

Abstract
We have investigated the expression and role of the 58‐kDa microspherule protein (MSP58) in colorectal carcinoma (CRC). By immuhistochemistry and immunofluorescence, we observed MSP58 in the nucleus and cytoplasm of CRC cells, and found MSP58 to be present in CRC specimens more often than in adjacent non‐tumor tissues (92.5 vs 36.3%, P < 0.01). The average staining score in adjacent non‐tumor tissues was significantly lower than in CRC tissues (2.05 ± 1.13 vs 5.23 ± 1.38, P < 0.01). Moreover, MSP58 mRNA and protein appeared to be upregulated in six fresh CRC samples compared to their adjacent non‐cancerous tissues. MSP58 expression was also detected in the human CRC‐derived cell lines LoVo, CoLo205, HCT116, HT‐29, SW620, and SW480. Downregulation of MSP58 inhibited in vitro growth and attenuated tumor growth in animal models by induction of cell cycle arrest, and was associated with reduced levels of cyclin D1, cyclin‐dependent kinase 4, phosphorylation‐Rb (p‐Rb), p21, and Retino blastoma (Rb) proteins. These results indicated that MSP58 might play an important role in the carcinogenesis of CRC via regulation of the cyclin D1–cyclin‐dependent kinase 4–p21 pathway. (Cancer Sci 2009; 100: 1585–1590)
Of all cancers, CRC has the third and fourth highest incidence, and the fifth and fourth highest mortality rates in women and men, respectively, throughout the world.( 1 , 2 ) In the USA, it is the fourth most common cancer and the second leading cause of cancer‐related death.( 3 , 4 ) Early diagnosis of CRC is critical.( 2 ) Initiating genes have been found for several CRC syndromes. For example, colorectal cancers almost invariably carry activating mutations in the Wnt pathway, which targets the tumor suppressors adenomatous polyposis coli (APC) or Axin 2, or the oncogene β‐catenin.( 5 ) Although much effort has focused on identifying genes responsible for CRC, and several genes have been found to be involved in CRC tumorigenesis, the molecular mechanism of the disease remains unknown.
Nucleolar MSP58 was initially identified through its interaction with the proliferation‐related nucleolar protein p120, and its overexpression leads to enlargement of nucleoli.( 6 , 7 ) MSP58 has also been reported to interact with several other proteins. It is reported to play a role in modulation of Daxx‐dependent transcriptional repression,( 8 ) and is associated with the transcriptional activity of the Basic‐helix‐loop helix (bHLH) transcription factor stimulated by retinoic acid (STRA)13.( 9 ) MSP58 is reported to interact with Mi‐2b in the nucleolus, and to upregulate ribosomal gene transcription.( 10 ) These findings suggest that MSP58 may have transcriptional regulation functions in the nucleus and nucleolus. Recently, MSP58 was shown to behave as an oncogene and its transformation activity was inhibited by physical interaction with the PTEN tumour suppressor.( 7 , 11 ) In addition, in our previous studies, we identified MSP58 as a novel Ndrg2‐interacting protein and found that Ndrg2 could alter the role of Msp58 in cell proliferation. Msp58 interaction with Ndrg2 might be an important clue in elucidating the role of Ndrg2 in tumor suppression.( 12 ) Nonetheless, despite these data, the functions of MSP58 in tumors remain largely unknown. In the present study, we investigated the expression and role of MSP58 in CRC.
Materials and Methods
Tissue collection and immunohistochemistry. Immunohistochemistry was carried out as described previously.( 13 ) One hundred and sixty formalin‐fixed paraffin‐embedded specimens of primary CRC and their adjacent non‐cancerous counterparts including only normal colonic mucosa were obtained from archival blocks of colectomy cases. These specimens were collected from the Department of Pathology at Xijing Hospital, Xi’an, China. All patients gave informed consent to use excess pathological specimens for research purposes. Six fresh CRC tumor samples with adjacent non‐cancerous tissues were collected from the Department of Gastrointestinal Surgery at Xijing Hospital. All tissues were stored at –70°C. Tissues were used after pathological diagnoses were definite. The protocols used in the study were approved by the hospital's Protection of Human Subjects Committee. The use of human tissues in this study was approved by the institutional review board of the Fourth Military Medical University and was done in accordance with international guidelines. Rabbit anti‐MSP58 was a gift from Dr Laetitia Davidovic.( 7 ) For analysis, 4‐µm sections of formalin‐fixed paraffin‐embedded specimens were made. Slides were dewaxed, rehydrated, and incubated in 0.3% (v/v) hydrogen peroxide in methanol for 20 min to quench endogenous peroxidase activity, before adding 10% (v/v) normal goat serum for 1 h. Sections were incubated at 4°C overnight with anti‐MSP58 at a 1:200 dilution in PBS containing 3% (w/v) bovine serum albumin. The sections were washed in PBS containing 0.02% (v/v) Tween‐20 three times for 5 min each. Sections were treated with biotinylated goat antirabbit antibody for 20 min at room temperature, rinsed with PBS, and incubated with streptavidin–horseradish peroxidase for 20 min at room temperature. Reaction products were visualized with diaminobenzidine at room temperature for 1 min. The sections were dehydrated and mounted after being counterstained with hematoxylin for 30 s and rinsed with tap water. The negative control used pre‐immune mouse serum in the primary antibody step. Data are the average of two independent scores.
