

Abstract
Signal transducers and activators of transcription‐3 (STAT3), a central cytoplasmic transcription factor, is frequently overexpressed and constitutively activated by tyrosine during malignant transformation. The overexpression and phosphorylation of STAT3 in pancreatic cancer has been described only recently, but the roles and mechanism still remain unclear. In this study, we elucidate the significance of the STAT3 signaling pathway in metastatic potentials of pancreatic cancer. We stably silence the expression of the STAT3 and p‐STAT3 by using RNA interference (RNAi) in the pancreatic cancer cell line SW1990, and then reduce its invasion capacity in vitro and metastasis capacity in vivo compared to parental cells or cells tansfected with a control vector. Furthermore, silencing SW1990 cells with the STAT3 gene by RNAi also led to a decrease of matrix metalloproteinases‐2 (MMP‐2) and vascular endothelial growth factor (VEGF) at the mRNA and protein level. Collectively, these studies suggest that activation of the STAT3 signaling pathway plays an important role in the progression of pancreatic cancer, and that silence of the STAT3 gene with RNAi may be a useful anti‐invasive therapeutic option in pancreatic cancer. (Cancer Sci 2007; 98: 1099–1106)
Pancreatic cancer remains a widespread and difficult disease to treat with an overall 5‐year survival of less than 5%. Thus, it represents one of the leading causes of cancer deaths in industrialized countries despite advances in medical therapy and surgical techniques.( 1 , 2 ) Because of the insidious and aggressive natural history of the disease, most patients have local or metastatic spread at the time of presentation that precludes a resection. Therefore, less than 10% of cases constitute candidates for surgical resection at the time of diagnosis. Even among patients undergoing a potentially curative resection, the long‐term outcome remains unsatisfactory due to early recurrence and metastatic disease.( 3 ) Effective systemic therapy capable of reversing the aggressive biology of this disease is currently not available. Thus, understanding the molecular mechanisms underlying the aggressive biology of pancreatic cancer is one of the most important issues for this disease.
STAT3, a member of the signal transduction and activation of transcription (STAT) family, is a central cytoplasmic transcription factor that is activated by phosphorylation of a conserved tyrosine residue in response to extracellular signals and oncogenes. Once tyrosine is phosphorylated, two STAT3 monomers form dimers through reciprocal phosphotyrosine–SH2 interactions. The dimers are phosphorylated STAT3 (p‐STAT3), which translocate to the nucleus and bind to cognate DNA sequences, regulate the transcription of target genes and modulate fundamental cellular processes, such as proliferation and differentiation.( 4 , 5 ) Lines of evidence demonstrate that elevated activity of STAT3 has been found frequently in a wide variety of human tumors including pancreatic cancer( 6 , 7 , 8 , 9 ) and STAT3 regulates a number of pathways important in tumorigenesis including cell cycle progression, apoptosis, tumor angiogenesis, and tumor cell evasion of the immune system.( 10 , 11 )
Recently, it was demonstrated that overexpression of p‐STAT3 correlated with the invasion and metastasis of colorectal adenocarcinoma and cutaneous squamous cell carcinoma.( 12 , 13 ) Furthermore, disrupted STAT3 signaling has been reported to lead to cell invasion through decreasing cell–cell homotypic adhesions and increasing cell motility and scattering.( 14 ) These studies suggest that the activation of STAT3 might play an important role in the invasion and metastasis of carcinomas. However, it is unknown whether and how aberrant STAT3 activation critically regulates the invasive and metastatic behavior of pancreatic tumors.
