

Abstract
Human papillomaviruses (HPV) are believed to be the primary causal agents for development of pre‐neoplastic and malignant lesions of the uterine cervix, and high‐risk types such as type 16 and 18 are associated with more than 90% of all cervical carcinomas. The E6 and E7 genes of HPV are thought to play causative roles, since E6 promotes the degradation of p53 through its interaction with E6AP, an E3 ubiquitin ligase, whereas E7 binds to the retinoblastoma protein (pRb) and disrupts its complex formation with E2F transcription factors. Although prophylactic vaccines have become available, it is still necessary to clarify the mechanisms of HPV‐induced carcinogenesis because of the widespread nature of HPV infection. Approximately 493 000 new cases of cervical cancer are diagnosed each year with approximately 274 000 mortalities due to invasive cervical cancer. In the present article, the mechanisms of HPV16 E6‐ and E7‐induced multistep carcinogenesis and recently identified functions of these onco‐proteins are reviewed. (Cancer Sci 2007; 98: 1505–1511)
HPV and cancer
The genome of human papillomavirus (HPV) is a circular double‐stranded DNA molecule of approximately 8000 base pairs (Fig. 1). More than 100 HPV genotypes have been described so far and approximately 40 of them infect the genital mucosa. These HPV are classified into low‐ or high‐risk types according to their presence in malignant lesions of the cervix, and high‐risk types (16, 18, 31, 33, 45, 51, 52, 58, etc.) are associated with more than 90% of cervical cancers. Of these, HPV16 accounts for approximately half of all cervical cancers while HPV18 is involved in another 10–20%,( 1 ). HPV infection has also been suggested to cause the majority of anal cancers as well as a subset of vulvar, vaginal and penile cancers.( 2 ) An association between the presence of HPV and the development of head and neck cancer has also been recently established.( 3 )
Figure 1.
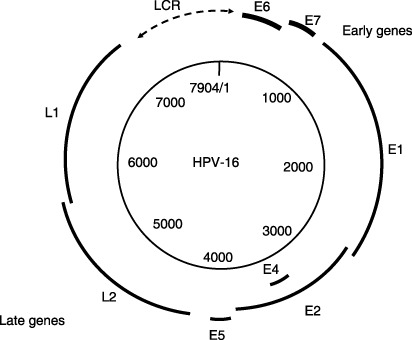
The genome of human papillomavirus (HPV) is a circular double‐stranded DNA molecule of approximately 8000 base pairs. The genetic map of HPV16 is illustrated. Open reading frames (ORF) are indicated by bold type. The six early ORF (E1, E2, E4, E5, E6 and E7 are expressed at different stages during epithelial differentiation. L1 and L2 ORF, grouped in late region, are expressed in cells replicating viral DNA in the upper epithelial cells.
The HPV viral oncogenes, E6 and E7, have been shown to be the main contributors to the development of HPV‐induced cervical cancer and increased expression, probably due to integration of the viral DNA in the host cell genome, has been detected in invasive cancers and a subset of high‐grade lesions.( 4 ) Inactivation of tumor suppressor p53 and/or retinoblastoma protein (pRb) is a common event for the carcinogenesis of human cells. Both E6 and E7 HPV oncogenes interact with and inhibit the activities of these tumor suppressors. Furthermore multifunctional properties of the two proteins have been revealed (see below).
The HPV life cycle
The HPV life cycle is tightly linked to their host cell biology (Fig. 2). Normal squamous epithelial cells grow as stratified epithelium, with those in the basal layers dividing as stem cells or transit amplifying cells. After division, one of the daughter cells migrates upward and begins to undergo terminal differentiation while the other remains in the basal layer as a slow‐cycling, self‐renewing population.( 5 ) HPV virions initially infect the basal layers of the epithelium, probably through microwounds and enter cells via interaction with certain receptors such as α‐6 integrin for HPV16.( 6 ) In infected cells at the basal layer, low levels of viral DNA are synthesized to an episomal copy number of approximately 50–100 genomes per cell. The early HPV genes E1 and E2 support viral DNA replication and its segregation so that the infected stem cells can be maintained in the lesion for a long period. As infected daughter cells migrate to the upper layers of the epithelium, viral late gene products are produced to initiate the vegetative phase of the HPV life cycle, resulting in high‐level amplification of the viral genome. As the viral DNA replication almost totally depends on host replication factors except for viral helicase E1, other early genes E5, E6 and E7 are considered to coordinate a host cell environment suitable for viral DNA replication, which sometimes induces host cellular DNA synthesis and prevents apoptosis. In the outer layers of the epithelium, viral DNA is packaged into capsids and progeny virions are released to re‐initiate infection. Because the highly immunogenic virions are synthesized at the upper layers of stratified squamous epithelia they undergo only relatively limited surveillance by cells of the immune system. In addition, E6 and E7 inactivate interferon (IFN) regulatory factor (IRF),( 7 , 8 ) so that HPV viruses can remain as persistent, asymptomatic infections.
Figure 2.
