

Abstract
We have recently synthesized a new platinum derivative, poly (γ, L‐glutamic acid)‐cisplatin conjugate (γ‐PGA‐CDDP), and shown that it displayed remarkable antitumor activity against breast tumor in a mouse model. The purpose of this study is to systematically compare this new drug with three platinum derivatives currently used in the clinic: cisplatin, carboplatin and oxaliplatin. Here, we show that γ‐PGA‐CDDP displays impressive antitumor activity over the current clinically used platinum drugs. More interestingly and more importantly, γ‐PGA‐CDDP conjugate significantly reduces cytotoxicity, mitigates oxidative stress and improves antioxidative capability in vivo. Animals treated with γ‐PGA‐CDDP display the same profile of body weight as the control animals, while the tumors in γ‐PGA‐CDDP‐treated animals are significantly suppressed compared with those treated with carboplatin and oxaliplatin. Our data suggest that γ‐PGA could be used as an effective carrier for drug delivery and that γ‐PGA‐CDDP conjugate may have potential therapeutic applications in human cancers that are sensitive to treatment with CDDP‐based chemotherapy such as ovarian cancer. (Cancer Sci 2010; 101: 2476–2482)
The cis‐dichlorodiammineplatinum (II) (CDDP or cisplatin) is one of the most widely used and most effective cytotoxic agents in the clinical treatment of many different malignancies. It can be used alone or in combination with other antitumor drugs to treat a wide spectrum of human tumors including breast, liver, lung, head and neck, ovarian, testicular, bladder, small‐cell and non‐small‐cell lung cancers.( 1 ) However, its therapeutic efficacy is restricted by highly toxic side effects, such as nausea, ear damage, vomiting and especially the persistence of severe nephrotoxicity.( 2 ) The severe side effects of cisplatin treatment have stimulated research toward developing less toxic CDDP analogue with enhanced antitumor activity. The modified platinum derivative carboplatin is more easily used in combination therapy and in ovarian cancer treatment.( 3 , 4 ) However, hematological adverse effects are more frequent with carboplatin than with cisplatin.( 5 ) In addition, carboplatin still needs to be administrated intravenously.( 6 ) Oxaliplatin is the third generation of platinum drugs and shows a wide antitumor effect both in vitro and in vivo,( 7 ) and is currently used in clinics as a first line treatment for metastatic colorectal cancer. The neuropathic( 8 , 9 ) and nephrotoxic( 10 ) side effects of oxaliplatin are milder than cisplatin and carboplatin. However, oxaliplatin may cause severe allergic reactions. These allergic reactions may happen within a few minutes and may even cause death.
So far, all of the improvements on cisplatin are reformed essentially by chemical methods and these platinum derivatives still exhibit severe side effects. An alternative and complementary method to increase efficiency is to conjugate cisplatin with macromolecular carriers such as water soluble polymers( 11 ) and nanoparticles.( 12 ) This strategy is based on observations that the macromolecular carriers could prolong blood circulation of the therapeutic agent. The conjugate can reduce nonspecific accumulation of the therapeutic agent in normal tissues and enhance preferentially the tumor accumulation.( 13 ) We have conjugated CDDP to biosynthesized poly (γ, L‐glutamic acid) (γ‐PGA) to develop more efficient platinum drug with less side effects.( 14 )γ‐PGA is water soluble, biodegradable and a nontoxic biopolymer; its free α‐carboxyl group in each repeating unit provides functionality for drug attachment. We have shown that the water soluble conjugate, poly (γ, L‐glutamic acid)‐cisplatin effectively inhibits tumor proliferation in vivo.( 14 )
In order to assess its potential use in clinics, we evaluated systematically the antitumor activity and toxicity of γ‐PGA‐CDDP conjugate with three clinically used platinum derivatives: cisplatin, carboplatin and oxaliplatin under the same experimental conditions. Our data in this study demonstrate that γ‐PGA‐CDDP conjugate displays potent antitumor activity and significantly reduces cytotoxicity, mitigates oxidative stress and improves antioxidative capability in vivo. Our research suggests that γ‐PGA produced by microbial fermentation may be used as an effective carrier for drug delivery and that γ‐PGA‐CDDP conjugate may have potential applications in clinical treatment.
Materials and Methods
Preparation of γ‐PGA‐CDDP conjugate. γ‐PGA‐CDDP conjugate was prepared according to our previous methods with some modifications (Data S1, and Fig. S1).( 14 , 15 )
In vivo toxicity of γ‐PGA‐CDDP conjugate. Forty female KM mice (5–7 weeks old, 20–22 g) were randomly divided into five groups of eight mice each. Animals were kept in a room at a constant temperature and had free access to diet and tap water. The mice were treated intraperitoneally (i.p.) every 2 days for three times with cisplatin, carboplatin and oxaliplatin at doses of 4 mg/kg; the dose of γ‐PGA‐CDDP conjugate was equivalent to free CDDP.( 14 ) The body weight change in each group was continuously measured and recorded. When animals were killed, serum samples were treated with white blood cell count solution and platelet count solution, and the number of white blood cells and blood platelets were recorded under a microscope. Kidney tissues were also removed immediately and stored at −70°C until analysis. As indicators of kidney function, serum creatinine (CRE) and blood urea nitrogen (BUN) were measured pectrophotometrically using an autoanalyzer (HITACHI‐BM704, Japan); the tissues were homogenized in nine volumes of ice‐cold 0.9% sodium chloride and centrifuged at 1000g for 10 min at 4°C. Supernatant was collected to determine the antioxidative level in vivo. Myeloperoxidase (MPO), malondialdehyde (MDA), glutathione (GSH) and glutathione peroxidase (GSH‐Px) were tested using a spectrophotometric method outlined in the kit.
