

Abstract
Arsenic trioxide (ATO), an ancient traditional Chinese medicine, has been successfully used as a therapeutic agent for leukemia. Drug resistance and toxicity are major concerns with the treatment. MicroRNAs (miRNAs) are endogenous small non‐coding RNA molecules that might modulate cellular sensitivity to anticancer drugs. miRNA‐21 (miR‐21) is one of the most prominent miRNAs involved in various aspects of human cancers. However, miR‐21 has been rarely characterized in chronic myelogenous leukemia (CML). Here, we used a specific anti‐miR‐21 oligonucleotide (AMO‐miR‐21) to sensitize K562 cells to ATO by degradation of miR‐21. The results showed that both AMO‐miR‐21 and ATO caused growth inhibition, apoptosis, and G1‐phase arrest in K562 cells. Meanwhile, AMO‐miR‐21 significantly promoted ATO‐mediated growth inhibition and apotosis without affecting the G1 phase. Apoptotic cells were confirmed morphologically with Giemsa’s staining. Furthermore, dual‐luciferase reporter vector, containing two tandem miR‐21 binding sites from PDCD4 3′UTR, validated that PDCD4 was directly regulated by miR‐21. Therefore, AMO‐miR‐21 sensitized leukemic K562 cells to ATO by inducing apoptosis partially due to its up‐regulation of PDCD4 protein level. The combination of ATO and AMO‐miR‐21 present therapeutic potential for CML.
(Cancer Sci 2010; 101: 948–954)
For a long time, arsenic trioxide (ATO), an ancient traditional Chinese medicine, was used as a therapeutic agent for some severe diseases in line with the ancient Chinese philosophy of ‘treating an evil with a toxic’ (S.Z. Li, Ming Dynasty, 1578).( 1 ) From the 1700s through the early 1900s, arsenic was a mainstay in the treatment of leukemia.( 2 ) In the mid‐1990s, the first controlled clinical trial of arsenic trioxide (ATO) further improved the clinical outcome of refractory or relapsed acute promyelocytic leukemia (APL).( 3 , 4 ) Contemporary studies show that arsenic is an effective therapeutic agent for the treatment of APL.( 5 , 6 )
The antitumor mechanism of ATO, mainly the induction of apoptosis and cell cycle arrest, suggests that it may be active against other malignancies. Actually, ATO has shown some activity in patients with accelerated phase chronic myelogenous leukemia (CML) and other hematologic malignancies.( 7 , 8 , 9 )
ATO used as a single agent at higher concentrations causes many side effects (ventricular arrhythmia, skin reaction, peripheral neuropathy, electrolyte changes, leukocytosis, hepatic dysfunction, gastrointestinal reactions, etc.). Therefore, low‐dose combination therapy is required.( 4 , 10 , 11 , 12 ) More recently it has been shown to be effective, particularly in combination with all‐trans retinoic acid, in the treatment of APL.( 4 ) It is not clear yet that whether there are better combination of drugs.
MicroRNAs (miRNAs) are a class of endogenous RNA molecules 19–25 nucleotides in length that are able to induce mRNA degradation, translational repression, or both, via pairing with partially complementary sites in the 3′UTR of the targeted genes.( 13 ) There are over 700 miRNAs estimated in humans,( 14 ) and 30% of all genes are regulated by miRNAs. However, only very few miRNAs have been functionally characterized and the general functions of miRNAs are not globally studied.( 15 ) Currently, miRNAs are strongly implicated in such processes as development, carcinogenesis, cell survival, and apoptosis.( 16 ) Recently, more studies have shown that miRNAs may have an effect on the chemosensitivity( 17 , 18 ) or chemoresistance of cancer cells.( 19 , 20 ) The miRNA‐21 gene has been identified as the only miRNA overexpressed in solid tumors of the lung, breast, stomach, prostate, colon, brain, head and neck, esophagus, and pancreas.( 21 , 22 ) Inhibition of miRNA‐21 has decreased cell growth in many cancer cells.
Therefore, we questioned whether miR‐21 may modulate the chemosensitivity of leukemic cells. In the current study, we evaluated the role of miR‐21 and the response to treatment with ATO by anti‐miR‐21 oligonucleotide (AMO‐miR‐21) suppression of miR‐21. We wanted to provide here mechanistic evidence for synergic effects of AMO‐miR‐21 and ATO in leukemic K562 cells. Consequently, the potential of AMO‐miR‐21 should be explored as a gene therapy for the treatment of leukemia.
Materials and Methods
Design and synthesis of oligonucleotide sequences.
The mature miRNA‐21 sequences are available from the miRNA Registry.( 14 ) The sequences of anti‐miR‐21 oligonucleotide (AMO‐miR‐21) were designed according to the principle of sequences complementary to mature miRNA‐21. The oligodeoxynucleotide sequences used in this study are as follows: AMO‐miR‐21, 5′‐TCAACATCAGTCTGATAAGCTA‐3′ (22 bp); Scramble (SCR), 5′‐CATTAATG TCGGACAACTCAAT‐3′ (22 bp). All oligodeoxynucleotides were chemically synthesized and modified with phosphorothioate by Shanghai Sangon Biological Engineering in China. The siRNA sequence of PDCD4 (siPDCD4) was as follows: sense, 5′‐AAGGUGGCUGGAACAUCUAUU‐3′; antisense, 5′‐AAUAGAUGUUCCAGCC ACCUU‐3′. The RNAs were synthesized and purified by HPLC by Shanghai GenePharma in China and stored at −20°C.
