acid and rubratoxin B (RB) on the phosphorylation of intracellular proteins. (a) Phosphorylation patterns of proteins in rubratoxins‐ or okadaic acid (OA)‐treated cells were detected by immunoblotting. Cells were treated with 20 μM rubratoxin A (RA), 20 or 50 nM OA, or vehicle (control) for 3 h. Since the rubratoxins and okadaic acid were stocked in 35% acetonitrile and dimethylsulfoxide (DMSO), respectively, final 0.07% acetonitrile and 0.25% DMSO were contained in all of the treatments to exclude the influence of using different vehicles. (b) Phosphorylation states of the proteins were detected as above, increasing the concentration of RB. Final 0.7% acetonitrile was used as the vehicle in all of the treatments. p‐ATF‐1, phosphorylated‐activating transcription factor‐1; p‐CREB, phosphorylated‐cyclic AMP response element binding protein; pPKA, phosphorylated substrates of protein kinase A (pPKA), pPKC phosphorylated substrates of protein kinase C.
Fig. S3. Differences in intramolecular hydrogen bonds between rubratoxins A and B. Structures of rubratoxin A (gray) and B (yellow in the structurally different part) in the binding model protein phosphatase 2A–RUB_AH are shown with intramolecular hydrogen bonds represented as green (rubratoxin A) and red (rubratoxin B) dashed lines with distances.
Fig. S4. Binding model of cytostatin to protein phosphatase (PP)2A. (a) Magnified view of the binding site. Cytostatin and amino acid residues of PP2A invoved in the interactions are depicted with thick and thinner lines, respectively. Atoms in the structure C, O, N, S, and P are gray, red, blue, yellow, and orange. Red sphere represents oxygen atom of the crystallization water. Hydrogen or ionic bonds are shown with green dashed lines, and CH/π interaction is shown with a brown line. (b) Binding site is shown from a different angle. Cytostatin is highlighted in yellow. (c) Illustration of the interactions between cytostatin and PP2A catalytic subunit. The cytostatin–PP2A binding model was constructed based on the structure protein data bank (PDB) code 2ie3. Although the absolute configuration of the lactone ring of cytostatin differ from that of rubratoxin A, it is still in the close vicinity of Cys269 and Arg268 and similar interactions are expected. In unrefined positioning, the phosphate of cytostatin was positioned, overlapping with the carbonyl of microcystin–LR interacting with Arg89. Therefore, the lactone ring was fixed to Cys269 by Michael addition, and an oxygen atom in the phosphate was fixed at the coordinate of the corresponding oxygen in the carboxyl of microcystin–LR. This preliminary model was energy minimized and successively refined as follows with energy minimization in each step: the carbonyl on the lactone ring set the distance restraint with an amino group of Arg268, and the restraint was released. Fixation of the oxygen in the phosphate was relaxed to the harmonic restraint, and the olefin tail of cytostatin was directed towards the hydrophobic pocket around Try200. Resultant cytostatin–PP2A binding model was obtained after the final energy minimization.
Fig. S5. Supplementary data for C57BL/6N mouse experiments. (a) Body weight changes of the mice employed in the first experiment (Mean±SE). (b–d) Results of the follow‐up experiment. The second experiment was carried out almost identically as the first experiment using doxorubicin and lentinan as the controls instead of cytostatin (CST). After the subcutaneous inoculation of Lewis lung carcinoma cells (day 0), 0.1, 0.4, and 1.6 mg/kg rubratoxin A (RA; days 1–21, excluding days 14 and 18), 2.5 mg/kg doxorubicin (days 1–21, excluding days 11, 18, and 20), and 2 mg/kg lentinan (days 1–11) were intraperitoneally injected. Primary tumor sizes (b) and body weights (e) were measured during the experiment. Mice were killed on day 21; the excised primary tumor weight was measured (c) and the number of lung metastases was counted (d). Nine mice were used as the control and seven were used for other treatments. Data shown are mean±SE. (*) Differences were significant between the control and drug treatment (P<0.01) in (c) and (d).