Cell proliferation assay. The capacity of the different platinum derivatives to inhibit cell growth was determined by the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐bromide (MTT) test. This method was based on MTT‐reduction into formazan by viable cells. Essentially, the tumor cell lines (Bcap‐37, BEL‐7404 and SH‐SY5Y) were plated (1 × l04 cells in 0.1 mL per well) in DMEM culture medium containing 10% bovine calf serum in a 96‐well plate. Following overnight incubation at 37°C in a 95% humidified atmosphere of 5% CO2, the growth medium was replaced with a fresh one containing cisplatin, carboplatin, oxaliplatin and γ‐PGA‐CDDP conjugate in CDDP equivalent concentrations ranging from 20 to 200 μM/L,( 14 ) and the cells were then treated for 24, 48 and 72 h of continuous exposure. At the end of incubation, 20 μL MTT (5 mg/mL) was added to each well and cells were incubated for an additional 4 h in a CO2 incubator at 37°C. The viable cells were quantified. Inhibition of cell viability was calculated in reference to cells incubated with culture medium alone.
Flow cytometric assay for measuring cell cycle and apop‐tosis. Human breast tumor cell lines (Bcap‐37) were seeded in six‐well plates (1 × 104 cells in 1 mL per well) and grown in DMEM culture medium containing 10% bovine calf serum at 37°C for 24 h in a 95% humidified atmosphere of 5% CO2. The growth medium was then replaced with a fresh one containing cisplatin, carboplatin, oxaliplatin and γ‐PGA‐CDDP conjugate in CDDP equivalent concentrations of 100 μM/L; optimal experimental conditions were previously determined by Ye et al. ( 14 ) In the end, harvest cells were collected and washed with D‐Hanks for 2–3 times. After centrifuging at 650g for 5 min and discarding the supernatant, the cell pellets were immediately suspended in 5 mL of pre‐cold 70% ethyl alcohol (ETOH), mixed quickly and kept at 4°C for 1 h in order to fix the cells. After the fixation, the cells were transferred to FACS tubes for flow cytometric sorting.
Release profiles of γ‐PGA‐CDDP conjugate and different plati‐num derivatives. The release profile of the γ‐PGA‐CDDP conjugate was evaluated by cellulose membrane with a cut off molecular weight at 3.5 kDa according to Ye et al. ( 14 , 16 , 17 )
Statistical analysis. Statistical analysis for the determination of differences in the measured properties between groups was done using a two‐sided log‐rank and Dunnett tests. A P value of <0.05 was considered statistically significant. All data are presented as the mean value with its standard deviation indicated (mean ± SD).
Results
Preparation and characterization of γ‐PGA‐CDDP conjugate. The preparation of γ‐PGA‐CDDP conjugate was achieved by a nucleophilic attack on one or two carboxyl groups of glutamate by the platinate derivative. Theoretically, three complex structures could be formed by this reaction (Fig. 1a). After purification by dialysis, the purified conjugate was analyzed by proton nuclear magnetic resonance (1H NMR) (Fig. 2). The results were further confirmed by carbon nuclear magnetic resonance (13C NMR) and Fourier transformed infrared (FT‐IR) analysis (Data S2 and Fig. S3).
Figure 1.
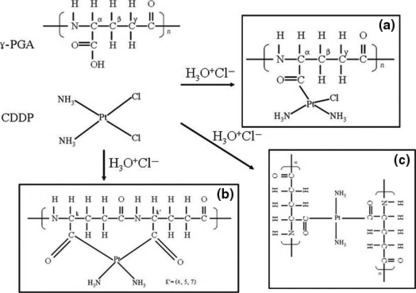
Structures of γ‐PGA, CDDP and three possible γ‐PGA‐CDDP conjugates: (a) monoadducts with one carboxyl group; (b) intrastrand coordination with carboxyl groups; and (c) interstrand coordination with carboxyl groups.
Figure 2.
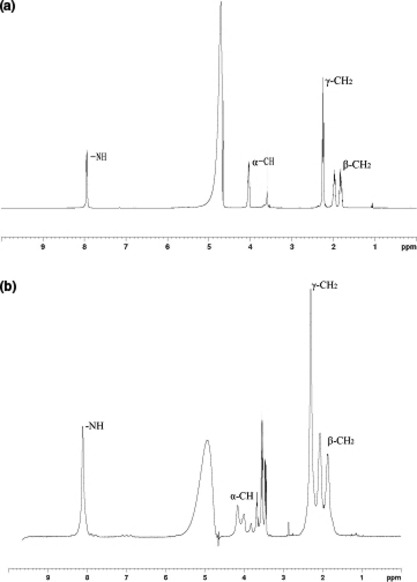
Identification of γ‐PGA and γ‐PGA‐CDDP conjugate by proton nuclear magnetic resonance (1H NMR). The 1H NMR spectra were recorded on a Bruker Avance 500 spectrometer, Switzerland. The solvent was D2O and the concentration of γ‐PGA in the samples was 10 mg/mL.