Cell line and AMO, siRNA transfection.
The K562 (chronic myelogenous leukemia) cell line (from the Shanghai Institute of Cell Biology, China) was grown in RPMI‐1640 containing 10% fetal calf serum (FCS) at 37°C in a humidified atmosphere with 5% CO2 (Thermo FORMA 3110; Marietta, OH, USA). K562 cells in the exponential phase of growth were seeded in 96‐ or 24‐well plates (Costar, Cambridge, MA, USA) and transfected with oligodeoxynucleotides mediated by Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) (2.0:1 volume/mass rate of Lipofectamine 2000 to the oligodeoxynucleotides) in serum‐free RPMI‐1640 for 6 h. At the end of transfection, the cells were incubated in medium containing 10% FCS. Transfection of PDCD4 siRNA (siPDCD4) into K562 cells in a final concentration of 50 nm was prepared according to the manufacturer’s instructions.
ATO‐sensitivity assay.
The viability of K562 cells was determined by the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2, 4‐diphenyl‐tetrazolium bromide (MTT) assay. Briefly, cells at a density of 5 × 104/mL were transfected with AMO‐miR‐21 or SCR control (0.4 μm) in the presence of Lipofectamine 2000 and serum‐free RPMI‐1640 media for 6 h. Then, the cells were plated in 96‐well plates in medium containing 10% FCS for another 48 h in the presence of varying concentrations of ATO (0.5, 1.0, 2.0, 3.0, and 4.0 μm). Twenty μL of MTT stock solution (5 mg/mL) was added to each well in the final MTT concentration of 0.45 mg/mL, and the plate was incubated for 4 h at 37°C. Media was then removed and dimethyl sulfoxide (DMSO) (150 μL) was added to dissolve the blue formazan crystals at room temperature for 30 min. The viability of cells was assessed by absorbance at 570 nm on a Bio‐Rad microtiter plate reader (Hercules, CA, USA). The IC50 values (μm) were determined with ICp software (Beijing, China).
Flow cytometry analysis of the cell cycle and apoptosis.
K562 cells were seeded at a density of 1.2 × 105/mL in 24‐well plates (Costar), 500 μL/well, transfected with 0.4 μm AMO‐miR‐21 by Lipofectamine 2000 reagent (Invitrogen) in serum‐free RPMI‐1640 for 6 h. After transfection, 500 μL of the appropriate growth medium containing 20% FCS were added to each well, total 1000 μL. Cells were continuously incubated for another 48 h in the presence of ATO (2 μm), then harvested, washed twice with PBS, and fixed with 70% ethanol, as well as treated with RNase A (1 mg/mL) after ethanol elimination. Finally, the cells were stained with propidium iodide (PI) solution (50 μg/mL). The cell cycles were analyzed by flow cytometry according to content of DNA (Coulter Elite; Fullerton, CA, USA).
Pretreatment of K562 cells was performed described as above. Viable cells were collected and double‐stained with FITC‐conjugated Annexin V and PI. For each sample, data from approximately 10 000 cells were recorded in the list mode on logarithmic scales. Apoptosis and necrosis were analyzed by quadrant statistics on PI‐negative, Annexin V‐positive cells and both positive cells, respectively.
Giemsa’s staining.
K562 cells tranfected with 0.4 μm AMO‐miR‐21 were incubated for 48 h. Meanwhile, K562 cells treated with ATO (2 μm) were also grown for 48 h. Then the cells were collected by centrifugation at 4°C for 5 min at 111.8g, and given a quick wash in PBS. The cells were sprayed on glass slides, air dried and fixed with methanol for 1 min, stained with 10% Giemsa’s stain for 15 min, washed with distilled water, and finally air dried again. The morphology of K562 cells was observed under light microscope.
Analysis of miR‐21 and PDCD4 mRNA level.
Pretreatment of K562 cells was performed described as above. The total RNA from treated cells was extracted in Trizol (Invitrogen) and was quantified by an ultraviolet spectrophotometer (UVP, Upland, CA, USA) at a wavelength of 260 nm. miR‐21 and U6 snRNA expression level were determined with a miRNA Real‐Time PCR Quantitation Kit (Shanghai GenePharma). The stem‐loop RT primer was hybridized to a miRNA molecule and then reverse transcribed with a MMLVReverse transcriptase. A 20 μL reverse transcription (RT) reaction was incubated at 25°C for 30 min.
Real‐time PCR was performed using a PCR amplifier (Bio‐Rad,). PCR cycles were as follows. There was an initial denaturation at 94°C for 3 min. The reaction was repeated for 45 cycles; each cycle consisted of denaturing at 94°C for 20 s, and annealing at 50°C for 25 s, synthesis at 72°C for 20 s as per the manufacturer’s instructions. U6 snRNAs was used as the internal control. The fold‐change for miR‐21 expression levels was calculated using △CT and 2−△△CT.