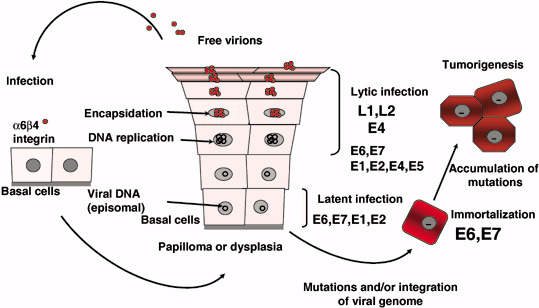
Human papillomavirus (HPV) use a unique strategy for propagation, limited to stratified flattened epithelial tissue of mucosa and skin. Initially, HPV must infect stem cells or basal cells of the tissue where a phase of latent infection is established in which viral DNA replicate without making virions. In the upper layer, as cells differentiate, vegetative replication of viral DNA coordinates with expression of capsid proteins to make virions that are then freed to search for new host cells. Expression levels of E6 and E7 in basal cells are considered to be quite low. However, as such infections can continue for years and even for decades, cells may acquire high‐level expression of E6 and E7 through mutations and integration of the viral genome. Such cells could become immortal and tumorigenic with further genetic and epigenetic events.
HPV infection and HPV‐induced transformation
Cervical cancers originate from the lining of the cervix, the lower part of the uterus. The squamocolumnar junction, where the stratified non‐keratinizing squamous epithelium from the exocervix and the columnar epithelium from the endocervix meet, is the most important cytologic and colposcopic landmark, as this is highly susceptible to HPV infection and is the site where more than 90% of lower genital tract neoplasia arises. Infection with high‐risk HPV is associated with cervical dysplasia or cervical intraepithelial neoplasia (CIN), and cervical cancers are thought to arise from these lesions after long persistent infection.( 9 , 10 ) CIN I (mild dysplasia) and CIN II (moderate dysplasia) lesions show relatively low levels of E6 and E7 expression in which the viral genomes replicate episomally, whereas CIN III (severe dysplasia, carcinoma in situ) and invasive cancer lesions often display high‐level expression of E6 and E7, in most cases with the integration of viral DNA into the host cell genome whereby neoplastic development is believed to be initiated.( 11 )
Although HPV infections are common and the life‐time risk of infection is approximately 80% for productive women, in most cases they are resolved spontaneously by an effective immune response. The ultimate development of cervical cancer is rarely accompanied by high expression of E6 and E7 proteins. Thus the authors speculate that the integration of the viral genome into the host cell is a very rare event, but after it has happened carcinogenic transformation progresses rapidly (Fig. 3). However, epidemiological studies and experimental data indicate that the viral presence is not enough to induce cervical cancer and additional genetic and epigenetic events are presumably required to alter the cellular factors.( 9 ) Amplifications of PIK3CA,( 12 ) c‐myc, ErbB2,( 13 ) and cIAP1,( 14 ) mutation of ras ( 13 ) and decreased expression of PTEN,( 15 ) and TSLC1,( 16 ) have been reported in cervical cancers.
Figure 3.
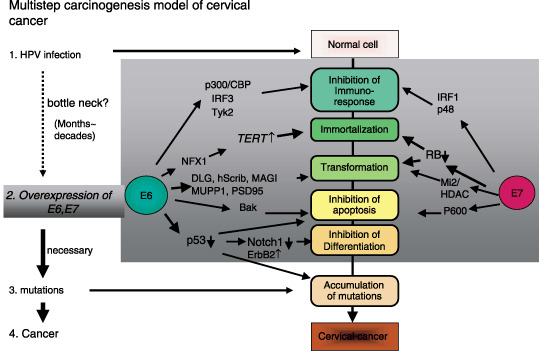
E6 and E7 cooperatively function in the development of cervical cancer. Multistep carcinogenesis for human papillomavirus (HPV)‐induced cervical cancer. The authors would like to emphasize that the bottleneck step to cancer is the overexpression of E6 and E7, which is usually achieved by accidental integration of a viral genome into a host chromosome. Once E6 and E7 genes are overexpressed, subsequent events (in the dark box) might be expected to occur within a short period of time because E6 and E7 can cooperatively induce chromosomal instability.
The functions of HPV onco‐proteins E6 and E7
Both E6 and E7 proteins are essential to induce and maintain cellular transformation, due to their interference with cell‐cycle control and apoptosis. Genomic instability is thought to be an essential part of the cellular transformation and it has been shown that E6 and E7 together cause polyploidy soon after they are introduced into cells. This appears to result from deregulation of Plk1 by the loss of p53 through E6, and pRb family members by E7, overcoming the safeguard arrest response.( 17 ) Acute loss of pRb family members by E7 has also shown to induce centrosome amplification and aneuploidy,( 18 ) In addition, E6 and E7 cause deregulation of cellular genes controlling the G2/M phase transition and progression through mitosis, such as the genes controlling centrosome homeostasis.( 19 , 20 )
The function of the E6 onco‐protein
Inactivation and degradation of p53 through the E6/E6AP complex. The most manifest function of the E6 protein is to promote the degradation of p53 through its interaction with a cellular protein, E6 associated protein (E6AP), an E3 ubiquitin ligase( 9 ) (Fig. 4). The affinity of E6AP for p53 is likely to be modified by the association with E6. The p53 tumor suppressor gene itself regulates growth arrest and apoptosis after DNA damage. When DNA damage is moderate, a prolonged p53‐dependent arrest and DNA repair are induced, but when the damage is severe, apoptosis is provoked. Although aberrant inactivation of pRb family members would also normally induce apoptosis through p53, HPV‐infected cells avoid such cell death by E6 inactivation of p53. In addition, E6 interferes with other pro‐apoptotic proteins, Bak, FADD and procaspase 8,( 21 , 22 ) (Table 1) to comprehensively prevent apoptosis. Alternatively, the susceptibility of E6‐induced degradation of p53 has been suggested to link the polymorphisms in codon 72 of p53.( 23 )
Figure 4.