Comparison of the 1H NMR spectrum between γ‐PGA and γ‐PGA‐CDDP (Fig. 2) showed that the γ‐PGA signals at 1.80, 1.96, 2.28, 4.01 and 7.95 p.p.m. (Fig. 2a) had been excursed to the low field. The γ‐PGA‐CDDP signals at 1.87, 1.97, 2.30, 4.16 and 8.11 in Figure 2b presented β‐CH2, γ‐CH2, α‐CH and ‐NH on the γ‐PGA‐CDDP conjugate. The ‐NH signal that influenced the most remarkably to the linkage reaction of CDDP, had a 0.16 p.p.m. low‐field excursion. More importantly, we observed the multiple cleavage of α‐CH (Fig. 2b), suggesting more conjugate patterns were formed between γ‐PGA and CDDP, as indicated in Figure 1a.
γ‐PGA‐CDDP conjugate exhibits potent antitumor activity. We have previously studied in vivo antitumor activity of γ‐PGA‐CDDP conjugate using human breast tumor cells (Bcap‐37) xenografted into BALB/cA nude mice.( 14 ) In order to compare systematically the antitumor efficacy of γ‐PGA‐CDDP conjugate with the clinically used platinum derivatives, parallel experiments were performed with cisplatin, carboplatin and oxaliplatin. Mice were treated i.p. three times at 2‐day intervals with 4 mg/kg of cisplatin, carboplatin, oxaliplatin and an equivalent dose of γ‐PGA‐CDDP conjugate. The change in tumor volume after first injection was followed for 2 weeks. As shown in Figure 3a,b, tumor size in the PBS group increased significantly with time, indicating that PBS had no effect on preventing tumor growth. The tumor growth was markedly inhibited in the mice treated with CDDP and γ‐PGA‐CDDP conjugate (P < 0.001), while there was no significant difference between the CDDP group and the γ‐PGA‐CDDP group (P > 0.05). Although carboplatin and oxaliplatin inhibited tumor growth, their efficiencies were significantly lower than that of γ‐PGA‐CDDP conjugate (P < 0.01 PGA‐CDDP vs carboplatin and P < 0.05 PGA vs oxaliplatin).
Figure 3.
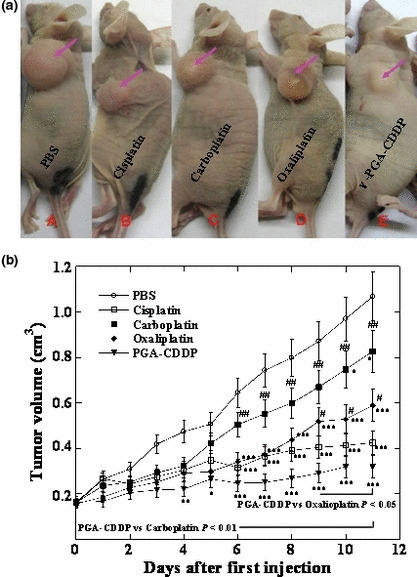
Antitumor activity of BALB/cA nude mice bearing human breast tumor Bcap‐37 cells following three repeated i.p. injections of cisplatin, carboplatin, oxaliplatin and γ‐PGA‐CDDP conjugate every 2 days for three doses. The control group received PBS. Data are presented as the mean ± SD (n = 7). (a) Typical pictures after 2 weeks of therapy with the drugs indicated in the figure. (b) Tumor volume changes following three repeated i.p. injections of platinum derivatives and γ‐PGA‐CDDP conjugate. The tumor volume was estimated by the following equation: V = π(a × b 2)/6, where a and b were major and minor axes of the tumor measured using a caliper. *P < 0.05 drugs vs control. **P < 0.01 drugs vs control. ***P < 0.001 drugs vs control. #P < 0.05 γ‐PGA‐CDDP vs other drugs. ##P < 0.01 γ‐PGA‐CDDP vs other drugs. ###P < 0.001 γ‐PGA‐CDDP vs other drugs.