RNA interference (RNAi) refers to a sequence‐specific, post‐transcriptional gene silencing mechanism.( 15 , 16 ) Gene silencing involving RNAi entails processing long double‐stranded RNA (dsRNA) into 19‐ to 21‐nt RNAs referred to as small interfering RNA (siRNA) by an endonuclease named Dicer. Subsequently, the siRNA molecules are incorporated into the RNA‐induced silencing complex (RISC). Then, the active complexes containing the guide RNA recognize and cleave the homologous mRNAs, thereby selectively inhibiting expression of the target gene.( 17 , 18 ) Currently, RNAi has been developed into a powerful and widely used technology to elucidate mammalian gene function in invertebrates and vertebrate animals.( 19 )
To investigate the roles STAT3 plays in the invasion and metastasis of pancreatic cancer, we have employed the RNA interference technology to knock down the STAT3 gene in SW1990, a metastatic human pancreatic cancer cell line in which STAT3 is highly expressed and analyzed its effect on the invasive and metastatic behavior, and explored some involved mechanisms. The metastatic phenotypic changes resulting from the reduction of STAT3 expression were studied both in vitro and in vivo. We found that silencing of the STAT3 gene by RNAi significantly suppressed the expression of matrix metalloproteinases‐2 (MMP‐2) and vascular endothelial growth factor (VEGF), which was accompanied by marked inhibition of tumor cell invasion and metastasis in vitro and in vivo. Our results demonstrate that activation of the STAT3 signaling pathway is critical for invasive and metastatic behavior of pancreatic cancer and silencing of the STAT3 gene with RNAi may offer a novel strategy for controlling the metastatic behavior of pancreatic cancers.
Materials and Methods
Cell lines and culture conditions. Human pancreatic cancer cell line SW1990 and uterine cervix cancer cell line HeLa were purchased from the American Type Culture Collection. They were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 units/mL penicillin, and 100 µg/mL streptomycin in a humidified incubator with an atmosphere of 5% CO2:95% air at 37°C.
Cell doubling time. Tumor cells were seeded in 24‐well plates (3 × 105/well) at day 0, and a hemocytometric count was carried out daily. All measurements were carried out in triplicate. The cell doubling time was determined from the logarithmic growth phase.
Immunocytochemistry. SW1990 cells were harvested by trypsinization and seeded onto poly L‐lysine‐treated glass slides for 24 h. The cells were then fixed for 5 min in methanol at –20°C. To deactivate the endogenous peroxidase activity, sections were treated with freshly prepared 0.3% (v/v) hydrogen peroxide in methanol in darkness, for 30 min, at room temperature (RT). Non‐specific antibody binding was then blocked with 5% goat serum in phosphate‐buffered saline (PBS) for 90 min at RT. The sections were then incubated with a rabbit antihuman STAT3 polyclonal antibody (Cell Signal, Beverly, MA, USA) or rabbit polyclonal IgG controls (Vector Laboratories, Burlingame, CA, USA) in blocking buffer overnight at 4°C. The sections were then rinsed in wash buffer (PBS containing 0.5% bovine serum albumin [BSA], 0.1% Tween‐20) and incubated for 30 min with biotinylated goat antirabbit IgG (ABC staining kit; Santa Cruz, Santa Cruz, CA, USA) diluted according to the manufacturer's protocol. Next, a solution of avidin‐conjugated horseradish peroxidase (ABC staining kit) was applied for 30 min, according to the manufacturer's instructions. Peroxidase activity was developed in 0.5% (v/v) 3,3′‐diaminobenzidine hydrochloride (DAB; Sigma, St Louis, MO, USA) in PBS containing 0.03% (v/v) hydrogen peroxide for 2 min. Sections were counterstained with Harris’ hematoxylin and mounted in gelatin (Sigma).