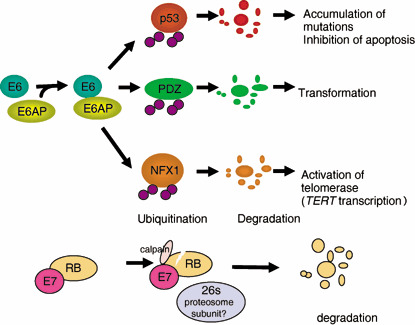
E6 and E7 proteins of the high risk human papillomaviruses (HPV) can inactivate tumor suppressors p53 and retinoblastoma protein (pRb), respectively. Although E6 protein itself cannot bind to p53, it can bind to a cellular ubiquitin ligase named E6AP, and make ternary complexes with p53 so that it becomes ubiquitinated. E6 protein also has functions independent of p53 inactivation. It is likely that ubiquitin ligase E6AP is a key player not only in the degradation of p53 but also in the activation of telomerase and cell transformation by E6. Most recently, it has been found that E7 promotes C‐terminal cleavage of pRb by the calcium‐activated cysteine protease calpain and that this cleavage is required before E7 can promote the proteasomal degradation of pRb (A. Suhrbier, personal communication, 2007). Most biological functions of E7 are achieved by inactivating pRb family proteins.
Table 1.
Target proteins of E6 and E7 and their functional relevance
| Target molecules of E6 | Implicated/observed biological effect |
|---|---|
| E6AP/p53 | Degradation of p53/suppression of apoptosis( 9 ) |
| PDZ‐domain‐containing proteins | Degradation of PDZ proteins/loss of cell polarity( 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 ) |
| CAL | Deregulation of the vesicular trafficking processes( 71 ) |
| NFX1‐91 | Degradation of NFX1‐91/activation of hTERT, immortalization( 36 ) |
| Paxillin | Interference in the association of paxillin and focal adhesion kinase( 72 ) |
| IRF3 | Inhibition of IRF‐3's transcriptional activity thereby inhibiting the IFN‐induced signaling( 7 ) |
| Bak | Degradation of Bak/suppression of apoptosis( 21 ) |
| FADD | Degradation of FADD/suppression of apoptosis( 22 ) |
| Procaspase 8 | Degradation of procaspase 8/suppression of apoptosis( 22 ) |
| GADD34/PP1 | Suppression of apoptosis( 73 ) |
| Tyk2 | Impairment of Tyk2 activation thereby inhibiting IFN‐induced signaling( 74 ) |
| CBP/p300 | Down‐regulation of p53 activity by targeting the transcriptional coactivator( 75 ) |
| MCM7 | Induction of chromosomal abnormalities( 76 ) |
| TSC2 (tubulin) | Activation of mTOR signaling( 53 ) |
| BRCA1 | Release the inhibition of ER signaling( 77 ) |
| Target molecules of E7 | Implicated/observed biological effect |
| pRb family proteins | Disruption of pRb–E2F complexes thereby initiating the E2F mediated transcription( 9 ) |
| Cyclin A | Regulation of cell cycle (binding through pRb)( 60 ) |
| Cyclin E | Regulation of cell cycle (binding through p107)( 61 ) |
| p27 | Binding to and subsequent inactivation of the CDK inhibitor p27( 62 ) |
| p21 | Binding to and subsequent inactivation of the CDK inhibitor p21( 63 ) |
| AP1 | Interaction with and transactivation of the AP1 family of transcription factors( 78 ) |
| TBP | Deregulation of the TBP mediated transcription( 79 ) |
| S4 subunit of the 26 S proteasome | Targeting of pRb for degradation( 80 ) |
| MPP2 | Activation of MPP2‐specific transcriptional activity( 81 ) |
| hTid 1 | Genome replication( 82 ) |
| p48 | Down‐regulation of IFN α‐mediated signal transduction( 64 ) |
| M2 pyruvate kinase | Modulation of type M2 pyruvate kinase activity( 83 ) |
| p600 | Contribution to anchorage‐independent growth and transformation( 65 , 66 ) |
| Mi2 | Form complex with HDAC to promote the E2F2‐mediated transcription( 59 ) |
| IRF1 | Abrogation of transactivation function of IRF1( 8 ) |
CDK, cyclin‐dependent kinase; ER, estrogen receptor; HDAC, histone deacetylases; IFN, interferon; IRF, interferon regulatory factor; pRb, retinoblastoma.
Newly identified target of p53: the Notch1 gene. Recently, the product of the Notch1 gene has been identified as a novel target of p53 (Fig. 5).( 24 , 25 ) Notch1 has been shown to function as an oncogene in the development of human T‐cell leukemia,( 26 ) but also acts as a determinant of keratinocyte differentiation,( 27 ) and a tumor suppressor in the mammalian epidermis.( 28 ) Although Notch1 expression has been found in neoplastic cervical lesions, particularly in well‐differentiated squamous cell carcinomas,( 29 ) it disappears in the late stages or poorly differentiated cervical cancer.( 30 ) Induction of Notch1 through p53 occurs in response to genotoxic stress. Therefore, its down regulation through p53 with E6/E6AP has been revealed as a novel tumor suppressor mechanism blocking development of HPV‐induced cervical carcinogenesis.( 25 )
Figure 5.