Expression of MSP58 was evaluated as the percentage of positive cells in a specimen, and by staining intensity as described previously.( 14 ) The percentage of positive cells was evaluated quantitatively and scored as 0 for staining of ≤1% of total cells counted, 1 for staining of 2–25%, 2 for staining of 26–50%, 3 for staining of 51–75%, and 4 for staining of >75% of the cells examined. Intensity was graded as follows: 0, no signal; 1, weak; 2, moderate; and 3, strong staining. A total ‘staining score’ of 0–12 was calculated and graded as negative (–, score 0–1), weak (+, score 2–4), moderate (++, score 5–8), or strong (+++, score 9–12).
Cell culture. The human CRC cell lines LoVo, CoLo205, HCT116, HT‐29, SW620, and SW480 were obtained from the American Type Culture Collection (Rockvill, MD, USA) and maintained as recommended. Cells were cultured in DMEM with 2 mM l‐glutamine and Earle's balanced salt solution (BSS) adjusted to contain 1.5 g/L sodium bicarbonate, 0.1 mM non‐essential amino acids, and 1.0 mM sodium pyruvate. All culture fluid was supplemented with 10% FCS. All cells were cultured with 5% CO2 at 37°C in a humidified chamber.
MSP58 subcellular localization analysis. Fluorescence microscopy analysis was carried out as described previously.( 13 ) Treated cells were fixed (4% paraformaldehyde in PBS, 30 min, room temperature), rinsed, and permeabilized with 0.1% Triton X‐100 in PBS for 15 min. Permeabilized cells were incubated with horse serum in PBS to block non‐specific binding. After thorough rinsing with PBS, cells were incubated overnight at 4°C with anti‐MSP58 at 1:200 dilution, rinsed with PBS, and incubated with FITC‐coupled antirabbit antibody at 1:1500 for 1 h, 37°C. Dual‐color detection was carried out by confocal laser scanning microscopy after treatment with 50 µg DAPI for 5 min to label nuclear DNA. Each experiment was carried out in triplicate.
RT‐PCR analysis. Total RNA was extracted from the tumor tissues by the acid guanidinium thiocyanate–phenol–chloroform extraction method.( 13 , 15 ) Total RNA was dissolved in 0.1% diethylpyrocarbonate‐treated water. The amount of RNA was determined by measuring spectrometric absorbance at 260 nm. The MSP58 forward and reverse primers were 5′‐aggctattgcagccatccagag‐3′ and 5′‐ctgtgcagcaggtcctggaa‐3′ and gave a 0.118‐kb product. The housekeeping gene β‐actin was used as an endogenous control for RNA normalization. The β‐actin forward and reverse primers were 5′‐atcatgtttgagaccttcaaca‐3′ and 5′‐catctcttgctcgaagtcca‐3′, and gave a 0.318‐kb product. PCR was carried out in a GeneAmp PCR system 2400 thermocycler (Perkin Elmer, Norwalk, CT, USA). The conditions were 40 s at 94°C, 30 s at 55°C, and 40 s at 72°C (24 cycles). PCR products were loaded onto a 1.5% agarose gel and electrophoretically separated. The gel was then visualised under ultraviolet light following ethidium bromide staining. Each experiment was carried out in triplicate.
Plasmid construction and cell transfection. pSilencer3.1 (Ambion, Austin, TX, USA) was used according to the manufacturer's protocol for construction of a human MSP58 siRNA vector. The oligonucleotide 5′‐GATC CGCA GCTC ATCA TCGA ACTT CTTC AAGA GAGA AGTT CGAT GATG AGCT GTTT TTTG GAAA‐3′ was used to encode siRNA against MSP58, as described previously.( 16 ) The non‐specific siRNA 5′‐GATC CGAC TTCA TAAG GCGC ATGC ACTT CAAG AGAG TGCA TGCG CCTT ATGA AGTC TTTT TTGT CGAC A‐3′ (ShanghaiGenePharma Co., Shanghai, China) was used as a negative control. The two resulting plasmids were designated pSilencer3.1‐MSP58 and pSilencer3.1‐NC. Cell transfection was carried out with Lipofectamine2000 (Invitrogen, Carlsbad, CA, USA) using the manufacturer's protocol. Briefly, cells were plated and grown to 70–90% confluence without antibiotics and transfected with 1 µg plasmid. For stable transfection, G418 (400 µg/mL) was added to the cells after 24 h of transfection. Mixed clones were screened and expanded for an additional 6 weeks. The colon carcinoma cell lines HT29 and SW480 transfected with pSilencer3.1‐MSP58, pSilencer3.1‐NC, and pSilencer3.1 were designated HT29‐ or SW480‐MSP58si, HT29‐ or SW480‐pSilencer3.1‐NC, and HT29‐ or SW480‐pSilencer3.1.