The PDCD4 mRNAs were determined by SYBR‐Green real‐time PCR assay conducted with 40 cycles of 10 s at 95°C, 30 s at 60°C, and 45 s at 72°C. The melting curve analysis was performed. The PDCD4 mRNAs were normalized to GAPDH.
Western blot analysis of PDCD4 protein level.
The cells were lyzed in RIPA buffer in the presence of proteinase inhibitor (Biocolor BioScience & Technology, Shanghai, China). Protein concentration was determined by bicinchoninic acid (BCA) (Bioss, Beijing, China). Thirty μg of protein was separated on 10% SDS‐PAGE and transferred to nitrocellulose membrane. Membranes were probed with primary antibodies against PDCD4 (rabbit polyclonal; Cell Biotech, Tianjin, China) at room temperature for 2 h, washed extensively with 0.1% Tween‐20 in PBS, and incubated with secondary antibodies conjugated with horseradish peroxidase at a dilution of 1:10 000. The signals were visualized with diaminobenzidine (DAB) (Boster, Wuhan, China).
Plasmid constructions and reporter assay.
Two primers, including one miR‐21 target site, were chemically synthesized (forward: XhoIF, 5′‐CCGctcgagCTCTTCTTAAGTGGAATATTC‐TAATAAGCTA CCTTTTGTCTCTTCTTAAG‐3′, reverse: NotIR, 5′‐ATAAGAATgcggccgcACAAAAGG TAGCTTATTAGAATA‐TTCCACTTAAGAAGAGACAAAAGGTAG‐3′).( 23 ) A 79 bp fragment of the 3′‐UTR of PDCD4, which contained NotI and XhoI restriction site overhangs and two tandem putative miR‐21 binding sites (indicated by the italic uppercase letters), was amplified by PCR. This PCR product was cloned into the psiCHECK‐2 vector (Promega, Madison, WI, USA) immediately downstream of the Renilla luciferase reporter gene between the XhoI and NotI sites (lower case) and named PDCD4‐UTR. The recognition sequence is underlined in the above sequence. The tandem mutations were introduced to the seed region of the miR‐21 binding site in the primers. ATAAGCTA was substituted by TAGCTACT, and named PDCD4‐mut‐UTR. Cells were co‐transfected with PDCD4‐UTR or PDCD4‐mut‐UTR and AMO‐miR‐21, then luciferase activity at 24 h post‐transfection were determined using the dual‐luciferase reporter assay system (E1910; Promega). Renilla luciferase activity was normalized to firefly luciferase activity for each sample.
Statistical analysis.
All the experiments were carried out in triplicate. The results were calculated using SPSS version 10 software (SPSS, Chicago, IL, USA), reported as x ± s and compared using anova. Statistical significance was defined as P < 0.05.
Results
AMO‐miR‐21 promotes ATO sensitivity of leukemic cells.
In this study, we determined the influence of AMO‐miR‐21 on cell viability (alone or in combination with ATO). As shown in Figure 1(A), AMO‐miR‐21 alone effectively inhibited cell viability significantly (*P < 0.01 as compared with SCR controls). Meanwhile, the data indicated that AMO‐miR‐21 was also able to increase the ATO‐induced inhibitory effects on K562 cells (Fig. 1A; #P < 0.05; & P < 0.01 compared with SCR‐ATO controls). Thus, AMO‐miR‐21 significantly decreased the IC50 values of ATO (Fig. 1B, #P < 0.01 as compared with SCR‐ATO controls).
Figure 1.
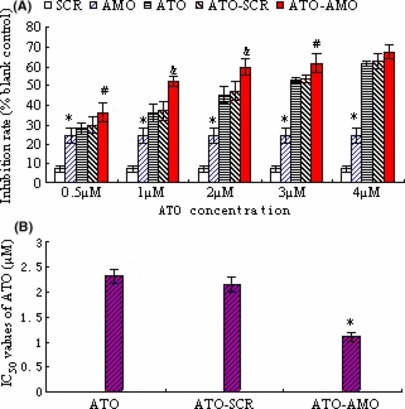
AMO‐miR‐21 promotes sensitivity of K562 cells to arsenic trioxide (ATO). (A) K562 cells were transfected with AMO‐miR‐21 or Scramble (SCR) control (0.4 μm) in the presence of Lipofectamine 2000 and serum‐free RPMI‐1640 for 6 h. At the end of transfection, the cells were plated in 96‐well plates in medium containing 10% FCS for another 48 h in the presence of varying concentrations of ATO (0.5, 1.0, 2.0, 3.0, and 4.0 μm). Cell viability was assessed by MTT assays performed in triplicate. The results indicate that AMO‐miR‐21 inhibited K562 cell viability (*P < 0.01 as compared with Scramble [SCR] control) and promoted ATO‐induced growth inhibition (#P < 0.01; &P < 0.05 compared with SCR‐ATO control). (B) The IC50 values of ATO were determined by ICp software. The data showed that AMO‐miR‐21 reduced the IC50 values of ATO. *P < 0.01 as compared with SCR‐ATO control.
AMO‐miR‐21 regulates the cell cycle using flow cytometry.