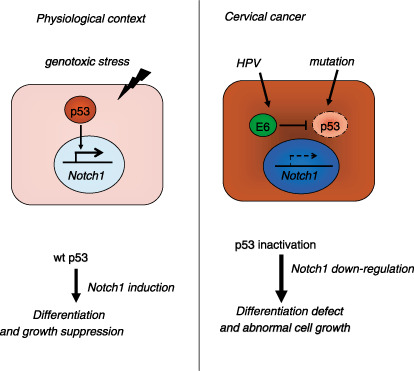
The Notch1 gene is a novel target of p53 at least in normal keratinocytes. Upon DNA damage, p53 transactivates the Notch1 gene so as to induce differentiation. This could be a barrier function against genotoxic stress. In cervical cancer, E6 can down‐regulate expression of Notch1 through inactivation of p53. As Notch1 could function as a tumor suppressor not only in skin but also in cervical keratinocytes, its down‐regulation by E6 might contribute to the development of cervical cancer. In other squamous cell carcinomas, mutation of p53 may have the same consequence. Modified from the authors’ latest publication.( 25 )
The ErbB2 protein expression level is also regulated by p53 degradation and interference with this by E6/E6AP complexes contributes to cervical carcinogenesis.( 31 )
E6‐mediated hTERT induction. Over the past dozen years or so, an increasing number of other proteins have also been revealed to be target proteins of E6 that might contribute to cellular transformation (Table 1), with telomerase as one probable important example. Human telomerase is a ribonucleoprotein complex composed of at least the reverse catalytic transcriptase (hTERT) and an RNA component (hTR). hTERT is expressed only in specific germ‐line cells, proliferative stem cells of renewal tissues, and cancer cells. The expression of hTERT in normal cells reconstitutes telomerase activity and suppresses senescence. Because telomerase activity is hardly detected in most somatic tissues, telomeres shorten with each cell division, eventually leading to senescence (aging), due to incomplete lagging DNA strand synthesis and end‐processing events. High telomerase activity is observed in more than 85% of human cancer cells, strongly indicating a key role in tumorigenesis.( 32 ) E6 induces telomerase activity, thereby contributing to the immortalization of epithelial cells by maintaining telomere length.( 33 ) E6 and Myc interaction has been shown to activate the telomerase reverse transcriptase promoter,( 34 ) and in the presence of E6, a repressor complex of TERT promoter, containing USF1 and USF2, is replaced by Myc, which corresponds to higher levels of TERT transcription and consequently, telomerase activity.( 35 ) NFX1‐91 is a recently identified novel cellular repressor of the hTERT promoter that is degraded in a E6/E6AP dependent manner so that myc binding to the hTERT promoter can occur and result in increased hTERT expression( 36 ) (Fig. 4). In contrast, NFX1‐123, a splice variant of NFX1, together with cytoplasmic poly(A) binding proteins, is suggested to be critical to hTERT activity in HPV16 E6‐expressing epithelial cells.( 37 )
Targeting of PDZ‐containing proteins by E6. High‐risk HPV E6 has been shown to interact with PDZ‐domain‐containing proteins through its C‐terminal motif,( 38 ) leading to their degradation (Fig. 4). This ability of E6, which is distinct from that to binding and degrading p53, is important for cell transformation because PDZ‐domain‐containing proteins are involved in a variety of cellular functions such as cell signaling and cell adhesion. E6 binding to these proteins appears to be particularly important for transformation and tumorigenesis in cultured cells,( 39 ) and hyperplasia and carcinogenesis in E6‐transgenic mice.( 40 ) Recently, several PDZ‐domain‐containing proteins have been identified to be targets of E6 proteins, including mammalian homologs of DLG (DLG1/hDLG) and Scribble (Scrib/Vartul), MUPP1, MAGI‐1, ‐2, and ‐3, GIPC, PATJ, PTPN3 and PSD95.( 38 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 ) E6‐induced degradation of these proteins potentially causes loss of cell–cell contacts mediated by tight junctions and thus contributes to the loss of cell polarity seen in HPV‐associated cervical cancers.( 45 ) The tumor suppressor properties of some of these proteins against the development of HPV‐associated cancers have been reported( 49 , 50 , 51 ) (Fig. 3). Meanwhile, interactions between β1‐adrenergic receptor and Class I PDZ‐domain‐containing proteins have been uncovered, indicating the regulation of the receptor signaling and trafficking by PDZ proteins.( 52 ) Thus E6 might affect the physiological status of G protein‐coupled receptors such as β1‐adrenergic receptor through the regulation of PDZ‐containing‐proteins.