Construction of STAT3 specific shRNA expression vector. The mammalian expression vector, pRNAT‐U6.1/Neo (Genscript, Piscataway, NJ, USA), was used for the expression of siRNA in SW1990 cells. Three coding regions corresponding to nucleotides 1819–1837, 1025–1043, 237–255 of STAT3 sequence in the GenBank (NM003150) were selected to form siRNA target sequences under the guide of siRNA designing software offered by Genscript. Then, three primer pairs were synthesized, one pair encoding nucleotide sites 1819–1837 (CTGCTAAGATTCAGTGAAA) followed by a 9 base ‘loop’ (TTCAAGAGA) and the inverted repeat (STAT3‐siRNA‐1), and the second encoding nucleotides sites 1025–1043 (GCGTCCAGTTCACTACTAA) also followed by the loop and the inverted repeat (STAT3‐siRNA‐2), and the third encoding nucleotides 237–255 (TCAGCACAATCTACGAAGA) again followed by the loop and the inverted repeat (STAT3‐siRNA‐3). Moreover, a negative control scrambled siRNA (TTCTCCGAACGTGTCACGT), which has no significant homology to mouse or human gene sequences, was taken as a control. These primer pairs were also designed to contain BamH I and Hind III restriction endonuclease sites at each end. Then, the primer pairs were annealed and subcloned into the BamH I and Hind III sites of pRNAT‐U6.1/Neo and transformed into DH5α competent cells (Promega, Madison, WI, USA). Positive clones were identified and verified by using restrictive cleavage. Positive clones were then sequenced and the recombinant plasmids were named as pRNAT‐STAT3‐siRNA‐I, pRNAT‐STAT3‐siRNA‐II, pRNAT‐STAT3‐siRNA‐III and pRNAT‐Con, respectively.
Transfection. For transient transfection, SW1990 cells were seeded in a 6‐well plate at a concentration of 2 × 105 cells/well and allowed to adhere for 24 h. Then, LipofectAMINE 2000 (Invitrogen, Carlsbad, CA, USA) was used for transfecting cells according to Invitrogen's instructions. Cells were cultured for 5–20 h and then replaced with fresh medium supplemented with 10% FBS and lyzed 48 h after transfection. For stable transfection, SW1990 cells were transfected with pRNAT‐STAT3‐RNAi‐II plasmid, which had the most obvious gene silencing effect. SW1990 cells were also transfected with the pRNAT‐Con vector. Cells were then selected with a standard medium containing 500 µg/mL G418 (Gibco, Carlsbad, CA, USA) for 14 days. Two independent transfections of pRNAT‐STAT3‐RNAi‐II expression vector were carried out in SW1990 cells, and G418‐resistant colonies were pooled to establish stable SW1990 STAT3‐RNAi transfectants (SW1990‐RNAi‐a and SW1990‐RNAi‐b). The stable transfection cells were then used for subsequent studies.
In vitro Invasion assay. Invasion assay was carried out using a specialized invasion chamber which included a 24‐well tissue culture plate with 12‐cell culture inserts (Chemicon, Temecula, CA, USA). The inserts contained an 8‐µm pore size polycarbonate membrane with a precoated thin layer of basement membrane matrix (ECMatrix). Briefly, media supplemented with 10% FBS was poured into the lower chamber as a hemoattractant. After reaching 60–70% subconfluence, tumor cells were trypsinized, re‐suspended in DMEM (1.0 × 106 cells/mL), and re‐seeded 0.3 mL into the upper chambers. After 48 h incubation at 37°C, non‐invasive cells were scrubbed off the upper surface of the membrane using a moist cotton‐tipped swab. Invasive cells on the lower surface of the membrane, which had invaded the ECMatrix and had migrated through the polycarbonate membrane, were stained by the staining solution for 20 min and rinsed with distilled water several times. Invasiveness was quantified by dissolving stained cells in 10% acetic acid and an equal amount of the dye/solution mixture was transferred onto a 96‐well plate for colorimetric reading of optical density (OD) at 560 nm. The same experiment was repeated three times and representative data are shown.
In vivo lung metastasis assay. Specific pathogen‐free male athymic BALB/c nude mice were obtained from the Animal Center of Chinese Academy of Science (Shanghai, China). The mice were housed in laminar flow cabinets under specific pathogen‐free conditions and used when they were 6–8 weeks old. The use of animals in this study complies with the Guide for the Care and Use of Laboratory Animals (NIH publication no. 86–23, revised 1985) and the current Chinese regulations and standards on the use of Laboratory Animals.