Monolayer growth rate. Monolayer culture growth rate was determined as described previously( 17 ) by conversion of MTT (Sigma Chemical Co., St. Louis, MD, USA) to water‐insoluble formazan by viable cells. Approximately 3000 cells in 200 µL medium were plated in 96‐well plates and grown under normal conditions. Cultures were assayed every day for 7 days and absorbance values were determined with an enzyme‐linked immunosorbent assay reader (DASIT, Milan, Italy) at 490 nm. Each experiment was carried out in triplicate.
Plate colony formation assay. Plate colony formation assay was as described previously.( 16 ) For colony formation assays, 1 × 103 cells were seeded into 60‐mm dishes with 5 mL DMEM supplemented with 10% FBS (Sigma Chemical Co.) and 400 µg/mL G418 (Merck, Darmsdadt, Germany). After 14 days, the resulting colonies were rinsed with PBS, fixed with methanol at –20°C for 5 min, and stained with Giemsa (Sigma‐Aldrich) for 20 min. Only clearly visible colonies (diameter > 50 µm) were counted.
Soft agar clonogenic assay. Soft agar clonogenic assays were carried out as described previously( 17 ) to assess anchorage‐independent growth, as a characteristic of in vitro tumorigenicity. Briefly, cells were detached and plated in 0.3% agarose with a 0.5% agarose underlay (1 × 104/well in six‐well plates). The number of foci >100 µm was counted after 17 days. Each experiment was carried out in triplicate.
Tumorigenicity in nude mice. Tumorigenicity in nude mice was measured as described previously.( 17 ) For tumorigenicity assays, four groups of five mice each were injected subcutaneously at a single site with stably transfected cells. Tumor onset was scored visually and by palpitation at the sight of injection by two trained members of the laboratory staff at different times on the same day. Average tumor size was estimated by physical measurement in cm of the excised tumor at the time of death. With the exception of mice with large tumor burdens, animals were killed 4 weeks after injection. Tumors were verified by HE staining. Blocks were stored for further analysis.
Cell cycle analysis. Flow cytometric analysis was carried out as described previously.( 17 ) Cells were seeded into 60 mm‐diameter plates in complete medium overnight, placed in serum‐free medium for 48 h to synchronize the cells, and returned to complete medium for 24 h before harvesting. After washing with ice‐cold PBS, cells were suspended in 0.5 mL 70% alcohol and kept at 4°C for 30 min. The suspension was filtered through 50 ml nylon mesh, and the DNA content of stained nuclei was analyzed using a flow cytometer (EPICS XL; Coulter, Miami, FL, USA). The cell cycle was analyzed using Multicycle‐DNA Cell Cycle Analyzed Software. The proliferation index (PI) was calculated as PI = (S + G2)/(S + G2 + G1). Each experiment was carried out in triplicate.
Western blot analysis. Protein extraction and immunoblot analysis were carried out as described previously.( 18 ) Cells were washed twice with Hanks's balanced salt solution and lysed directly in RIPA buffer (50 mM Tris–HCl pH 7.4, 1%[v/v] Triton X‐100, 1 mM EDTA, 1 mM leupeptin, 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, 1 mM Na3VO4). Lysates were centrifuged at 10878 g for 30 min at 4°C and supernatants were collected. Cell lysate (60 µg) was separated by SDS‐PAGE, blotted onto nitrocellulose membrane, and incubated with monoclonal antibody against cyclin D1, CDK4, or p21 (diluted 1:500; BD Biosciences, San Jose, CA, USA), Rb, or phospho‐Rb (diluted 1:500; Cell Signaling Technology, Beverly, MA, USA) and polyclonal antibody against MSP58 (at 1:200 dilution). Anti‐β‐actin antibody was used for all western blots as a loading control (diluted 1:5000; Sigma). After three washes for 15 min in TBS‐Tween, the membranes were incubated at room temperature for 2 h with horseradish peroxidase‐conjugated antimouse or antirabbit secondary antibody (dilution 1:2000; Santa Cruz Biotechnology, CA, USA). The membranes were visualized using the enhanced chemiluminescence system (Amersham Pharmacia Biotech, CA, USA). Each experiment was carried out in triplicate.
Statistical analysis. The statistical SPSS software package (SPSS, Chicago, IL, USA) was used to analyze data. Stastical analysis was carried out with the Kruskal–Wallis rank test, and the Mann–Whitney U‐test was used to calculate P‐values and to compare differences between groups in immunohistochemistry. Assays for characterizing cell phenotype were analyzed by Student's t‐test. Differences were considered statistically different at P < 0.05.