Others. The tumor suppressor gene TSC2 product, Tuberin, has been proposed as a possible target of E6, implying a contribution to E6‐induced oncogenesis.( 53 ) Because TSC complexes play an important regulatory role in the survival signaling involving mTOR, further exploration of this possibility is needed. More recently, induction by E6 of E2F‐responsive genes, MCM7 and cyclin E, has been reported, implying the existence of dysregulation of the p16/pRb pathway with mechanisms distinct from those involving E7.( 54 )
The function of the E7 onco‐protein
Inactivation of pRb. E7 is a small nuclear phosphoprotein separated into three conserved regions denoted in an analogous fashion to adenovirus E1A as CR1, CR2 and CR3.( 55 ) E7 is known to bind to the retinoblastoma tumor suppressor gene product, pRb, and its family members, p107 and p130, via a LXCXE (where X represents any amino acid) binding motif conserved in its CR2 region. In the hypophosphorylated state, pRb family proteins can bind to transcription factors such as E2F family members and repress the transcription of particular genes involved in DNA synthesis and cell‐cycle progression.( 56 ) Phosphorylation of pRb by G1 cyclin‐dependent kinases releases E2F leading to cell cycle progression into the S phase. Because E7 is able to bind to unphosphorylated pRb, it may prematurely induce cells to enter the S phase by disrupting pRb–E2F complexes. Most recently, it was found that E7 promotes C‐terminal cleavage of pRb by the calcium‐activated cysteine protease calpain and that this cleavage is required before E7 can promote the proteasomal degradation of pRb (A. Suhrbier, personal communication, 2007; Fig. 4). The E7 protein function enables HPV replication in the upper layers of the epithelium where uninfected daughter cells normally differentiate and completely exit the cell cycle (Fig. 2). One cyclin‐dependent kinase inhibitor, p16INK4a, which prevents the phosphorylation of pRb family members, is overexpressed when pRb is inactivated by HPV E7.( 57 ) Normally, overexpression of p16INK4a results in cell cycle arrest but with E7 expression, this is overcome. Thus overexpression of p16INK4a is suggested to be a useful bio‐marker for evaluating HPV pathogenic activity in cervical lesions.
Others. In addition to the inactivation of pRb family members, numerous functions of E7 have been reported (Table 1). The histone deacetylases (HDAC), the transcriptional co‐repressors, have been reported to associate with E7 via Mi2 to promote cell growth,( 58 , 59 ) The interaction of E7 with cyclin‐dependent kinase (CDK)2/cyclin A,( 60 ) as well as CDK2/cyclin E,( 61 ) has also been reported. These cyclin–kinase complexes exhibit kinase activities that can phosphorylate the pRb proteins. Furthermore, E7 binds the CDK‐inhibitor (CKI), p27,( 62 ) and CKI p21,( 63 ) confirming the abrogation of cell‐cycle inhibition. For evasion of the immune system, E7 also interacts with p48,( 64 ) as well as IRF1.( 8 ) Recently, p600 has been identified as a cellular target of E7 that contributes to anchorage‐independent growth and cellular transformation.( 65 ) Because the knockdown of p600 sensitizes cells to apoptosis induced by cell detachment irrespective of the presence of E7,( 66 ) it is suggested as a novel target of cancer treatment. Binding of E7 with both the 35‐kDa catalytic and 65‐kDa structural subunits of PP2A has been reported, resulting in the sequestration of these subunits and inhibition of their interaction with PKB/Akt, thereby maintaining its signaling by blocking its dephosphorylation.( 67 ) Despite these multifunctional properties of E7 protein, however, the expression of a mutant form of pRb (DeltaLXCXE) that is selectively defective for binding E7 revealed that most of the effects of E7 on epidermal differentiation are indeed due to pRb inactivation.( 68 )
Conclusions
HPV onco‐proteins E6 and E7 are essential factors for HPV‐induced cellular immortalization, transformation and carcinogenesis (Fig. 3). RNA interference with E6 and E7,( 69 ) as well as their functions in inhibiting the actions of various molecules,( 70 ) is a promising approach for the treatment of cervical cancers. Because prophylactic vaccination is still in its early stages, concentration of attention on such therapeutic measures is still warranted.
References
- 1. Munoz N, Bosch FX, De Sanjose S et al . Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 2003; 348: 518–27. [DOI] [PubMed] [Google Scholar]
- 2. Steenbergen RD, De Wilde J, Wilting SM, Brink AA, Snijders PJ, Meijer CJ. HPV‐mediated transformation of the anogenital tract. J Clin Virol 2005; 32 (Suppl 1): S25–33. [DOI] [PubMed] [Google Scholar]
- 3. Nair S, Pillai MR. Human papillomavirus and disease mechanisms: relevance to oral and cervical cancers. Oral Dis 2005; 11: 350–9. [DOI] [PubMed] [Google Scholar]
- 4. Klaes R, Woerner SM, Ridder R et al . Detection of high‐risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res 1999; 59: 6132–6. [PubMed] [Google Scholar]
- 5. Watt FM. Epidermal stem cells: markers, patterning and the control of stem cell fate. Philos Trans R Soc Lond B Biol Sci 1998; 353: 831–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoon CS, Kim KD, Park SN, Cheong SW. alpha(6) Integrin is the main receptor of human papillomavirus type 16 VLP. Biochem Biophys Res Commun 2001; 283: 668–73. [DOI] [PubMed] [Google Scholar]