Fig. S6. Antitumor and antimetastatic effects of rubratoxin A (RA) on immunodeficient mice. Five‐week‐old female SCID (CB17/Icr–Prkdcscid/CrlCrlj) and non‐obese diabetic (NOD)–SCID (NOD.CB17‐Prkdcscid/J) mice were subcutaneously inoculated with 1×106 cells/100 μL saline of Lewis lung carcinoma cells in the left flank (day 0). Rubratoxin A (RA, 1.6 mg/kg) or vehicle (control, 5% dimethylsulfoxide, 0.5% Tween‐80) were intraperitoneally injected on days 1–12, 13–15, 18, and 20 to SCID mice, and on days 1–12, 13–15, and 20 to NOD mice. Primary tumor volumes (a) and body weights (d) were measured during the experiment. Mice were killed on day 21; excised primary tumor weight was measured (b) and the number of lung metastatic foci in each size was counted (c). Data shown are mean±SE. (*) Differences were significant between the control and RA treatment (P<0.01) in (b) and (c).
Fig. S7. Effects of rubratoxins A and B on natural killer (NK) cell activity. Six‐week‐old female BDF1 mice were intraperitoneally injected with saline (control), 0.4 or 1.6 mg/kg rubratoxin A (RA), 1.6 mg/kg rubratoxin B (RB), 1.6 mg/kg cytostatin (CST), or a cytostatin analog, leustroducsin H (LH) daily for 3 days. One day after the last injection, cytolytic activity in splenocytes was assessed against B16–BL6 (a) and YAC‐1 (b) target cells. Data presented are mean±SE. E/T, effector/target ratio.
Appendix S1. Mass spectrometry and nuclear magnetic resonance rubratoxin spectrometric data.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We thank Professor Maurice Moss (University of Surrey, UK), Mr That Nguyen, and Dr Ryuichi Sawa (Microbial Chemistry Research Foundation, Japan) for providing information about rubratoxins and experimental techniques and for the assistance with the mass spectrometric analysis. We also thank Professor Sachi Sri Kantha (Gifu Pharmaceutical University, Japan) for critically reading this manuscript. This work was supported by a Grant‐in‐Aid for Cancer Research from the Ministry of Education, Science, Sports, and Culture of Japan.
References
- 1. Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 2001; 353: 417–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta 2009; 1795: 1–15. [DOI] [PubMed] [Google Scholar]
- 3. Amemiya M, Ueno M, Osono M et al. Cytostatin, a novel inhibitor of cell adhesion to components of extracellular matrix produced by Streptomyces sp. MJ654‐NF4. I. Taxonomy, fermentation, isolation and biological activities. J Antibiot 1994; 47: 536–40. [DOI] [PubMed] [Google Scholar]
- 4. Amemiya M, Someno T, Sawa R, Naganawa H, Ishizuka M, Takeuchi T. Cytostatin, a novel inhibitor of cell adhesion to components of extracellular matrix produced by Streptomyces sp. MJ654‐NF4. II. Physico‐chemical properties and structure determination. J Antibiot 1994; 47: 541–4. [DOI] [PubMed] [Google Scholar]
- 5. Kawada M, Amemiya M, Ishizuka M, Takeuchi T. Cytostatin, an inhibitor of cell adhesion to extracellular matrix, selectively inhibits protein phosphatase 2A. Biochim Biophys Acta 1999; 1452: 209–17. [DOI] [PubMed] [Google Scholar]
- 6. Lewy DS, Gauss CM, Soenen DR, Boger DL. Fostriecin: chemistry and biology. Curr Med Chem 2002; 9: 2005–32. [DOI] [PubMed] [Google Scholar]
- 7. Matsuzawa S, Suzuki T, Suzuki M et al. Thyrsiferyl 23‐acetate is a novel specific inhibitor of protein phosphatase PP2A. FEBS Lett 1994; 356: 272–4. [DOI] [PubMed] [Google Scholar]
- 8. Usui T, Marriott G, Inagaki M, Swarup G, Osada H. Protein phosphatase 2A inhibitors, phoslactomycins. Effects on the cytoskeleton in NIH/3T3 cells. J Biochem 1999; 125: 960–5. [DOI] [PubMed] [Google Scholar]
- 9. Kawada M, Kawatsu M, Masuda T et al. Specific inhibitors of protein phosphatase 2A inhibit tumor metastasis through augmentation of natural killer cells. Int Immunopharmacol 2003; 3: 179–88. [DOI] [PubMed] [Google Scholar]