γ‐PGA‐CDDP conjugate exhibits significant low toxicity. In order to evaluate the systemic toxicity of γ‐PGA‐CDDP conjugate in vivo, we first examined the body weight changes in both normal KM mice and the mice bearing human breast cancer Bcap‐37 xenografts (Fig. 4). Both types of mice dropped 35% of original body weight (P < 0.001) when treated with cisplatin (Fig. 4). This result indicated that cisplatin had a severe systemic toxicity in vivo. The KM mouse group treated with oxaliplatin had a slight increase in body weight while the Bcap‐37 xenografted mice treated with oxaliplatin had a subtle decrease in body weight. However, contrary to the PBS control group, oxaliplatin had a negative impact on the body weight change profile in both groups of mice (P < 0.001). In striking contrast, the groups treated with carboplatin and γ‐PGA‐CDDP conjugate exhibited similar body weight change profiles as those of the PBS control group (P > 0.05). Most interestingly, a comparison between tumor volume (Fig. 3b) and body weight change (Fig. 4b) indicated that γ‐PGA‐CDDP conjugate displayed significant antitumor activity with the least effect on mouse body weight change (Fig. 5). We next performed a hematological analysis and evaluated the nephrotoxicity on animals treated with and without different platinum derivatives by monitoring white blood cell and platelet number, BUN, CRE in serum and MPO, MDA, GSH, GSH‐Px in kidney tissue. All groups treated respectively with cispaltin, carboplatin and oxaliplatin had a significant decrease in white blood cell and platelet counts (P < 0.01) (Table 1). However, the γ‐PGA‐CDDP group had no significant change in the white blood cell number compared with the PBS control group (P > 0.05). Platelets in the γ‐PGA‐CDDP group were also reduced (P < 0.05), but it was a moderate decrease compared with the other three platinum derivative drugs. BUN and CRE, both of which represented the level of renal function, were evaluated (Table 1). Kidney tissues were also collected for the estimation of MPO, MDA, GSH and GSH‐Px, which reflected the antioxidative level in vivo. The data of serum analysis revealed that cisplatin caused a marked reduction in renal function, as characterized by significant increase in serum BUN and CRE levels (P < 0.05). There was no significant difference between the groups treated with carboplatin, oxaliplatin, γ‐PGA‐CDDP conjugate and the control group (P > 0.05), indicating that these drugs displayed a lower toxicity. Kidney tissue analysis showed that cisplatin and oxaliplatin induced a substantial decrease in the antioxidative level, indicated by marked increase in MPO (P < 0.05 in the cisplatin group and P < 0.01 in the oxaliplatin group) and MDA (P < 0.01 in the cisplatin group and P < 0.05 in the oxaliplatin group) levels with a concomitant decrease in GSH (P < 0.01) and GSH‐Px levels (P < 0.05) (Table 1). In contrast, groups treated with carboplatin and γ‐PGA‐CDDP conjugate were found to have no significant changes in the reduction of antioxidative level as values were similar to those of the PBS control group (P > 0.05). Interestingly, the GSH level in the γ‐PGA‐CDDP group was increased moderately compared with the PBS control group (P < 0.05). The above results were further confirmed by the Dunnett test. Taking all of these observations into consideration, we conclude that γ‐PGA‐CDDP conjugate is equally efficacious as cisplatin, but yields significantly less systemic toxicity than cisplatin, carboplatin and oxaliplatin.
Figure 4.
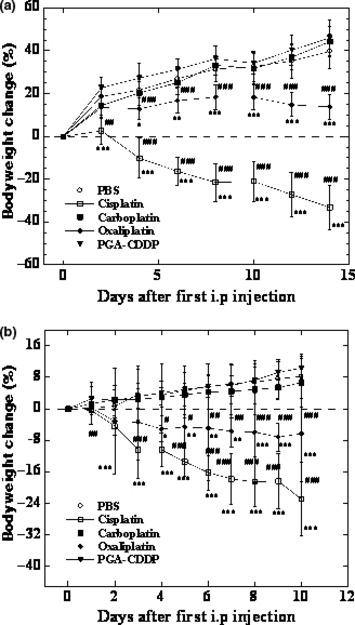
Body weight change in normal KM mice (a) and BALB/cA nude mice (b) following three repeated i.p. injections of cisplatin, carboplatin, oxaliplatin and γ‐PGA‐CDDP conjugate at 4 mg/kg every 2 days for three doses. The control group received PBS. Data are presented as the mean ± SD (n = 8 in KM mice and n = 7 in BALB/cA nude mice). *P < 0.05 drugs vs control. **P < 0.01 drugs vs control. ***P < 0.01 drugs vs control. #P < 0.05 γ‐PGA‐CDDP vs other drugs. ##P < 0.01 γ‐PGA‐CDDP vs other drugs. ###P < 0.001 γ‐PGA‐CDDP vs other drugs.
Figure 5.
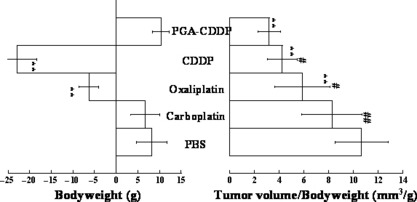
Tumor volume/body weight in BALB/cA nude mice following 2 weeks of therapy. Proportionality in tumor volume/body weight changes was evaluated as gross efficiency after tumor therapy; each of the data in tumor volume (Fig. 3b) was divided with each of the data in body weight (Fig. 4b) in the BALB/cA nude mice. Data are presented as the mean ± SD (n = 7). *P < 0.05 drugs vs control. **P < 0.01 drugs vs control. #P < 0.05 γ‐PGA‐CDDP vs other drugs. ##P < 0.01 γ‐PGA‐CDDP vs other drugs.
Table 1.