- 7. Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16, E6 oncoprotein binds to interferon regulatory factor‐3 and inhibits its transcriptional activity. Genes Dev 1998; 12: 2061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Um SJ, Rhyu JW, Kim EJ, Jeon KC, Hwang ES, Park JS. Abrogation of IRF‐1 response by high‐risk HPV E7 protein in vivo . Cancer Lett 2002; 179: 205–12. [DOI] [PubMed] [Google Scholar]
- 9. Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2002; 2: 342–50. [DOI] [PubMed] [Google Scholar]
- 10. Munger K, Baldwin A, Edwards KM et al . Mechanisms of human papillomavirus‐induced oncogenesis. J Virol 2004; 78: 11 451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Von Knebel Doeberitz M. New markers for cervical dysplasia to visualise the genomic chaos created by aberrant oncogenic papillomavirus infections. Eur J Cancer 2002; 38: 2229–42. [DOI] [PubMed] [Google Scholar]
- 12. Bertelsen BI, Steine SJ, Sandvei R, Molven A, Laerum OD. Molecular analysis of the PI3K‐AKT pathway in uterine cervical neoplasia: frequent PIK3CA amplification and AKT phosphorylation. Int J Cancer 2006; 118: 1877–83. [DOI] [PubMed] [Google Scholar]
- 13. Zhang A, Maner S, Betz R et al . Genetic alterations in cervical carcinomas: frequent low‐level amplifications of oncogenes are associated with human papillomavirus infection. Int J Cancer 2002; 101: 427–33. [DOI] [PubMed] [Google Scholar]
- 14. Imoto I, Tsuda H, Hirasawa A et al . Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res 2002; 62: 4860–6. [PubMed] [Google Scholar]
- 15. Minaguchi T, Yoshikawa H, Nakagawa S et al . Association of PTEN mutation with HPV‐negative adenocarcinoma of the uterine cervix. Cancer Lett 2004; 210: 57–62. [DOI] [PubMed] [Google Scholar]
- 16. Steenbergen RD, Kramer D, Braakhuis BJ et al . TSLC1 gene silencing in cervical cancer cell lines and cervical neoplasia. J Natl Cancer Inst 2004; 96: 294–305. [DOI] [PubMed] [Google Scholar]
- 17. Incassati A, Patel D, McCance DJ. Induction of tetraploidy through loss of p53 and upregulation of Plk1 by human papillomavirus type‐16, E6. Oncogene 2006; 25: 2444–51. [DOI] [PubMed] [Google Scholar]
- 18. Iovino F, Lentini L, Amato A, Di Leonardo A. RB acute loss induces centrosome amplification and aneuploidy in murine primary fibroblasts. Mol Cancer 2006; 5: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walboomers JM, Jacobs MV, Manos MM et al . Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189: 12–19. [DOI] [PubMed] [Google Scholar]
- 20. Patel D, Incassati A, Wang N, McCance DJ. Human papillomavirus type 16, E6 and E7 cause polyploidy in human keratinocytes and up‐regulation of G2‐M‐phase proteins. Cancer Res 2004, 64: 1299–306. [DOI] [PubMed] [Google Scholar]
- 21. Thomas M, Banks L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J General Virol 1999; 80: 1513–17. [DOI] [PubMed] [Google Scholar]
- 22. Garnett TO, Filippova M, Duerksen‐Hughes PJ. Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16, E6 impairs TRAIL‐mediated apoptosis. Cell Death Differ 2006; 13: 1915–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Storey A, Thomas M, Kalita A et al . Role of a p53 polymorphism in the development of human papillomavirus‐associated cancer. Nature 1998; 393: 229–34. [DOI] [PubMed] [Google Scholar]
- 24. Lefort K, Mandinova A, Ostano P et al . Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCK{alpha} kinases. Genes Dev 2007; 21: 562–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yugawa T, Handa K, Narisawa‐Saito M, Ohno SI, Fujita M, Kiyono T. Regulation of Notch1 gene expression by p53 in epithelial cells. Mol Cell Biol 2007; 27: 3732–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ellisen LW, Bird J, West DC et al . TAN‐1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991; 66: 649–61. [DOI] [PubMed] [Google Scholar]
- 27. Rangarajan A, Talora C, Okuyama R et al . Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. Embo J 2001; 20: 3427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nicolas M, Wolfer A, Raj K et al . Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 2003; 33: 416–21. [DOI] [PubMed] [Google Scholar]
- 29. Zagouras P, Stifani S, Blaumueller CM, Carcangiu ML, Artavanis‐Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc Natl Acad Sci USA 1995; 92: 6414–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Talora C, Sgroi DC, Crum CP, Dotto GP. Specific down‐modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV‐E6/E7 expression and late steps of malignant transformation. Genes Dev 2002; 16: 2252–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Narisawa‐Saito M, Handa K, Yugawa T, Ohno S, Fujita M, Kiyono T. HPV16 E6‐mediated stabilization of ErbB2 in neoplastic transformation of human cervical keratinocytes. Oncogene 2007; 26: 2988–96. [DOI] [PubMed] [Google Scholar]
- 32. Pendino F, Tarkanyi I, Dudognon C et al . Telomeres and telomerase: Pharmacological targets for new anticancer strategies? Curr Cancer Drug Targets 2006; 6: 147–80. [DOI] [PubMed] [Google Scholar]
- 33. Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996; 380: 79–82. [DOI] [PubMed] [Google Scholar]