To explore the effects of AMO‐miR‐21 on the cell cycle, AMO‐miR‐21 treatment alone or in combination with 2 μm ATO was investigated in K562 cells. Cells were stained with PI solution. Cell cycles were analyzed by flow cytometry according to DNA content. As shown in Table 1, AMO‐miR‐21 and ATO were able to independently induce G1 phase arrest. Meanwhile, increased sub‐G1‐phase cells indicated that both could induce apoptosis in K562 cells, which was confirmed by the enhancement of the subdiploid peak in the DNA content histograms (Fig. 2).
Table 1.
AMO‐miR‐21 regulates K562 cell cycles (x ± s%, n = 3)
| Sub G1 | G0/G1 | S | G2/M | |
|---|---|---|---|---|
| Blank | 1.47 ± 0.08 | 55.42 ± 3.41 | 22.21 ± 1.42 | 21.22 ± 2.12 |
| SCR | 1.55 ± 0.07 | 54.02 ± 4.23 | 21.55 ± 1.52 | 18.50 ± 1.78 |
| AMO | 7.05 ± 0.33* | 74.20 ± 3.98* | 10.86 ± 0.94 | 10.46 ± 1.38 |
| ATO | 8.53 ± 0.96* | 72.56 ± 3.4* | 11.40 ± 1.12 | 8.71 ± 1.02 |
| ATO‐SCR | 9.54 ± 1.02 | 71.15 ± 3.73 | 12.72 ± 0.98 | 9.22 ± 1.08 |
| ATO‐AMO | 14.36 ± 1.09** | 69.46 ± 3.02 | 8.94 ± 0.96 | 8.41 ± 0.53 |
*P < 0.01, as compared with blank or Scramble (SCR) controls.
**P < 0.01, as compared with ATO‐SCR control.
Figure 2.
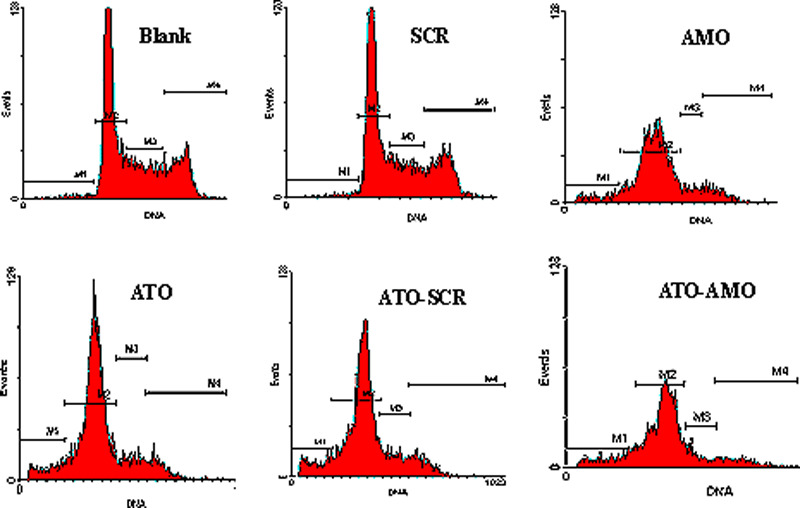
AMO‐miR‐21 affects cell cycles in K562 cells. Cell cycles were analyzed by flow cytometry at 48 h post transfection. The results indicated that both AMO‐miR‐21 and arsenic trioxide (ATO) were able to increase the G1 and sub‐G1 phases in cells. AMO‐miR‐21 promoted ATO‐induced sub‐G1 phase cells without affecting G1 phase cells.
AMO‐miR‐21 induces cell apoptosis using flow cyto‐metry.
AMO‐miR‐21 was used alone or in combination with 2 μm ATO in K562 cells. Apoptosis of K562 cells was detected by flow cytometry using double‐staining with Annexin V and PI. The results demonstrated that AMO‐miR‐21 alone induced cell apoptosis, and promoted ATO‐induced apoptosis of cells as compared with controls (Fig. 3A).
Figure 3.
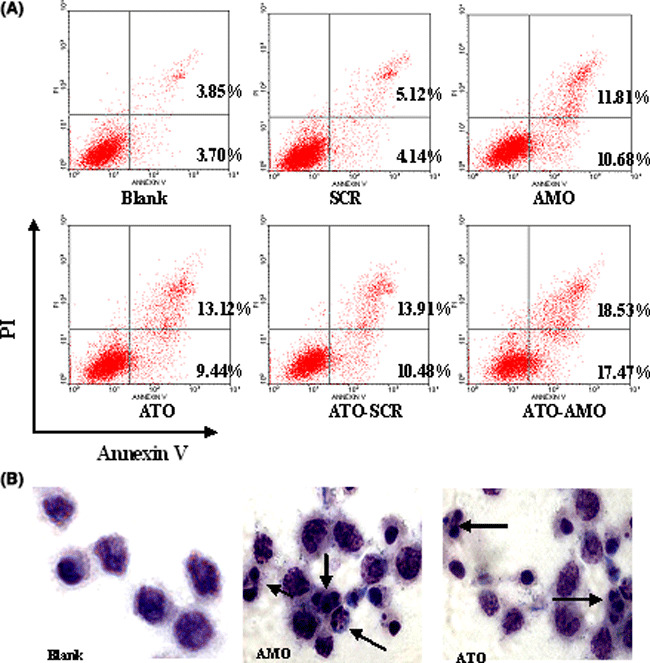
AMO‐miR‐21 affects cell apoptosis in K562 cells. Viable cells were collected at 48 h post transfection. (A) K562 cells were double‐stained with FITC‐conjugated Annexin V and propidium iodide (PI), then detected by flow cytometry. The results demonstrated that both AMO‐miR‐21 and arsenic trioxide (ATO) induced cell apoptosis, Meanwhile, AMO‐miR‐21 significantly promoted ATO‐induced apoptosis in K562 cells. (B) K562 cells were stained with 10% Giemsa’s stain for 15 min at 48 h post transfection. The morphologic aspect of nuclei was observed under light microscope. Arrows indicate fragmented nuclei and apoptotic bodies.