Biochemical parameters with the platinum derivatives treated animals
| Group | WBC (/mm3) | Platelet (/cm3) | BUN (mM) | CRE (mM) | MPO (U/g) | MDA (nmol/mg) | GSH (mgGSH/g) | GSH‐Px (U/g) |
|---|---|---|---|---|---|---|---|---|
| Control | 1537.5 ± 324.9 | 690 ± 85 | 6.9 ± 0.7 | 9.4 ± 1.3 | 0.6 ± 0.2 | 1.6 ± 0.5 | 60.1 ± 4.9 | 71.9 ± 10.3 |
| Cisplatin | 550.0 ± 200.0**## | 200 ± 51**## | 9.5 ± 2.8* | 15.8 ± 6.8*# | 0.9 ± 0.3* | 2.6 ± 0.3**# | 37.5 ± 6.4**## | 40.6 ± 13.4*# |
| Carboplatin | 831.3 ± 96.1**## | 570 ± 62** | 7.1 ± 0.8 | 9.7 ± 1.0 | 0.7 ± 0.1 | 1.8 ± 0.6 | 60.2 ± 9.4 | 62.8 ± 6.2 |
| Oxaliplatin | 581.3 ± 158.0**## | 300 ± 51**## | 7.1 ± 0.9 | 10.3 ± 2.0 | 0.9 ± 0.2** | 2.0 ± 0.4* | 39.4 ± 6.1**## | 53.5 ± 14.0* |
| γ‐PGA‐CDDP | 1362.5 ± 148.2 | 590 ± 59* | 7.8 ± 0.9 | 9.9 ± 1.5 | 0.7 ± 0.1 | 1.6 ± 0.5 | 75.7 ± 11.1* | 64.0 ± 4.1 |
White blood cell count (WBC), platelet count, changes of blood urea nitrogen (BUN) and creatinine (CRE) in serum and in vivo antioxidation level of myeloperoxidase (MPO), malondialdehyde (MDA), glutathione (GSH) and glutathione peroxidase (GSH‐Px) were analyzed as indicated in the “Materials and Methods”. All data are presented as the mean ± SD (n = 8). *P < 0.05 drugs vs control. **P < 0.01 drugs vs control. #P < 0.05 γ‐PGA‐CDDP vs other drugs. ##P < 0.01 γ‐PGA‐CDDP vs other drugs.
Cell proliferation assay further confirms the low toxicity of γ‐PGA‐CDDP conjugate. Low toxicity and high antitumor activity were further evaluated at the cellular level. Cell proliferate inhibition in the presence and absence of different platinum derivatives were tested by MTT assay. Three cell lines were used for in vitro cytotoxicity assay. Typical representative concentration‐growth inhibition curves with cisplatin, carboplatin, oxaliplatin and γ‐PGA‐CDDP conjugate on the growth of the Bcap‐37 cell line are shown in Figure 6 and the IC50 values of all of the drugs obtained with different cell lines are summarized in Table 2. All of the four antitumor drugs inhibited cell growth in a dose‐dependent manner, but γ‐PGA‐CDDP conjugate was consistently less toxic and had a higher IC50 value at each incubation time than that of cisplatin at an equivalent dose (Table 2). It is worth noting that consistent with the in vivo animal experiments, carboplatin displayed low tumor inhibition activity. All together, these results show that γ‐PGA‐CDDP conjugate retains the antitumor activity and displays a low toxicity.
Figure 6.
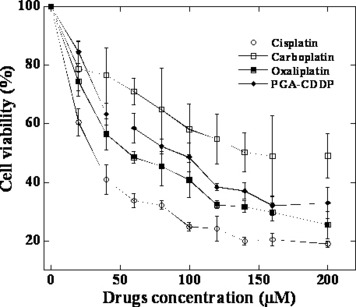
Growth inhibition against the human breast tumor Bcap‐37 cell line. Cells were incubated with cisplatin, carboplatin, oxaliplatin and γ‐PGA‐CDDP conjugate for 48 h. Cytotoxicity was measured by MTT assay. Data are shown as the mean ± SD (n = 4).
Table 2.
In vitro cytotoxicity of cisplatin, carboplatin, oxaliplatin and γ‐PGA‐CDDP conjugate against the Bcap‐37, BEL7404 and SH‐SY5Y cell lines
| Cell line | Drugs | IC50 (μM) determined at different times | ||
|---|---|---|---|---|
| 24 h | 48 h | 72 h | ||
| Bcap‐37 | Cisplatin | 20.5 ± 0.8 | 2.3 ± 0.6 | 1.9 ± 0.4 |
| Carboplatin | 265.0 ± 5.2** | 152.9 ± 12.1** | 78.8 ± 16.3** | |
| Oxaliplatin | 100.8 ± 13.0** | 38.6 ± 1.6** | 13.5 ± 1.2* | |
| γ‐PGA‐CDDP | 102.6 ± 6.1** | 25.2 ± 2.6** | 5.5 ± 0.6 | |
| BEL‐7404 | Cisplatin | 62.0 ± 11.4 | 11.1 ± 0.6 | 9.5 ± 0.5 |
| Carboplatin | 505.6 ± 108.3** | 190.8 ± 17.8** | 180.2 ± 18.2** | |
| Oxaliplatin | 92.2 ± 15.9 | 24.7 ± 3.3 | 22.4 ± 2.0 | |
| γ‐PGA‐CDDP | 149.9 ± 12.5 | 44.2 ± 6.8** | 30.0 ± 3.4* | |
| SH‐SY5Y | Cisplatin | 62.3 ± 13.1 | 28.6 ± 4.1 | 4.2 ± 0.5 |
| Carboplatin | 308.9 ± 63.0** | 150.0 ± 43.5** | 37.52 ± 7.6** | |
| Oxaliplatin | 138.1 ± 12.4* | 59.8 ± 7.8 | 2.8 ± 0.6 | |
| γ‐PGA‐CDDP | 217.6 ± 42.0** | 86.0 ± 10.0* | 17.6 ± 1.2** | |
Fifty percent inhibitory concentration was evaluated by MTT assay. Each value was the mean of four data points. *P < 0.05 drugs vs CDDP. **P < 0.01 drugs vs CDDP.