Results
MSP58 expression is increased in CRC. MSP58 expression was evaluated by immunohistochemistry in 160 cases of CRC and adjacent non‐cancerous counterparts. We observed, qualitatively, that MSP58 was predominantly located in the nucleus and cytoplasm of CRC cells (Fig. 1a). Positive expression was quantitated, and CRC cells (92.5%) were found to express MSP58 more frequently than adjacent non‐cancerous tissue cells (36.25%, P < 0.01). By average staining score, non‐cancerous cells had significantly lower MSP58 expression than adjacent CRC cells (2.05 ± 1.13 vs 5.23 ± 1.38, P < 0.01) (Fig. 1b). As shown in Table 1, in CRC specimens, the expression of MSP58 protein in highly differentiated tumor tissues was lower than in moderately or poorly differentiated tissues (P < 0.05), indicating a correlation between MSP58 expression and CRC differentiation grade. In addition, MSP58 expression in samples from patients with lymph node and/or distant metastasis (TNM stages III, IV) was significantly higher in than those without metastasis (TNM stages I, II; P < 0.01), indicating a relationship between MSP58 expression and CRC metastasis.
Figure 1.
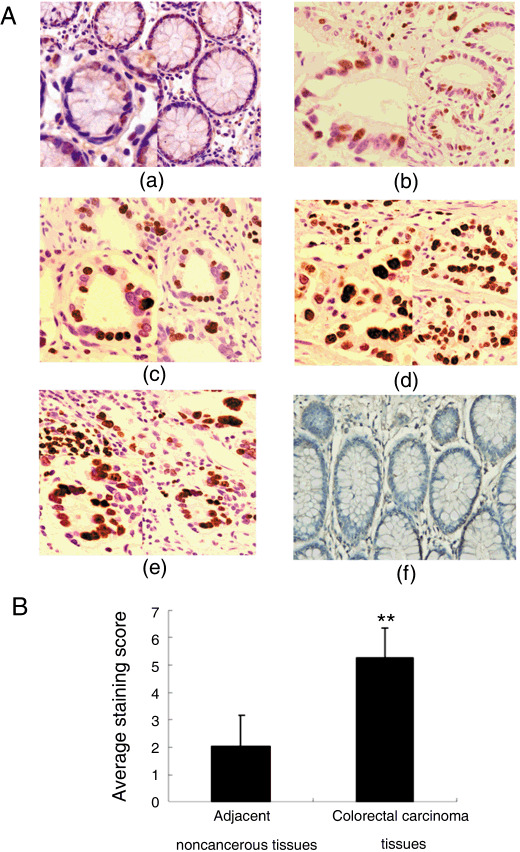
Immunohistochemical staining of 58‐kDa microspherule protein (MSP58) in colorectal carcinoma (CRC) tissue and adjacent non‐cancerous tissue at different stages of differentiation. (A) Rabbit anti‐MSP58 antibodies were used to stain paraffin sections. (a) Non‐cancerous tissue exhibiting weak MSP58 immunostaining, (b,c) well differentiated, (d) moderately differentiated, and (e) poorly differentiated CRC tissues showing weak, moderate, or high MSP58 immunosignals in most cells. (f) Negative control slides using anti‐6His as the primary antibody. (B) Average staining scores of MSP58 immunostaining in non‐cancerous and CRC tissue specimens. **P < 0.01.
Table 1.
Clinicopathological associations of 58‐kDa microspherule protein (MSP58) expression in patients with colorectal carcinoma
| Clinical samples | Total no. cases | MSP58 immunostaining | |||
|---|---|---|---|---|---|
| – | + | ++ | +++ | ||
| Adjacent non‐cancerous tissue | 160 | 102 (63.75%) | 30 (18.75%) | 18 (11.25%) | 10 (6.25%) |
| Colorectal carcinoma | 160 | 12 (7.5%) | 24 (15%) | 48 (30%) | 76 (47.5%) |
| Differentiation | |||||
| High | 42 | 6 | 12 | 16 | 8 |
| Moderate | 52 | 6 | 8 | 14 | 24 |
| Poor | 66 | 0 | 4 | 18 | 44 |
| TNM | |||||
| I + II | 70 | 10 | 18 | 22 | 20 |
| III + IV | 90 | 2 | 6 | 26 | 56 |
| Metastasis | |||||
| With | 72 | 0 | 4 | 22 | 46 |
| Without | 88 | 12 | 20 | 26 | 30 |
MSP58 staining was graded as negative (–, score 0–1), weak (+, score 2–4), moderate (++, score 5–8), or strong (+++, score 9–12).
To further investigate these observations, RT‐PCR and western blots were carried out for six fresh CRC tumor tissue samples and adjacent non‐cancerous tissues. We found that tumor tissue specimens tended to show higher MSP58 expression when compared with the adjacent non‐cancerous tissues (Fig. 2a,b), consistent with the MSP58 protein expression seen in immunohistochemical staining. MSP58 gene expression was tested in the human CRC cell lines LoVo, CoLo205, HCT116, HT‐29, SW620, and SW480. As shown in Figure 2(c), MSP58 expression was detected in all CRC cell lines.
Figure 2.