Cell morphological validates cell apoptosis using Giemsa’s staining.
Giemsa’s staining (Fig. 3B) also showed that K562 cells in the AMO‐miR‐21 and ATO groups acquired typical features of apoptosis, including cell shrinkage, nuclear pyknosis, and apoptotic body at 48 h post transfection. The results validated that both AMO‐miR‐21 and ATO were able to induce cell apoptosis.
AMO‐miR‐21 degrades miR‐21 and up‐regulates PDCD4 mRNA level by real‐time PCR.
To evaluate the effects of AMO‐miR‐21 on miR‐21 and PDCD4 mRNA level on the cells, K562 cells transfected with AMO‐miR‐21 were processed and analyzed for the mRNAs. miR‐21 and U6 snRNA expression was determined by quantitative real‐time PCR. U6 snRNAs were used as the internal control. The fold‐change for miR‐21 expression level was calculated using 2−△△CT, as described in the Materials and Methods. As shown in Figure 4(A), the 2−△△CT value of K562 cells treated with AMO‐miR‐21 was only 0.022. The results indicated AMO‐miR‐21 effectively down‐regulated miR‐21 levels and up‐regulated PDCD4 mRNA (Fig. 4B) protein levels in K562 cells.
Figure 4.
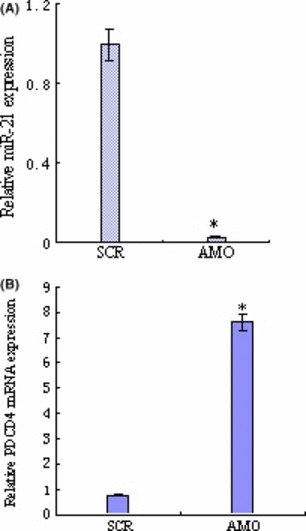
AMO‐miR‐21 regulates miRNA‐21 (miR‐21) and PDCD4 mRNA levels. The total RNA from treated K562 cells was extracted in Trizol and quantified by ultraviolet spectrophotometry at 48 h after transfection. (A) AMO‐miR‐21 degrades miR‐21. miR‐21 and U6 snRNA expression was determined by quantitative real‐time PCR. (B) AMO‐miR‐21 up‐regulates PDCD4 mRNA level. PDCD4 mRNA relative expression level by SYBR‐Green real‐time PCR. The data showed that AMO‐miR‐21 down‐regulated miR‐21 levels and up‐regulated PDCD4 mRNA expression in K562 cells. *P < 0.01 as compared with control.
AMO‐miR‐21 up‐regulates protein levels by Western blott‐ing.
To evaluate the effects of AMO‐miR‐21 on PDCD4 protein levels, K562 cells transfected with AMO‐miR‐21 were processed and analyzed for PDCD4 protein (by Western blotting) as described in the Materials and Methods. The results indicated that AMO‐miR‐21 significantly up‐regulated protein levels (Fig. 5) in K562 cells.
Figure 5.
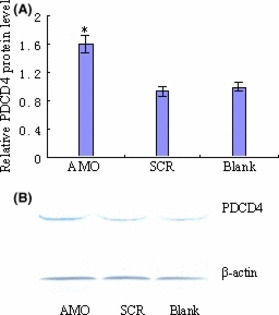
AMO‐miR‐21 up‐regulates PDCD4 protein levels. The cells were lyzed at 48 h after transfection. Protein concentration was determined by bicinchoninic acid (BCA). PDCD4 protein level was determined by Western blotting. (A) Relative PDCD4 protein level. PDCD4 protein expression levels were up‐regulated by AMO‐miR‐21. *P < 0.01 as compared with blank or Scramble (SCR) control. (B) Representative images of PDCD4 Western blotting.
AMO‐miR‐21 up‐regulates the PDCD4‐3′UTR level.
By the computer‐based sequence analysis software TargetScan,( 24 ) the first eight nucleotides (seed sequence) from the 5′ end of miR‐21 were complementary to the sequence of the PDCD4‐3′UTR (Fig. 6A). miRNAs exert their effects by targeting the 3′UTR of the protein‐coding genes and thus induce mRNA degradation and/or translational repression.
Figure 6.