- 34. Veldman T, Liu X, Yuan H, Schlegel R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc Natl Acad Sci USA 2003; 100: 8211–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McMurray HR, McCance DJ. Human papillomavirus type 16, E6 activates TERT gene transcription through induction of c‐Myc and release of USF‐mediated repression. J Virol 2003; 77: 9852–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gewin L, Myers H, Kiyono T, Galloway DA. Identification of a novel telomerase repressor that interacts with the human papillomavirus type‐16, E6/E6‐AP complex. Genes Dev 2004; 18: 2269–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Katzenellenbogen RA, Egelkrout EM, Vliet‐Gregg P, Gewin LC, Gafken PR, Galloway DA. NFX1‐123 and Poly(A) binding proteins synergistically augment activation of telomerase in human papillomavirus type 16E6 expressing cells. J Virol 2007; 81: 3786–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M. Binding of high‐risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci USA 1997; 94: 11 612–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Watson RA, Thomas M, Banks L, Roberts S. Activity of the human papillomavirus E6 PDZ‐binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J Cell Sci 2003; 116: 4925–34. [DOI] [PubMed] [Google Scholar]
- 40. Nguyen ML, Nguyen MM, Lee D, Griep AE, Lambert PF. The PDZ ligand domain of the human papillomavirus type 16, E6 protein is required for E6's induction of epithelial hyperplasia in vivo . J Virol 2003; 77: 6957–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Favre‐Bonvin A, Reynaud C, Kretz‐Remy C, Jalinot P. Human papillomavirus type 18, E6 protein binds the cellular PDZ protein TIP‐2/GIPC, which is involved in transforming growth factor beta signaling and triggers its degradation by the proteasome. J Virol 2005; 79: 4229–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Glaunsinger BA, Lee SS, Thomas M, Banks L, Javier R. Interactions of the PDZ‐protein MAGI‐1 with adenovirus E4‐ORF1 and high‐risk papillomavirus E6 oncoproteins. Oncogene 2000; 19: 5270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee SS, Glaunsinger B, Mantovani F, Banks L, Javier RT. Multi‐PDZ domain protein MUPP1 is a cellular target for both adenovirus E4‐ORF1 and high‐risk papillomavirus type 18, E6 oncoproteins. J Virol 2000; 74: 9680–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee SS, Weiss RS, Javier RT. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci USA 1997; 94: 6670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakagawa S, Huibregtse JM. Human scribble (Vartul) is targeted for ubiquitin‐mediated degradation by the high‐risk papillomavirus E6 proteins and the E6AP ubiquitin‐protein ligase. Mol Cell Biol 2000; 20: 8244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Storrs CH, Silverstein SJ. PATJ, a tight junction‐associated PDZ protein is a novel degradation target of high‐risk HPV E6 and the alternatively spliced isoform 18, E6. J Virol 2007; 81: 4080–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jing M, Bohl J, Brimer N, Kinter M, Vande Pol SB. Degradation of tyrosine phosphatase PTPN3 (PTPH1) by association with oncogenic human papillomavirus E6 proteins. J Virol 2007; 81: 2231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Handa K, Yugawa T, Narisawa‐Saito M, Ohno S, Fujita M, Kiyono T. E6AP‐dependent degradation of DLG4/PSD95 by high‐risk human papillomavirus type 18, E6 protein. J Virol 2007; 81: 1379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ishidate T, Matsumine A, Toyoshima K, Akiyama T. The APC‐hDLG complex negatively regulates cell cycle progression from the G0/G1 to S phase. Oncogene 2000; 19: 365–72. [DOI] [PubMed] [Google Scholar]
- 50. Massimi P, Gammoh N, Thomas M, Banks L. HPV E6 specifically targets different cellular pools of its PDZ domain‐containing tumour suppressor substrates for proteasome‐mediated degradation. Oncogene 2004; 23: 8033–9. [DOI] [PubMed] [Google Scholar]
- 51. Gabius S, Joshi SS, Gabius HJ, Sharp JG. Establishment, characterization and determination of cell surface sugar receptor (lectin) expression by neoglycoenzymes of a human myeloid marker‐expressing B lymphoblastoid cell line. Anticancer Res 1991; 11: 793–800. [PubMed] [Google Scholar]
- 52. He J, Bellini M, Inuzuka H et al . Proteomic analysis of beta1‐adrenergic receptor interactions with PDZ scaffold proteins. J Biol Chem 2006; 281: 2820–7. [DOI] [PubMed] [Google Scholar]
- 53. Lu Z, Hu X, Li Y et al . Human papillomavirus 16, E6 oncoprotein interferences with insulin signaling pathway by binding to tuberin. J Biol Chem 2004; 279: 35 664–70. [DOI] [PubMed] [Google Scholar]
- 54. Shai A, Brake T, Somoza C, Lambert PF. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res 2007; 67: 1626–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu X, Clements A, Zhao K, Marmorstein R. Structure of the human Papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J Biol Chem 2006; 281: 578–86. [DOI] [PubMed] [Google Scholar]
- 56. Dyson N. The regulation of E2F by pRB‐family proteins. Genes Dev 1998; 12: 2245–62. [DOI] [PubMed] [Google Scholar]
- 57. Ishikawa M, Fujii T, Saito M et al . Overexpression of p16 INK4a as an indicator for human papillomavirus oncogenic activity in cervical squamous neoplasia. Int J Gynecol Cancer 2006; 16: 347–53. [DOI] [PubMed] [Google Scholar]
- 58. Longworth MS, Laimins LA. The binding of histone deacetylases and the integrity of zinc finger‐like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol 2004; 78: 3533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Brehm A, Nielsen SJ, Miska EA et al . The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. Embo J 1999; 18: 2449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arroyo M, Bagchi S, Raychaudhuri P. Association of the human papillomavirus type 16 E7 protein with the S‐phase‐specific E2F‐cyclin A complex. Mol Cell Biol 1993; 13: 6537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McIntyre MC, Ruesch MN, Laimins LA. Human papillomavirus E7 oncoproteins bind a single form of cyclin E in a complex with cdk2 and p107. Virology 1996; 215: 73–82. [DOI] [PubMed] [Google Scholar]