Mechanism of action of γ‐PGA‐CDDP conjugate. The antitumor mechanism of γ‐PGA‐CDDP conjugate was further investigated by analyzing the cell cycle using flow cytometric assay. The cell ratio of the apoptosis amount and DNA content in S‐phase are shown in Table 3. These results demonstrated that γ‐PGA‐CDDP conjugate induced cell apoptosis, but the mechanism needs to be investigated further. The ratio of apoptosis amount indicated the relatively lower toxicity of γ‐PGA‐CDDP conjugate compared with free CDDP.
Table 3.
Results of an apoptosis assay on the Bcap‐37 cell line with four platinum derivatives
| Drugs | Apoptosis (%) | S phase (%) |
|---|---|---|
| Control | 0.0 | 43.9 |
| Cisplatin | 44.9** | 0.0** |
| Carboplatin | 0.6 | 44.3 |
| Oxaliplatin | 10.2** | 31.8 |
| γ‐PGA‐CDDP | 14.4** | 33.6 |
Human breast tumor Bcap‐37 cells were treated with cisplatin, carboplatin, oxaliplatin and γ‐PGA‐CDDP conjugate at 100 μM/L for 24 h. The cell cycle was analyzed by FCM assay. **P < 0.01 drugs vs control.
To gain further insight into the possible mechanism of low toxicity of γ‐PGA‐CDDP conjugate, the release profiles of the different platinum derivatives have been analyzed. γ‐PGA‐CDDP conjugate was stable in distilled water for over 1 month, with no detectable dissociation and precipitation. However, CDDP could be released in an environment containing chloride ions by the mechanism of a substitution reaction between chloride ions and the carboxylic groups of γ‐PGA in the conjugate (Fig. S2). The release curves of CDDP from γ‐PGA‐CDDP conjugate in normal saline buffer and plasma at 37°C are shown in Figure 7. The release profile of CDDP in normal saline buffer (Fig. 7a) showed that CDDP in the γ‐PGA‐CDDP conjugate was released gradually, and then slowly, compared with CDDP, carboplatin and oxaplatin, which arrived at approximately 60–70% of the total amount in the first 2 h. In the release profile of the drugs in plasma (Fig. 7b), CDDP in the γ‐PGA‐CDDP conjugate was released stably and slowly all of the time; otherwise, CDDP increased expeditiously in the first 6 h and then decreased gradually, probably because some of the remaining CDDP eventually bound irreversibly to plasma proteins and resulted in drug inactivation. Because of technical limitations, the release profiles of carboplatin and oxaplatin in plasma can not be determined. Taken together, comparison of the release profiles of the different platinum derivatives indicates that γ‐PGA‐CDDP conjugate could be released mildly and maintains sufficient stability.
Figure 7.
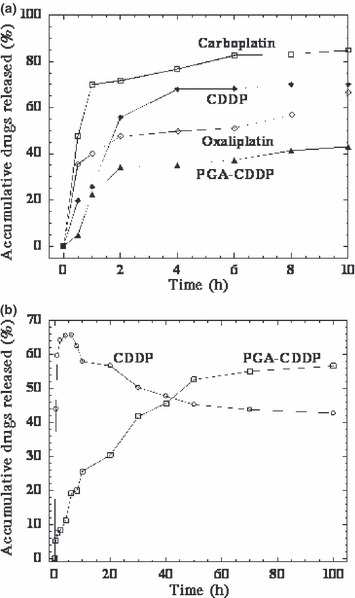
Release profiles of cisplatin, carboplatin, oxaliplatin and γ‐PGA‐CDDP conjugate in normal saline (a) and plasma (b) at 37°C. The amount of CDDP in the solutions was measured by HPLC. Experiments were performed in triplicate and similar results were obtained. The data shown are for one representative experiment.
Discussion
The main aim of this study was to compare the antitumor activity and cytotoxicity of our previously developed poly(γ, L‐glutamic acid)‐cisplatin bioconjugate with three clinically used platinum derivatives. We showed that the γ‐PGA‐CDDP conjugate produced an impressive antitumor response and yielded significantly less systemic toxicity than cisplatin, carboplatin and oxaliplatin that are currently used in clinics.