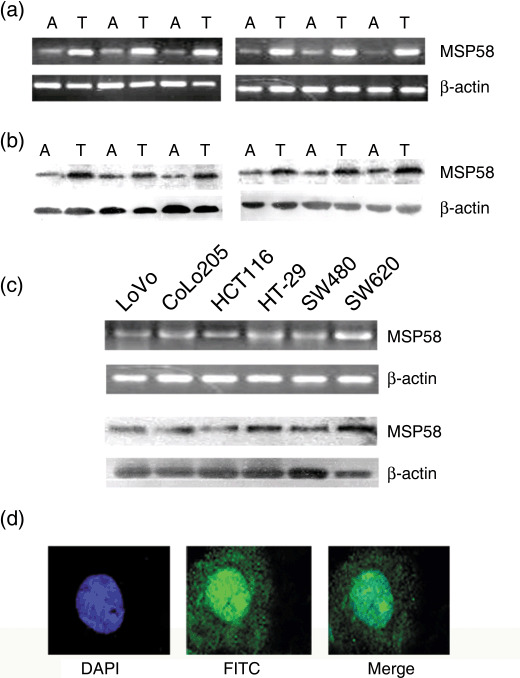
58‐kDa microspherule protein (MSP58) expression in colorectal carcinoma (CRC) (T) and adjacent non‐cancerous (A) tissues in CRC cell lines. The data shown are representative of three replicates that gave similar results. (a) RT‐PCR and (b) western blotting results showing MSP58 transcripts and protein. (c) RT‐PCR and western blotting results showing MSP58 transcripts and protein in CRC cell lines. β‐Actin was used as an internal control. (d) MSP58 was visualized with secondary antibody conjugated to FITC (green) and nuclei were counterstained with DAPI (blue).
Subcellular localization of MSP58. In previous studies, MSP58 was found to be either exclusively nucleolar or cytoplasmic,( 6 , 8 , 9 , 10 , 11 ) or detected in the nucleus, nucleolus, and cytoplasm.( 7 ) Here, the subcellular localization of MSP58 in HT‐29 cells was determined using anti‐MSP58 antibody and fluorescent staining that could be visualized under a confocal laser scanning fluorescence microscopy. Dual‐color detection of MSP58 and DAPI demonstrated that MSP58 was mainly localized to the nucelus with a faint cytoplasmic staining (Fig. 2d). On the basis of observations in tissue samples and cultured cells, we concluded that MSP58 lies predominantly in the nucleus, with a small fraction detectable in the cytoplasm.
Downregulation of MSP58 inhibits in vitro proliferation and growth and in vivo tumorigenicity of colon carcinoma cells. To downregulate the expression of MSP58 in CRC cells, the MSP58‐specific siRNA vector MSP58si was constructed.( 16 ) After cell transfection and antibiotic screening for 6 weeks, the expression of MSP58 in stably transfected cells was determined by western blotting. MSP58si appeared to reduce the expression of MSP58 in HT29 and SW480 effectively, whereas no effect was observed with the negative control pSilencer3.1‐NC vector (Fig. 3a). Expression of MSP58 was similar in HT29 and SW480 cells transfected with empty vector and pSilencer3.1‐NC vector. Stably transfected HT29‐MSP58si and SW480‐MSP58si were chosen for further cellular assay. When the growth curves of these cell lines were compared in medium containing 10% FCS, the curves for MSP58si cells were significantly lower than control cells (P < 0.05 on days 4–7; Fig. 3b).
Figure 3.
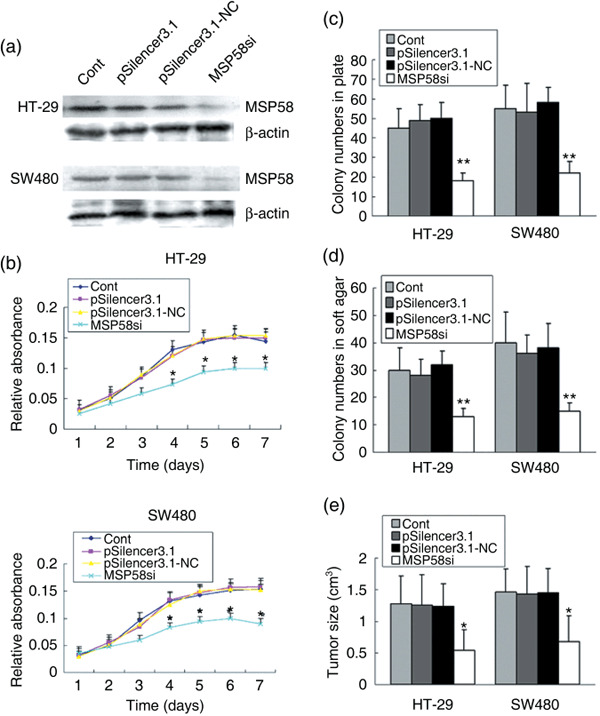
Effect of 58‐kDa microspherule protein (MSP58) siRNA on in vitro proliferation and growth and in vivo tumorigenicity of colorectal carcinoma (CRC) cells. (a) After stable transfection, the expression of MSP58 in HT‐29 and SW480 cells was evaluated by western blotting. β‐Actin was used as an internal control. (b) Monolayer growth rates of HT‐29 and SW480 cells transfected with the indicated plasmids were determined by MTT assay. Values represent the mean from at least three separate experiments. Error bars are standard error of the mean (SEM). *P < 0.05. (c) To test plate colony formation of HT‐29 and SW480 cells transfected with the indicated plasmids, cells were placed in wells with media and incubated for 15 days before counting the number of foci. Values are the mean from at least three separate experiments, each conducted in triplicate, and error bars show SEM. **P < 0.01. *P < 0.05. (d) Colony formation of HT‐29 and SW480 cells transfected with the indicated plasmids was carried out by placing cells in media containing soft agar for 17 days. Foci >100 µm were counted. Values represent the mean of at least three separate experiments, each conducted in triplicate. Error bars are SEM. **P < 0.01. *P < 0.05. (e) Effect of MSP58 siRNA on tumorigenicity of HT‐29 and SW480 cells in nude mice was calculated by measurement of excised tumors at the time of death. Tumors were verified as colon cancer by HE staining (data not shown). *P < 0.05.