- 10. Kawada M, Yoshimoto Y, Kumagai H et al. PP2A inhibitors, harzianic acid and related compounds produced by fungus strain F‐1531. J Antibiot 2004; 57: 235–7. [DOI] [PubMed] [Google Scholar]
- 11. Yamaoka M, Sato K, Kobayashi M et al. FR177391, a new anti‐hyperlipidemic agent from Serratia. IV. Target identification and validation by chemical genetic approaches. J Antibiot 2005; 58: 654–62. [DOI] [PubMed] [Google Scholar]
- 12. Swingle M, Ni L, Honkanen RE. Small‐molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol 2007; 365: 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Masuda T, Watanabe S, Amemiya M, Ishizuka M, Takeuchi T. Inhibitory effect of cytostatin on spontaneous lung metastases of B16‐BL6 melanoma cells. J Antibiot 1995; 48: 528–9. [DOI] [PubMed] [Google Scholar]
- 14. De Jong RS, Mulder NH, Uges DR et al. Phase I and pharmacokinetic study of the topoisomerase II catalytic inhibitor fostriecin. Br J Cancer 1999; 79: 882–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burnside JE, Sippel WL, Forgacs J, Carll WT, Atwood MB, Doll ER. A disease of swine and cattle caused by eating moldy corn. II. Experimental production with pure cultures of molds. Am J Vet Res 1957; 18: 817–24. [PubMed] [Google Scholar]
- 16. Moss MO. The rubratoxins, toxic metabolites of Penicillium rubrum Stoll. In: Ciegler A, Kadis S, Ajl SJ, eds. Microbial Toxins. New York and London: Academic Press, 1971; 381–407. [Google Scholar]
- 17. Moss MO. Microbial food toxicants: rubratoxins. In: Recheigl M, ed. Handbook of Foodborne Diseases of Biological Origin. Florida: CRC Press, 1983; 167–79. [Google Scholar]
- 18. Abbott SP. Mycotoxins and indoor molds. Indoor Environment Connections 2002; 3: 14–24. [Google Scholar]
- 19. Iwashita K, Nagashima H. Rubratoxin B induces interleukin‐6 secretion in mouse white adipose tissues and 3T3‐L1 adipocytes. Toxicol Lett 2008; 182: 79–83. [DOI] [PubMed] [Google Scholar]
- 20. Wada S, Niimi M, Niimi K et al. Candida glabrata ATP‐binding cassette transporters Cdr1p and Pdh1p expressed in a Saccharomyces cerevisiae strain deficient in membrane transporters show phosphorylation‐dependent pumping properties. J Biol Chem 2002; 277: 46809–21. [DOI] [PubMed] [Google Scholar]
- 21. Wada S, Tanabe K, Yamazaki A et al. Phosphorylation of candida glabrata ATP‐binding cassette transporter Cdr1p regulates drug efflux activity and ATPase stability. J Biol Chem 2005; 280: 94–103. [DOI] [PubMed] [Google Scholar]
- 22. Xing Y, Xu Y, Chen Y et al. Structure of protein phosphatase 2A core enzyme bound to tumor‐inducing toxins. Cell 2006; 127: 341–53. [DOI] [PubMed] [Google Scholar]
- 23. Paul IC, Sim GA, Hamor TA, Robertson JM. Fungal metabolites. Part II. The structure of byssochlamic acid: X‐ray analysis of byssochlamic acid bis‐p‐bromphenylhydrazide. J Chem Soc 1963; 5502–12. [Google Scholar]
- 24. Büchi G, Snader KM, White JD, Gougoutas JZ, Singh S. Structures of rubratoxins A and B. J Am Chem Soc 1970; 92: 6638–41. [DOI] [PubMed] [Google Scholar]
- 25. Lawhorn BG, Boga SB, Wolkenberg SE et al. Total synthesis and evaluation of cytostatin, its C10‐C11 diastereomers, and additional key analogues: impact on PP2A inhibition. J Am Chem Soc 2006; 128: 16720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wadzinski BE, Wheat WH, Jaspers S et al. Nuclear protein phosphatase 2A dephosphorylates protein kinase A‐phosphorylated CREB and regulates CREB transcriptional stimulation. Mol Cell Biol 1993; 13: 2822–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yeh E, Cunningham M, Arnold H et al. A signalling pathway controlling c‐Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 2004; 6: 308–18. [DOI] [PubMed] [Google Scholar]
- 28. Margolis SS, Walsh S, Weiser DC, Yoshida M, Shenolikar S, Kornbluth S. PP1 control of M phase entry exerted through 14‐3‐3‐regulated Cdc25 dephosphorylation. EMBO J 2003; 22: 5734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell 2007; 25: 917–31. [DOI] [PubMed] [Google Scholar]