- 62. Zerfass‐Thome K, Zwerschke W, Mannhardt B, Tindle R, Botz JW, Jansen‐Durr P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16, E7 oncoprotein. Oncogene 1996; 13: 2323–30. [PubMed] [Google Scholar]
- 63. Funk JO, Waga S, Harry JB, Espling E, Stillman B, Galloway DA. Inhibition of CDK activity and PCNA‐dependent DNA replication by p21 is blocked by interaction with the HPV‐16 E7 oncoprotein. Genes Dev 1997; 11: 2090–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Barnard P, McMillan NA. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon‐alpha. Virology 1999; 259: 305–13. [DOI] [PubMed] [Google Scholar]
- 65. Huh KW, DeMasi J, Ogawa H, Nakatani Y, Howley PM, Munger K. Association of the human papillomavirus type 16 E7 oncoprotein with the 600‐kDa retinoblastoma protein‐associated factor, p600. Proc Natl Acad Sci USa 2005; 102: 11 492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nakatani Y, Konishi H, Vassilev A et al . p600, a unique protein required for membrane morphogenesis and cell survival. Proc Natl Acad Sci U S A 2005; 102: 15 093–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pim D, Massimi P, Dilworth SM, Banks L. Activation of the protein kinase B pathway by the HPV‐16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005; 24: 7830–8. [DOI] [PubMed] [Google Scholar]
- 68. Balsitis S, Dick F, Lee D et al . Examination of the pRb‐dependent and pRb‐independent functions of E7 in vivo . J Virol 2005; 79: 11 392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gu W, Putral L, Hengst K et al . Inhibition of cervical cancer cell growth in vitro and in vivo with lentiviral‐vector delivered short hairpin RNA targeting human papillomavirus E6 and E7 oncogenes. Cancer Gene Ther 2006; 13: 1023–32. [DOI] [PubMed] [Google Scholar]
- 70. Griffin H, Elston R, Jackson D et al . Inhibition of papillomavirus protein function in cervical cancer cells by intrabody targeting. J Mol Biol 2006; 355: 360–78. [DOI] [PubMed] [Google Scholar]
- 71. Jeong KW, Kim HZ, Kim S, Kim YS, Choe J. Human papillomavirus type 16, E6 protein interacts with cystic fibrosis transmembrane regulator‐associated ligand and promotes E6‐associated protein‐mediated ubiquitination and proteasomal degradation. Oncogene 2007; 26: 487–99. [DOI] [PubMed] [Google Scholar]
- 72. Mattiussi S, Matsumoto K, Illi B, Martelli F, Capogrossi MC, Gaetano C. Papilloma protein E6 abrogates shear stress‐dependent survival in human endothelial cells. evidence for specialized functions of paxillin. Cardiovasc Res 2006; 70: 578–88. [DOI] [PubMed] [Google Scholar]
- 73. Kazemi S, Papadopoulou S, Li S et al . Control of alpha subunit of eukaryotic translation initiation factor 2 (eIF2 alpha) phosphorylation by the human papillomavirus type 18, E6 oncoprotein: implications for eIF2 alpha‐dependent gene expression and cell death. Mol Cell Biol 2004; 24: 3415–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li S, Labrecque S, Gauzzi MC et al . The human papilloma virus (HPV)‐18, E6 oncoprotein physically associates with Tyk2 and impairs Jak‐STAT activation by interferon‐alpha. Oncogene 1999; 18: 5727–37. [DOI] [PubMed] [Google Scholar]
- 75. Zimmermann H, Degenkolbe R, Bernard HU, O’Connor MJ. The human papillomavirus type 16, E6 oncoprotein can down‐regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J Virol 1999; 73: 6209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kukimoto I, Aihara S, Yoshiike K, Kanda T. Human papillomavirus oncoprotein E6 binds to the C‐terminal region of human minichromosome maintenance 7 protein. Biochem Biophys Res Commun 1998; 249: 258–62. [DOI] [PubMed] [Google Scholar]
- 77. Zhang Y, Fan S, Meng Q et al . BRCA1 interaction with human papillomavirus oncoproteins. J Biol Chem 2005; 280: 33 165–77. [DOI] [PubMed] [Google Scholar]
- 78. Antinore MJ, Birrer MJ, Patel D, Nader L, McCance DJ. The human papillomavirus type 16, E7 gene product interacts with and trans‐activates the AP1 family of transcription factors. Embo J 1996; 15: 1950–60. [PMC free article] [PubMed] [Google Scholar]
- 79. Phillips AC, Vousden KH. Analysis of the interaction between human papillomavirus type 16, E7 and the TATA‐binding protein, TBP. J General Virol 1997; 78 (4): 905–9. [DOI] [PubMed] [Google Scholar]
- 80. Berezutskaya E, Bagchi S. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26 S proteasome. J Biol Chem 1997; 272: 30 135–40. [DOI] [PubMed] [Google Scholar]
- 81. Luscher‐Firzlaff JM, Westendorf JM, Zwicker J et al . Interaction of the fork head domain transcription factor MPP2 with the human papilloma virus 16, E7 protein: enhancement of transformation and transactivation. Oncogene 1999; 18: 5620–30. [DOI] [PubMed] [Google Scholar]
- 82. Schilling B, De‐Medina T, Syken J, Vidal M, Munger K. A novel human DnaJ protein, hTid‐1, a homolog of the Drosophila tumor suppressor protein Tid56, can interact with the human papillomavirus type 16, E7 oncoprotein. Virology 1998; 247: 74–85. [DOI] [PubMed] [Google Scholar]
- 83. Zwerschke W, Mazurek S, Massimi P, Banks L, Eigenbrodt E, Jansen‐Durr P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16, E7 oncoprotein. Proc Natl Acad Sci USA 1999; 96: 1291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]