To determine the effect of MSP58 on the colony‐forming ability of colon cancer cells, we carried out in vitro plate colony formation assays. Compared to control cells, fewer clones were observed in HT29‐MSP58si and SW480‐MSP58si cells (P < 0.01) (Fig. 3c). We then evaluated the effect of MSP58 suppression on anchorage‐independent colony formation in soft agar as an additional assessment of tumorigenicity in vitro. MSP58 downregulation significantly impaired anchorage‐independent colony growth, reducing both colony number and size. In contrast, no loss of colony‐forming ability was observed with either pSilencer3.1‐NC or pSilencer3.1 (P < 0.01) (Fig. 3d). Collectively, our results indicated that inhibition of MSP58 expression impaired the colony‐forming ability of HT29 and SW480 cells in vitro.
An in vivo subcutaneous tumor formation assay was used to examine the proliferative ability of HT29‐ and SW480‐MSP58si cells in nude mice. Injected HT29‐ or SW480‐MSP58si cells led to significantly smaller tumors than control cells carrying empty vector (Fig. 3e; P < 0.05). Thus, both in vitro and in vivo assays suggested that downregulation of MSP58 might inhibit proliferation, growth, and tumorigenicity of colon cancer.
Downregulation of MSP58 induces cell cycle arrest of colon cancer cells. To further investigate the mechanism by which downregulation of MSP58 inhibits colon cancer cell growth, we used FACS analysis to study the effects of MSP58 expression on the cell cycle (Table 2). The results of cell cycle analysis showed that 24 h after the release of synchronized cultures, 16.42% of HT29‐MSP58si cells were in S‐phase, compared to 25.49% of untransfected HT‐29 cells; and 15.53% of SW480‐MSP58si cells were in S‐phase compared to 27.49% of untransfected SW480 cells (P < 0.05).
Table 2.
Cell cycle analysis of 58‐kDa microspherule protein (MSP58) expression in colon cancer cells
| HT‐29 and SW480 clones expressing MSP58 protein | HT‐29 | SW480 | ||||
|---|---|---|---|---|---|---|
| G1 | G2 | S | G1 | G2 | S | |
| Cont | 64.17 | 10.34 | 25.49 | 61.43 | 11.08 | 27.49 |
| pSilencer3.1 | 62.78 | 10.73 | 26.49 | 64.51 | 10.11 | 25.38 |
| pSilencer3.1‐NC | 63.06 | 10.03 | 26.91 | 62.54 | 10.49 | 26.97 |
| MSP58si | 73.34 | 10.24 | 16.42 | 75.29 | 9.18 | 15.53 |
| Statistical significance | * | NS | * | * | NS | * |
Enumeration (%) of cell cycle analysis in HT‐29 and SW480 clones. NS, not significant. *P < 0.05.
Cell cycle initiation and progression are regulated by several classic cyclins and CDK, which associate to form hetrodimeric complexes (cyclin–CDK). The activated cyclin–CDK complexes sequentially phosphorylate the retinoblastoma protein (pRb).( 19 , 20 ) Here, we investigated these cell cycle effectors by western blotting. Our results indicated that downregulation of MSP58 protein tended to be associated with the reduction of cyclin D1, CDK4, and p‐Rb proteins, but with an increase in p21 and Rb protein levels (Fig. 4). The levels of the β‐actin loading control appeared to be similar over all samples.
Figure 4.
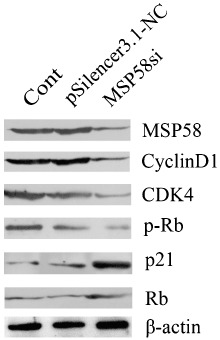
Effect of 58‐kDa microspherule protein (MSP58) siRNA on cell cycle effector levels by western blotting. Cyclin D1, cyclin‐dependent kinase (CDK) 4, phosphorylation Retino blastoma (p‐Rb), retinoblastoma (Rb), and p21 proteins were evaluated in HT‐29 cells transfected with the indicated plasmids. β‐Actin was used as an internal control.
Discussion
Although mortality from CRC can be often be prevented by surgical resection of the involved bowel before tumor cell dissemination,( 3 , 21 , 22 , 23 ) 61% of patients have metastatic disease at presentation, and 90% of those patients die within 5 years of diagnosis. During CRC carcinogenesis, genetic and epigenetic alterations accumulate, facilitating cell transformation and enhancing aggressive behavior. The current model is that the three main pathways that occur in CRC are chromosomal instability, microsatellite instability, and epigenetic silencing through the CpG island methylator phenotype.( 24 ) Nonetheless, the precise mechanisms of CRC tumorigenesis and progression have not been fully elucidated.