- 30. Keyse SM. Protein phosphatases and the regulation of mitogen‐activated protein kinase signalling. Curr Opin Cell Biol 2000; 12: 186–92. [DOI] [PubMed] [Google Scholar]
- 31. Dickinson RJ, Keyse SM. Diverse physiological functions for dual‐specificity MAP kinase phosphatases. J Cell Sci 2006; 119: 4607–15. [DOI] [PubMed] [Google Scholar]
- 32. Teruya T, Simizu S, Kanoh N, Osada H. Phoslactomycin targets cysteine‐269 of the protein phosphatase 2A catalytic subunit in cells. FEBS Lett 2005; 579: 2463–8. [DOI] [PubMed] [Google Scholar]
- 33. Inoue H, Someno T, Kato T, Kumagai H, Kawada M, Ikeda D. Ceramidastin, a novel bacterial ceramidase inhibitor, produced by Penicillium sp. Mer‐f17067. J Antibiot 2009; 62: 63–7. [DOI] [PubMed] [Google Scholar]
- 34. Nishio M, Hirota M, Umezawa Y. The CH/π Interaction. Evidence, Nature, and Consequences. New York: Wiley‐VCH, 1998. [Google Scholar]
- 35. Fujiki H, Suganuma M. Tumor promotion by inhibitors of protein phosphatases 1 and 2A: the okadaic acid class of compounds. Adv Cancer Res 1993; 61: 143–94. [DOI] [PubMed] [Google Scholar]
- 36. Katsiari CG, Kyttaris VC, Juang YT, Tsokos GC. Protein phosphatase 2A is a negative regulator of IL‐2 production in patients with systemic lupus erythematosus. J Clin Invest 2005; 115: 3193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nagashima H, Maeda‐Nakamura K, Iwashita K, Goto T. Induced secretion of tissue inhibitor of metalloproteinases‐1 (TIMP‐1) in vivo and in vitro by hepatotoxin rubratoxin B. Food Chem Toxicol 2006; 44: 1138–43. [DOI] [PubMed] [Google Scholar]
- 38. Nagashima H, Nakamura K, Goto T. Hepatotoxin rubratoxin B induced the secretion of TNF‐alpha, IL‐8, and MCP‐1 in HL60 cells. Biochem Biophys Res Commun 2001; 287: 829–32. [DOI] [PubMed] [Google Scholar]
- 39. Nagashima H, Nakamura K, Goto T. Rubratoxin B induced the secretion of hepatic injury‐related colony stimulating factors in human hepatoma cells. Toxicol Lett 2003; 145: 153–9. [DOI] [PubMed] [Google Scholar]
- 40. Kujbida P, Hatanaka E, Campa A, Curi R, Poliselli Farsky SH, Pinto E. Analysis of chemokines and reactive oxygen species formation by rat and human neutrophils induced by microcystin‐LA, ‐YR and ‐LR. Toxicon 2008; 51: 1274–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Chemical structures of rubratoxins A and B, cantharidin and its dicarboxylic acid form cantharidic acid, and cytostatin analogs.
Fig. S2. Effects of okadaic acid and rubratoxin B (RB) on the phosphorylation of intracellular proteins. (a) Phosphorylation patterns of proteins in rubratoxins‐ or okadaic acid (OA)‐treated cells were detected by immunoblotting. Cells were treated with 20 μM rubratoxin A (RA), 20 or 50 nM OA, or vehicle (control) for 3 h. Since the rubratoxins and okadaic acid were stocked in 35% acetonitrile and dimethylsulfoxide (DMSO), respectively, final 0.07% acetonitrile and 0.25% DMSO were contained in all of the treatments to exclude the influence of using different vehicles. (b) Phosphorylation states of the proteins were detected as above, increasing the concentration of RB. Final 0.7% acetonitrile was used as the vehicle in all of the treatments. p‐ATF‐1, phosphorylated‐activating transcription factor‐1; p‐CREB, phosphorylated‐cyclic AMP response element binding protein; pPKA, phosphorylated substrates of protein kinase A (pPKA), pPKC phosphorylated substrates of protein kinase C.
Fig. S3. Differences in intramolecular hydrogen bonds between rubratoxins A and B. Structures of rubratoxin A (gray) and B (yellow in the structurally different part) in the binding model protein phosphatase 2A–RUB_AH are shown with intramolecular hydrogen bonds represented as green (rubratoxin A) and red (rubratoxin B) dashed lines with distances.