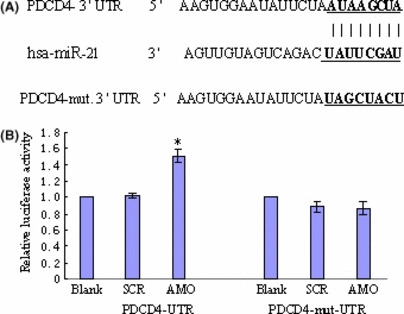
miRNA‐21 (miR‐21) directly targets the PDCD4‐3′UTR. K562 cells were co‐transfected with PDCD4‐3′UTR vector or PDCD4‐mut‐3′UTR luciferase reporter and AMO‐miR‐21. Transfection with scramble control was done in parallel. The Renilla luciferase activity and firefly luciferase activity was measured by dual‐luciferase reporter assay (Promega). Renilla luciferase activity was normalized to firefly luciferase activity for each sample. Data represents the mean value of three independent experiments. *P < 0.01. (A) Predicted binding site of miR‐21 with PDCD4‐3′UTR by using TargetScan. The matched sequences are shown in bold. (B) Dual‐luciferase reporter assay.
To confirm PDCD4 as a direct target of miR‐21 in leukemic K562 cells, we constructed the dual‐luciferase reporter (psi‐CHECK) containing either the miR‐21 recognition or mutated sequences from the 3′UTR of PDCD4 mRNA immediately downstream of the luciferase gene. As shown in Figure 6(B), transfection with AMO‐miR‐21 increased the activity of PDCD4‐UTR reporter in K562 cells without affecting the activity of PDCD4‐mut‐UTR reporter. These results suggested that PDCD4 is a target of miR‐21 in K562 cells.
PDCD4 siRNA prevents growth inhibition and apotosis induced by AMO‐miR‐21.
To elucidate the contribution of PDCD4 to the inhibitory effects of AMO‐miR‐21 on leukemia, K562 cells were co‐transfected with AMO‐miR‐21 (0.4 μm) or siPDCD4 (50 nm). MTT and flow cytometric assay demonstrated that siPDCD4 was able to prevent growth inhibition and apoptosis induced by AMO‐miR‐21 (Fig. 7). The result validated that AMO‐miR‐21 inhibited cell viability and induced apoptosis at least partially by up‐regulation of the PDCD4 protein level.
Figure 7.
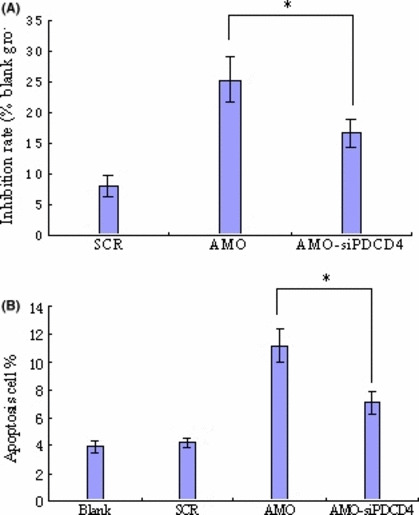
PDCD4 siRNA prevents growth inhibition and apoptosis induced by AMO‐miR‐21. Cells were co‐transfected with AMO‐miR‐21 (0.4 μm) or siPDCD4 (50 nm). MTT (A) and flow cytometry (B) demonstrated that PDCD4 siRNA (siPDCD4) could prevent growth inhibition and apotosis induced by AMO‐miR‐21. There was statistical significance, *P < 0.05, as compared with the AMO group.
Discussion
Recently, increasing evidence has suggested that significantly over‐expressed miRNAs in tumors may be considered to be a novel class of oncogenes. These oncogene miRNAs, called ‘oncomirs,’ usually promote tumor development by negatively regulating tumor suppressor genes that control biological processes. Therefore, altering the expression of oncomirs might be a valuable strategy for cancer treatment.( 25 , 26 )
Diverse studies have shown that miR‐21 is overexpressed in different tumor types, and is one of the most prominent miRNAs implicated in the various aspects of tumors, including growth, proliferation, antiapoptosis,( 21 , 22 , 27 ) and response to chemotherapy( 17 ) in many human cancers. However, the role of miR‐21 has been rarely characterized and studied with regard to CML.
Currently, antisense oligonucleotides targeting mRNAs have been successfully used to identify miRNA functions and for the development of therapeutic agents.( 28 , 29 , 30 )
As miRNAs are small nucleic acids (19–24 nt), antisense inhibition is considered the best and possibly the only practical approach to specific pharmacological inhibition of their function.( 28 ) In this work, we attempted to study the role of miRNA‐21 in K562 cells using antisense inhibition.
Our data reveals that AMO‐miR‐21 significantly down‐regulated mature miRNA‐21 expression level due to RNase H‐mediated degradation of miRNA in K562 cells (Fig. 4A). The alteration of miRNA levels in cells was detected by real‐time PCR. Meanwhile, the knockdown of endogenous miR‐21 expression contributed to sensitizing leukemic K562 cells to ATO (Fig. 1) mainly through increasing apoptosis (Fig. 3A). Apoptotic cells were confirmed morphologically by Giemsa’s staining.
The mechanisms of drug insensitivity or resistance is involved in multiple factors, especially the apoptotic capacity of cancer cells.( 31 ) Almost all cytotoxic antitumor drugs in clinical use exert their antitumor activity by inducing apoptosis.( 32 ) Many tumors may develop by escaping apoptotic death signals by expressing anti‐apoptotic proteins, such as Bcl‐2.( 31 , 32 ) In our study, AMO‐miR‐21 significantly promoted apoptosis and contributed to sensitization of K562 cells to ATO (1, 3).