The nucleolar protein MSP58 was identified initially through its interaction with the proliferation‐related nucleolar protein p120.( 6 , 7 ) Recently, the transforming activity of MSP58 was found to be inhibited by the PTEN tumour suppressor,( 7 , 11 ) suggesting that MSP58 might have an important function as an oncogene. The activation of most oncogenes, like c‐Myc, Src, and K‐ras, is related to gene mutation.( 13 , 25 , 26 , 27 ) In the present study, we first investigated the expression of MSP58 in human CRC tissues and cell lines with the aim of investigating the molecular mechanisms of CRC tumorigenesis, and ultimately, to find new therapeutic targets. Our results showed that the MSP58 protein was expressed in the nucleus and cytoplasm of renal tubular epithelial cells, and MSP58 expression was significantly increased in CRC specimens compared with their adjacent non‐cancerous tissues. The results of immunohistochemistry, RT‐PCR, and western blotting suggested that CRC tissues were more likely to express MSP58 than adjacent normal tissues. We observed MSP58 expression, at both the mRNA and protein levels, in the human CRC cell lines LoVo, CoLo205, HCT116, HT‐29, SW620, and SW480. Our results suggest that MSP58 gene expression might be different in CRC and normal tissue, and MSP58 might be involved in colon carcinogenesis and development, as well as functioning as an oncogene in CRC.
To determine whether altering MSP58 levels could modulate the proliferation of colon cancer cells, we introduced pSilencer‐MSP58si into HT29 and SW480 cells. We found that downregulation of MSP58 significantly inhibited CRC cell proliferation and growth in vitro, and tumorigenicity in animal models. The suppressive effect of the pSilencer‐MSP58si on growth of HT29 and SW480 cells, and on neoplastic development, indicated that MSP58 might be an oncogene in CRC cells that acts directly or indirectly to control proliferation. Cell cycle analysis revealed that downregulation of MSP58 rapidly led to G1 arrest of HT29 and SW480 colon carcinoma cells.
Cell cycle initiation and progression are regulated by several classic cyclins and CDK, which associate to form hetrodimeric complexes (cyclin–CDK). The activated cyclin–CDK complexes sequentially phosphorylate Rb.( 19 , 20 ) Furthermore, the restriction point through the cell cycle is negatively regulated by association with CDKI, such as p21.( 28 ) Deregulation of these CDKI is a common feature in tumor cells and contributes to the disruption of cell cycle control. p21 is a CDKI that exerts its inhibitory activity on many steps of the cell cycle.( 17 ) In the present study, we found that downregulation of MSP58 was associated with changes in the protein levels of several of these cell cycle regulatory components. With the reduction of MSP58, cyclin D1, CDK4, and p‐Rb protein levels appeared to decrease, but the levels of p21 and Rb appeared to increase. From these preliminary observations, we propose a hypothesis to guide further investigations, in which downregulation of MSP58 regulates the cell cycle through decreasing expression of cyclins or CDK, increasing CDKI, or mediating CDKI redistribution. Understanding the function and role of MSP58 in CRC may establish new targets for CRC gene therapy strategies.
Abbreviations
| CDK | cyclin‐dependent kinase |
| CDKI | cyclin‐dependent kinase inhibitor |
| CRC | colorectal carcinoma |
| MSP58 | 58‐kDa microspherule protein |
| Ndrg | N‐myc downstream‐regulated gene |
| p‐RB | phosphorylation Retino Blastoma |
| PTEN | phosphatase and tensin homolog |
| Rb | Retinoblastoma |
References
- 1. Parkin DM, Bray F, Ferlay J et al . Global cancer statistics. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Flamini E, Mercatali L, Nanni O et al . Free DNA and carcinoembryonic antigen serum levels: an important combination for diagnosis of colorectal cancer. Clin Cancer Res 2006; 12: 6985–8. [DOI] [PubMed] [Google Scholar]
- 3. Jemal A, Tiwari RC, Murray T et al . American Cancer Society. Cancer statistics. CA Cancer J Clin 2004; 54: 8–29. [DOI] [PubMed] [Google Scholar]
- 4. Kaz AM, Brentnall TA. Genetic testing for colon cancer. Nat Clin Prac Gastrol Hepat 2006; 3: 670–9. [DOI] [PubMed] [Google Scholar]
- 5. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005; 434: 843–50. [DOI] [PubMed] [Google Scholar]
- 6. Ren Y, Busch RK, Perlaky L et al . The 58‐kDa microspherule protein (MSP58), a nucleolar protein, interacts with nucleolar protein p120. Eur J Biochem 2005; 253: 734–42. [DOI] [PubMed] [Google Scholar]