Fig. S4. Binding model of cytostatin to protein phosphatase (PP)2A. (a) Magnified view of the binding site. Cytostatin and amino acid residues of PP2A invoved in the interactions are depicted with thick and thinner lines, respectively. Atoms in the structure C, O, N, S, and P are gray, red, blue, yellow, and orange. Red sphere represents oxygen atom of the crystallization water. Hydrogen or ionic bonds are shown with green dashed lines, and CH/π interaction is shown with a brown line. (b) Binding site is shown from a different angle. Cytostatin is highlighted in yellow. (c) Illustration of the interactions between cytostatin and PP2A catalytic subunit. The cytostatin–PP2A binding model was constructed based on the structure protein data bank (PDB) code 2ie3. Although the absolute configuration of the lactone ring of cytostatin differ from that of rubratoxin A, it is still in the close vicinity of Cys269 and Arg268 and similar interactions are expected. In unrefined positioning, the phosphate of cytostatin was positioned, overlapping with the carbonyl of microcystin–LR interacting with Arg89. Therefore, the lactone ring was fixed to Cys269 by Michael addition, and an oxygen atom in the phosphate was fixed at the coordinate of the corresponding oxygen in the carboxyl of microcystin–LR. This preliminary model was energy minimized and successively refined as follows with energy minimization in each step: the carbonyl on the lactone ring set the distance restraint with an amino group of Arg268, and the restraint was released. Fixation of the oxygen in the phosphate was relaxed to the harmonic restraint, and the olefin tail of cytostatin was directed towards the hydrophobic pocket around Try200. Resultant cytostatin–PP2A binding model was obtained after the final energy minimization.
Fig. S5. Supplementary data for C57BL/6N mouse experiments. (a) Body weight changes of the mice employed in the first experiment (Mean±SE). (b–d) Results of the follow‐up experiment. The second experiment was carried out almost identically as the first experiment using doxorubicin and lentinan as the controls instead of cytostatin (CST). After the subcutaneous inoculation of Lewis lung carcinoma cells (day 0), 0.1, 0.4, and 1.6 mg/kg rubratoxin A (RA; days 1–21, excluding days 14 and 18), 2.5 mg/kg doxorubicin (days 1–21, excluding days 11, 18, and 20), and 2 mg/kg lentinan (days 1–11) were intraperitoneally injected. Primary tumor sizes (b) and body weights (e) were measured during the experiment. Mice were killed on day 21; the excised primary tumor weight was measured (c) and the number of lung metastases was counted (d). Nine mice were used as the control and seven were used for other treatments. Data shown are mean±SE. (*) Differences were significant between the control and drug treatment (P<0.01) in (c) and (d).
Fig. S6. Antitumor and antimetastatic effects of rubratoxin A (RA) on immunodeficient mice. Five‐week‐old female SCID (CB17/Icr–Prkdcscid/CrlCrlj) and non‐obese diabetic (NOD)–SCID (NOD.CB17‐Prkdcscid/J) mice were subcutaneously inoculated with 1×106 cells/100 μL saline of Lewis lung carcinoma cells in the left flank (day 0). Rubratoxin A (RA, 1.6 mg/kg) or vehicle (control, 5% dimethylsulfoxide, 0.5% Tween‐80) were intraperitoneally injected on days 1–12, 13–15, 18, and 20 to SCID mice, and on days 1–12, 13–15, and 20 to NOD mice. Primary tumor volumes (a) and body weights (d) were measured during the experiment. Mice were killed on day 21; excised primary tumor weight was measured (b) and the number of lung metastatic foci in each size was counted (c). Data shown are mean±SE. (*) Differences were significant between the control and RA treatment (P<0.01) in (b) and (c).
Fig. S7. Effects of rubratoxins A and B on natural killer (NK) cell activity. Six‐week‐old female BDF1 mice were intraperitoneally injected with saline (control), 0.4 or 1.6 mg/kg rubratoxin A (RA), 1.6 mg/kg rubratoxin B (RB), 1.6 mg/kg cytostatin (CST), or a cytostatin analog, leustroducsin H (LH) daily for 3 days. One day after the last injection, cytolytic activity in splenocytes was assessed against B16–BL6 (a) and YAC‐1 (b) target cells. Data presented are mean±SE. E/T, effector/target ratio.
Appendix S1. Mass spectrometry and nuclear magnetic resonance rubratoxin spectrometric data.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item