The identification of target genes is a key step in assessing the role of aberrantly expressed miRNA in human cancer and for the further development of miRNA‐based gene therapy. miR‐21 has been found to have strongly elevated expression in a variety of human neoplastic disorders and regulatory potential in targeting a number of important tumor suppressor genes (e.g. programmed cell death 4 [PDCD4], phosphatase and tensin [PTEN], tropomyosin [TPM1], sprouty 2 [SPRY], reversion‐inducing cysteine‐rich protein with kazal [RECK], nuclear factor I/B [NFIB]) that have attracted the attention of researchers in various fields.( 33 , 34 ) Recent research has demonstrated that miR‐21 post‐transcriptionally down‐regulates tumor suppressor PDCD4( 21 , 22 ) which is widely involved in growth, apoptosis, invasion and the cell cycle.( 35 ) Therefore, the blockade of miR‐21, which at least partially up‐regulates PDCD4, may suppress the bioactivity of K562 cells.
It is unknown whether PDCD4 is also directly targeted by miR‐21 in K562 cells. Our research indicated that PDCD4 expression level was up‐regulated by AMO‐miR‐21 (4, 5), which possibly degraded miR‐21 (Fig. 4A). Further research validated that suppression of miR‐21 could specifically enhance the expression of the reporter gene that carries two tandem PDCD4 3′UTRs without changing the expression level of the mutations’ reporter gene in the seed sites (Fig. 6). So we can initially surmise that PDCD4 is directly regulated by miR‐21, and suppression of cell growth by AMO‐miR‐21 can be explained in part by PDCD4 inducing apoptosis.
The study also indicated that arsenic trioxide alone inhibited cell growth and increased the G1 phase in K562 cells following 48 h of exposure. This is consistent with the report by Hyun Park.( 36 ) As shown in 1, 3, ATO and AMO‐miR‐21 have somewhat synergistic effects in growth inhibition and apoptosis promotion. Namely, AMO‐miR‐21 could sensitize K562 cells to ATO possibly by inducing apoptosis. In this study, both AMO‐miR‐21 and ATO caused G1 phase arrest, but without synergistic action.
At present, several antisense therapeutics in phase III clinical trials have a promising future. It is possible that these drugs could eventually gain approval by the United States Food and Drug Administration.
AMO‐miR‐21 significantly sensitized K562 cells to ATO by inducing apoptosis. When AMO‐miR‐21 and ATO were used in combination, AMO‐miR‐21 reduced the therapeutic dose of ATO; this could lead to low non‐specific effects and high tolerance, and also possibly reverse drug resistance in leukemia. Theoretically, the strategy may be feasible. Therefore, exploiting synergistic effects between AMO‐miR‐21 and ATO might be an effective clinical strategy for leukemia chemotherapy, and miR‐21 might be a potential drug target in K562 cells.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This study was supported by grants from the Guangdong Administration of Traditional Chinese Medicine Research Project (2008098), Natural Science Foundation of Guangdong Province (no. 5300488), and Science and Technology Plan Projects of Guangdong Province (nos. 2006B35502010, 2005B33101005).
References
- 1. Li SZ. The Compendium of Materia Medica [originally published in the Ming Dynasty, 1578]. Beijing: People’s Medical Publishing House, 1982. [Google Scholar]
- 2. Aronson SM. Arsenic and old myths. R I Med 1994; 77: 233–4. [PubMed] [Google Scholar]
- 3. Sun HD, Ma L, Hu XC, Zhang TD. Ai‐lin 1 treated 32 cases of acute promyelocytic leukemia. Chin J Integrat Chin West Med 1992; 12: 170–1. [Google Scholar]
- 4. Shen ZX, Chen GQ, Ni JH et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL). II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997; 89: 3354–60. [PubMed] [Google Scholar]
- 5. Niu C, Yan H, Yu T et al. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow‐up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood 1999; 94: 3315–24. [PubMed] [Google Scholar]
- 6. Soignet SL, Frankel SR, Douer D et al. United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol 2001; 19: 3852–60. [DOI] [PubMed] [Google Scholar]
- 7. Zhang QY, Mao JH, Liu P et al. A systems biology understanding of the synergistic effects of arsenic sulfide and Imatinib in BCR/ABL‐associated leukemia. Proc Natl Acad Sci U S A 2009; 106: 3378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yin T, Wu YL, Sun HP et al. Combined effects of As4S4 and imatinib on chronic myeloid leukemia cells and BCR‐ABL oncoprotein. Blood 2004; 104: 4219–25. [DOI] [PubMed] [Google Scholar]
- 9. Chen Z, Chen GQ, Shen ZX et al. Expanding the use of arsenic trioxide: leukemias and beyond. Semin Hematol 2002; 39 (2 Suppl 1): 22–6. Review. [DOI] [PubMed] [Google Scholar]