- 7. Davidovic L, Bechara E, Gravel M et al . The nuclear microspherule protein 58 is a novel RNA‐binding protein that interacts with fragile X mental retardation protein in polyribosomal mRNPs from neurons. Mol Genet 2006; 15: 1525–38. [DOI] [PubMed] [Google Scholar]
- 8. Lin DY, Shih HM. Essential role of the 58‐kDa microspherule protein in the modulation of Daxx‐dependent transcriptional repression as revealed by nucleolar sequestration. J Biol Chem 2002; 277: 25 446–56. [DOI] [PubMed] [Google Scholar]
- 9. Ivanova AV, Ivanov SV, Lerman ML. Association, mutual stabilization, and transcriptional activity of the STRA13 and MSP58 proteins. Cell Mol Life Sci 2005; 62: 471–84. [DOI] [PubMed] [Google Scholar]
- 10. Shimono K, Shimono Y, Shimokata K et al . Microspherule protein 1, Mi‐2beta, and RET finger protein associate in the nucleolus and up‐regulate ribosomal gene transcription. J Biol Chem 2005; 280: 39 436–47. [DOI] [PubMed] [Google Scholar]
- 11. Okumura K, Zhao M, Depinho RA et al . Cellular transformation by the MSP58 oncogene is inhibited by its physical interaction with the PTEN tumor suppressor. Proc Natl Acad Sci USA 2005; 102: 2703–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang J, Liu JY, Li X et al . The physical and functional interaction of NDRG2 with MSP58 in cells. Biochem Biophys Res Commun 2007; 352: 6–11. [DOI] [PubMed] [Google Scholar]
- 13. Ma J, Jin H, Wang H et al . Expression of NDRG2 in clear cell renal cell carcinoma. Biol Pharm Bull 2008; 31: 1316–20. [DOI] [PubMed] [Google Scholar]
- 14. Maaser K, Däubler P, Barthel B et al . Oesophageal squamous cell neoplasia in head and neck cancer patients: upregulation of COX‐2 during carcinogenesis. Br J Cancer 2003; 88: 1217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi SC, Kim KD, Kim JT et al . Expression and regulation of NDRG2 (N‐myc downstream regulated gene 2) during the differentiation of dendritic cells. FEBS Lett 2003; 553: 413–18. [DOI] [PubMed] [Google Scholar]
- 16. Lin W, Zhang J, Zhang J et al . RNAi‐mediated inhibition of MSP58 decreases tumor growth, migration and invasion in a human glioma cell line. J Cell Mol Med 2008; 15 (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin HF, Pan YL, Zhao LN et al . p75 neurotrophin receptor suppresses the proliferation of human gastric cancer cells. Neoplasia 2007; 9: 471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jin HF, Pan YL, He LJ et al . p75 neurotrophin receptor inhibits invasion and metastasis of gastric cancer. Mol Cancer Res 2007; 5: 423–33. [DOI] [PubMed] [Google Scholar]
- 19. Simmons Kovacs LA, Orlando DA, Haase SB. Transcription networks and cyclin/CDKs: the yin and yang of cell cycle oscillators. Cell Cycle 2008; 7: 2626–9. [DOI] [PubMed] [Google Scholar]
- 20. Brooks G. Cyclins, cyclin‐dependent kinases, and cyclin‐dependent kinase inhibitors: detection methods and activity measurements. Methods Mol Biol 2005; 296: 291–8. [DOI] [PubMed] [Google Scholar]
- 21. Cresanta JL. Epidemiology of cancer in the United States. Prim Care 1992; 19: 419–41. [PubMed] [Google Scholar]
- 22. Greenwald P. Colon cancer overview. Cancer 1992; 70: 1206–15. [DOI] [PubMed] [Google Scholar]
- 23. Pihl E, Hughes ES, McDermott FT et al . Disease‐free survival and recurrence after resection of colorectal carcinoma. J Surg Oncol 1981; 16: 333–41. [DOI] [PubMed] [Google Scholar]
- 24. Kjetil S, Bjørn SN, Jens‐Christian K et al . Evolving molecular classification by genomic and proteomic biomarkers in colorectal cancer: Potential implications for the surgical oncologist. Surg Oncol 2008; 6: 1–20. [DOI] [PubMed] [Google Scholar]
- 25. Wilkins JA, Sansom OJ. C‐Myc is a critical mediator of the phenotypes of Apc loss in the intestine. Cancer Res 2008; 68: 4963–6. [DOI] [PubMed] [Google Scholar]
- 26. Chen J. Is Src the key to understanding metastasis and developing new treatments for colon cancer? Nat Clin Pract Gastroenterol Hepatol 2008; 5: 306–7. [DOI] [PubMed] [Google Scholar]
- 27. Eng C. K‐Ras and sensitivity to EGFR inhibitors in metastatic colorectal cancer. Clin Adv Hematol Oncol 2008; 6: 174–5. [PubMed] [Google Scholar]
- 28. Peschiaroli A, Figliola R, Coltella L et al . MyoD induces apoptosis in the absence of RB function through a p21(WAF1)‐dependent re‐localization of cyclin/cdk complexes to the nucleus. Oncogene 2002; 21: 8114–27. [DOI] [PubMed] [Google Scholar]