The most interesting and most important discovery in this study is that while γ‐PGA‐CDDP retains its potent antitumor activity, its toxic side effects are greatly reduced: (i) analysis of mice show that the body weight of animals treated with CDDP and oxaliplatin are severely reduced. In sharp contrast with free CDDP, the body weight of animals treated with γ‐PGA‐CDDP increase significantly. Furthermore, the evaluation profiles of the body weight of animals treated with and without γ‐PGA‐CDDP are the same, clearly showing that γ‐PGA‐CDDP is nontoxic to animals. Although carboplatin displays less toxicity than CDDP and oxaliplatin, its clinical dose is almost 10 times higher. More importantly, the comparison between tumor volume and body weight change of animals receiving different drugs reveals that γ‐PGA‐CDDP is the most potent antitumor drug with a significant low toxicity (Fig. 5). (ii) Cytotoxicity studies with cell lines further confirm the above conclusion. (iii) In accordance with the above in vitro and in vivo studies, biochemical analysis of hematology and renal metabolism of animals treated with different platinum derivatives confirm that γ‐PGA‐CDDP has substantially lower toxicity than the three clinically used platinum drugs. Cisplatin‐induced nephrotoxicity is closely associated with a lipid peroxidation increase in kidney tissues. Formation of free radicals, which leads to oxidative stress, has been shown to be one of the main pathogenic mechanisms of these toxicities and side effects.
The molecular mechanism of γ‐PGA‐CDDP displaying marked antitumor activity and a significant low toxicity is not fully understood in this study. It is very interesting to note that our in vivo toxicity study shown that γ‐PGA‐CDDP significantly increased GSH levels (Table 1, P < 0.01). Because the GSH level is important to protect cellular function and integrity, this may partially explain why γ‐PGA‐CDDP displayed reduced toxicity when compared with the other three drugs. Moreover, the flow cytometric analysis shows that γ‐PGA‐CDDP may use the same mechanism as free CDDP in achieving tumor eradication, because both CDDP and γ‐PGA‐CDDP induce apoptosis by inhibiting replication of DNA in the S‐phase. The release profiles of γ‐PGA‐CDDP and other platinum derivatives in normal saline and in plasma indicate that γ‐PGA‐CDDP may enhance antitumor activity with low toxicity by accumulating the polymer‐drug conjugate in tumor tissue through enhanced permeability and retention (EPR).( 18 ) It is well known that the EPR effects play an important role in tumor‐selective targeting therapy. Because of the high permeability and long retention in disordered capillary endothelia, γ‐PGA‐CDDP conjugate, which has a molecular weight of 40 kDa, accumulates in malignant tumor tissues much easier than that of other normal tissues. This kind of passive target can effectively inhibit tumor growth while significantly reducing side effects.( 19 ) In addition, in malignant tumor tissues, CDDP is released slowly from γ‐PGA‐CDDP conjugate accompanying enzymolysis of γ‐PGA; this gradual release not only inhibits the proliferation of tumor tissues, but also extends the half‐life of drugs in vivo and mitigates the damage to normal tissues due to one time, high dose administration.( 20 ) Finally, although the level of apoptosis and S phase DNA contents between oxaliplatin and γ‐PGA‐CDDP‐treated cells are similar (Table 3), it is unlikely that both drugs function through a common pathway because oxaliplatin decreased both GSH and WBC levels while γ‐PGA‐CDDP increased GSH or had no effect on the WBC level, respectively (Table 1). Further studies are needed to clarify this issue.
The elegant work of Avichezer et al. ( 21 ) in γ‐PGA‐CDDP has shown that this complex is biologically active and effective in suppressing tumor growth in vivo. Our previous( 14 ) and present studies are consistent with their observations. However, it is worth noting that the PGA that Avichezer used in his study was chemically synthesized compounds containing essentially α‐PGA or a mixture of α‐ and γ‐PGA, while the PGA that we used in our study was produced by fermentation containing highly pure γ‐PGA. It is also interesting to note that there are three major forms in the linkage reaction between free CDDP and γ‐PGA (Fig. 1). The three linkage forms happen in the coordinate probability and the reaction products are 33%, respectively. If we consider that only one or two forms displayed bioactivity, the effective γ‐PGA‐CDDP conjugate may have much more antitumor activity than evaluated in this and in the previous study.
In conclusion, our results clearly demonstrate that γ‐PGA‐CDDP conjugate has significant therapeutic activity against breast tumor. The impressive antitumor activity with significantly low side effects of γ‐PGA‐CDDP conjugate clearly warrants clinical investigation of this novel potential drug for possible use against human solid tumor and use in combination therapy. Additional studies will be required to investigate its biodistribution and characterize its interaction with various tumor and host factors, as well as the nature of the tumor uptake of the conjugate.
Supporting information
Fig. S1. Agarose gel electrophoresis profiles of γ‐PGA.
Fig. S2. Stability of γ‐PGA‐CDDP conjugate.
Fig. S3. Identification of γ‐PGA and γ‐PGA‐CDDP conjugate by carbon nuclear magnetic resonance (13C NMR) and Fourier transformed infrared (FT‐IR) analyses.
Data S1. Preparation of γ‐PGA‐CDDP conjugate.
Data S2. Analysis of γ‐PGA‐CDDP conjugate.