- 10. Hu J, Liu YF, Wu CF et al. Long‐term efficacy and safety of all‐trans retinoic acid/arsenic trioxide‐based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A 2009; 106: 3342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mathews V et al. Single‐agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: durable remissions with minimal toxicity. Blood 2006; 107: 2627–32. [DOI] [PubMed] [Google Scholar]
- 12. Havamzadeh A et al. Treatment of acute promyelocytic leukemia with arsenic trioxide without ATRA and/or chemotherapy. Ann Oncol 2006; 17: 131–4. [DOI] [PubMed] [Google Scholar]
- 13. Zhao Y, Srivastava D. A developmental view of microRNA function. Trends Biochem Sci 2007; 32: 189–97. [DOI] [PubMed] [Google Scholar]
- 14. Griffiths‐Jones S, Grocock RJ, van Dongen S. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 2006; 34: 140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu ZB, Jian ZF, Shen SH. Global analysis of microRNA target gene expression reveals that miRNA targets are lower expressed in mature mouse and Drosophila tissues than in the embryos. Nucleic Acids Res 2007; 35: 152–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–97. [DOI] [PubMed] [Google Scholar]
- 17. Blower PE, Chung JH, Verducci JS. MicroRNAs modulate the chemo‐sensitivity of tumor cells. Mol Cancer Ther 2008; 7: 1–9. [DOI] [PubMed] [Google Scholar]
- 18. Salerno E, Scaglione BJ, Coffman FD et al. Correcting miR‐15a/16 genetic defect in New Zealand Black mouse model of CLL enhances drug sensitivity. Mol Cancer Ther 2009; 8: 268–92. [DOI] [PubMed] [Google Scholar]
- 19. Zhu H, Wu H, Liu X, Evans BR, Medina DJ, Liu CG, Yang JM. Role of MicroRNA miR‐27a and miR‐451 in the regulation of MDR1/P‐glycoprotein expression in human cancer cells. Biochem Pharmacol 2008; 76: 582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boren T, Xiong Y, Hakam A et al. MicroRNAs and their target messenger RNAs associated with ovarian cancer response to chemotherapy. Gynecol Oncol 2009; 113: 249–55. [DOI] [PubMed] [Google Scholar]
- 21. Lu Z, Liu M, Stribinskis V. MicroRNA‐21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 2008; 27: 4373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Asangani IA, Rasheed SA, Nikolova DA et al. MicroRNA‐21 (miR‐21) post‐transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008; 27: 2128–36. [DOI] [PubMed] [Google Scholar]
- 23. Gu S, Jin L, Zhang F, Sarnow P, Kay MA. Biological basis for restriction of microRNA targets to the 3′ untranslated region in mammalian mRNAs. Nat Struct Mol Biol 2009; 16: 144–50. Epub February 1, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lewis BP, Shih IH, Jones‐Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell 2003; 115: 787–98. [DOI] [PubMed] [Google Scholar]
- 25. Manikandan J, Aarthi JJ, Kumar SD, Pushparaj PN. Oncomirs: the potential role of non‐coding microRNAs in understanding cancer. Bioinformation 2008; 2: 330–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Esquela‐Kerscher A, Slack FJ. Oncomirs‐microRNAs with a role in cancer. Nat Rev Cancer 2006; 6: 259–69. [DOI] [PubMed] [Google Scholar]
- 27. Chan JA, Krichevsky AM, Kosik KS. MicroRNA‐21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005; 65: 6029–33. [DOI] [PubMed] [Google Scholar]
- 28. Esau CC. Inhibition of microRNA with antisense oligonucleotides. Methods 2008; 44: 55–60. [DOI] [PubMed] [Google Scholar]
- 29. Cheng AM, Byrom MW, Shelton J. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 2005; 33: 1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weiler J, Hunziker J, Hall J. Anti‐miRNA oligonucleotides (AMOs): ammunition to target miRNAs implicated in human disease? Gene Ther 2005; 13: 496–502. [DOI] [PubMed] [Google Scholar]
- 31. Fraser M, Leung BM, Yan X, Dan HC, Cheng JQ, Tsang BK. Tsang. p53 is a determinant of X‐linked inhibitor of apoptosis protein/Akt‐mediated chemoresistance in human ovarian cancer cells. Cancer Res 2003; 63: 7081–8. [PubMed] [Google Scholar]
- 32. Fraser M, Leung B, Jahani‐Asl A, Yan X, Thompson WE, Tsang BK. Chemoresistance in human ovarian cancer: the role of apoptotic regulators. Reprod Biol Endocrinol 2003; 1: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krichevsky AM, Gabriely G. miR‐21: a small multi‐faceted RNA. J Cell Mol Med 2009; 13: 39–53. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Selcuklu SD, Donoghue MT, Spillane C. miR‐21 as a key regulator of oncogenic processes. Biochem Soc Trans 2009; 37 (Pt 4): 918–25. Review. [DOI] [PubMed] [Google Scholar]
- 35. Lankat‐Buttgereit B, Göke R. The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol Cell 2009; 101: 309–17. [DOI] [PubMed] [Google Scholar]
- 36. Hyun Park W, Hee Cho Y, Won Jung C et al. Arsenic trioxide inhibits the growth of A498 renal cell carcinoma cells via cell cycle arrest or apoptosis. Biochem Biophys Res Commun 2003; 300 (1): 230–5. [DOI] [PubMed] [Google Scholar]