Supporting info item
References
- 1. Boulikas T, Vougiouka M. Cisplatin and platinum drugs at the molecular level. (Review). Oncol Rep 2003; 10: 1663–82. [PubMed] [Google Scholar]
- 2. McWhinney SR, Goldberg RM, McLeod HL. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther 2009; 8: 10–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arafa HM. Carnitine deficiency aggravates carboplatin nephropathy through deterioration of energy status, oxidant/anti‐oxidant balance, and inflammatory endocoids. Toxicology 2008; 254: 51–60. [DOI] [PubMed] [Google Scholar]
- 4. Kraus KS, Ding D, Zhou Y, Salvi RJ. Central auditory plasticity after carboplatin‐induced unilateral inner ear damage in the chinchilla: up‐regulation of GAP‐43 in the ventral cochlear nucleus. Hear Res 2009; 255: 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reck M, Macha HN, Del Barco S et al. Phase II study of oral vinorelbine in combination with carboplatin followed by consolidation therapy with oral vinorelbine as single‐agent in unresectable localized or metastatic non‐small cell lung carcinoma. Lung Cancer 2009; 64: 319–25. [DOI] [PubMed] [Google Scholar]
- 6. Igawa S, Murakami H, Takahashi T et al. Efficacy of chemotherapy with carboplatin and paclitaxel for unresectable thymic carcinoma. Lung Cancer 2010; 67: 194–7. [DOI] [PubMed] [Google Scholar]
- 7. Jerremalm E, Wallin I, Ehrsson H. New insights into the biotransfor‐mation and pharmacokinetics of oxaliplatin. J Pharm Sci 2009; 98: 3879–85. [DOI] [PubMed] [Google Scholar]
- 8. Kagiava A, Tsingotjidou A, Emmanouilides C, Theophilidis G. The effects of oxaliplatin, an anticancer drug, on potassium channels of the peripheral myelinated nerve fibres of the adult rat. Neurotoxicology 2008; 29: 1100–6. [DOI] [PubMed] [Google Scholar]
- 9. Argyriou AA, Polychronopoulos P, Iconomou G, Chroni E, Kalofonos HP. A review on oxaliplatin‐induced peripheral nerve damage. Cancer Treat Rev 2008; 34: 368–77. [DOI] [PubMed] [Google Scholar]
- 10. Capdevila J, Elez E, Peralta S, Macarulla T, Ramos FJ, Tabernero J. Oxaliplatin‐based chemotherapy in the management of colorectal cancer. Expert Rev Anticancer Ther 2008; 8: 1223–36. [DOI] [PubMed] [Google Scholar]
- 11. Kim WJ, Kang MS, Kim HK et al. Water‐soluble porphyrin‐polyethylene glycol conjugates with enhanced cellular uptake for photodynamic therapy. J Nanosci Nanotechnol 2009; 9: 7130–5. [DOI] [PubMed] [Google Scholar]
- 12. Chen H, Wang L, Yeh J et al. Reducing non‐specific binding and uptake of nanoparticles and improving cell targeting with an antifouling PEO‐b‐PgammaMPS copolymer coating. Biomaterials 2010; 31: 5397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hobbs SK, Monsky WL, Yuan F et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA 1998; 95: 4607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ye H, Jin L, Hu R et al. Poly(gamma,L‐glutamic acid)‐cisplatin conjugate effectively inhibits human breast tumor xenografted in nude mice. Biomaterials 2006; 27: 5958–65. [DOI] [PubMed] [Google Scholar]
- 15. Hu R, Ye H, Jin L, Wu C, Wu Z. Screening and optimizing fermentation condition of Bacillus strain with high‐productive poly‐gamma‐glutamic acid. China Biotechnology 2005; 25: 62–5. [Google Scholar]
- 16. Hanada K, Nagai N, Ogata H. Quantitative determination of unchanged cisplatin in rat kidney and liver by high‐performance liquid chromatography. J Chromatogr B Biomed Appl 1995; 663: 181–6. [DOI] [PubMed] [Google Scholar]
- 17. Augey V, Cociglio M, Galtier M, Yearoo R, Pinsani V, Bressolle F. High‐performance liquid chromatographic determination of cis‐dichloro‐diammineplatinum(II) in plasma ultrafiltrate. J Pharm Biomed Anal 1995; 13: 1173–8. [DOI] [PubMed] [Google Scholar]
- 18. Noguchi Y, Wu J, Duncan R et al. Early phase tumor accumulation of macromolecules: a great difference in clearance rate between tumor and normal tissues. Jpn J Cancer Res 1998; 89: 307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst 2006; 98: 335–44. [DOI] [PubMed] [Google Scholar]
- 20. Jain RK. Delivery of molecular and cellular medicine to solid tumors. J Control Release 1998; 53: 49–67. [DOI] [PubMed] [Google Scholar]
- 21. Avichezer D, Schechter B, Arnon R. Functional polymers in drug delivery: carrier‐supported CDDP (cis‐platin) complexes of polycarboxylates – effect on human ovarian carcinoma. React Funct Polym 1998; 36: 59–69. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Agarose gel electrophoresis profiles of γ‐PGA.
Fig. S2. Stability of γ‐PGA‐CDDP conjugate.
Fig. S3. Identification of γ‐PGA and γ‐PGA‐CDDP conjugate by carbon nuclear magnetic resonance (13C NMR) and Fourier transformed infrared (FT‐IR) analyses.
Data S1. Preparation of γ‐PGA‐CDDP conjugate.
Data S2. Analysis of γ‐PGA‐CDDP conjugate.
Supporting info item