To evaluate experimental lung metastasis, 0.2 mL of the tumor‐cell suspensions (5 × 105 cells/mouse) were injected into the lateral tail veins of anesthetized nude mice. The mice were killed 21 days after tumor‐cell injection or when they had become moribund. Their lungs were then removed and fixed in Bouin's solution for 24 h to permit differentiation of the neoplastic lesions from the organ parenchyma. The lung metastases were counted in a double‐blind manner with the aid of a dissecting microscope as described previously,( 20 ) and the metastasis nodus were proved by histopathological examination with H&E staining. For H&E staining, nude mice lung tissues were fixed in 4% formaldehyde for 24–48 h and then embedded in paraffin. Five‐micrometer sections were cut and stained with hematoxylin and eosin (HE).
To confirm the expression of STAT3, MMP2 and VEGF in the in vivo study using nude mice, we carried out the immunohistochemistry analysis as follows: formalin‐fixed paraffin‐embedded tissue sections of 5‐µm thickness were dewaxed in 100% xylene and rehydrated by serial incubations in 100, 90, and 80% ethanol, followed by PBS. Antigen retrieval was carried out by microwaving slides in 10 mM sodium citrate buffer (pH 6.0) for 1 min at full power followed by 9 min at medium power, according to the manufacturer's instructions. To deactivate the endogenous peroxidase activity, sections were treated with freshly prepared 0.3% (v/v) hydrogen peroxide in methanol in darkness for 30 min, at RT. Non‐specific antibody binding was then blocked with 5% goat serum in PBS for 90 min at RT. The sections were then incubated with rabbit antihuman STAT3 polyclonal antibody (Cell Signal), mouse antihuman MMP‐2 monoclonal antibody (Maixin, Fuzhou, China) rabbit antihuman VEGF polyclonal antibody (Maixin) or rabbit polyclonal IgG controls (Vector Laboratories) in blocking buffer overnight at 4°C. The sections were then rinsed in wash buffer (PBS containing 0.5% BSA, 0.1% Tween‐20) and incubated for 30 min with biotinylated goat antirabbit IgG (ABC staining kit, Santa Cruz) diluted according to the manufacturer's protocol. Next, a solution of avidin‐conjugated horseradish peroxidase (ABC staining kit) was applied for 30 min, according to the manufacturer's instruction. Peroxidase activity was developed in 0.5% (v/v) 3,3′‐diaminobenzidine hydrochloride (DAB, Sigma) in PBS containing 0.03% (v/v) hydrogen peroxide for 2 min. Sections were counterstained with Harris’ hematoxylin and mounted in gelatin (Sigma).
Reverse transcription‐polymerase chain reaction. Total RNA extraction from tumor cells was carried out with Trizol Reagent (Life Technologies, Rockville, MD, USA). Then, 2 µg of total RNA was reverse‐transcribed with the first strand cDNA synthesis kit (Promega) to synthesize cDNA samples. Subsequently, 2 µL of cDNA product was then subjected to polymerase chain reaction (PCR) amplification with Taq DNA polymerase (Sangon, Shanghai, China) on a thermal cycler using the following primers. The oligo‐nucleotide primers for STAT3 was constructed under the help of primer‐design software ‘Primer Premier 5.0’. The oligo‐nucleotide primers for MMP‐2, VEGF and β‐actin were constructed on the basis of the published sequence. The PCR primers used to detect each factor were as follows: STAT3, sense strand 5′‐CACCAAGCGAGGACTGAGCAT‐3′, antisense strand 5′‐GCCAGACCCAGAAGGAGAAGC‐3′, with a product length of 149 bp; MMP‐2, sense strand 5′‐GTGCTGAAGGACACACTAAAGAAGA‐3′, antisense strand 5′‐TTGCCATCCTTCTCAAAGTTGTAGG‐3′, with a product length of 605 bp;( 21 ) VEGF, sense strand 5′‐CCTGGTGGACATCTTCCAGGAGTACC‐3′, antisense strand 5′‐GAAGCTCATCTCTCCTATGTGCTGGC‐3′, with a product length of 196 bp;( 22 )β‐actin, sense strand 5′‐ATCTGGCACCACACCTTCTACAATGAGCTGCG‐3′, antisense strand 5′‐CGTCATACTCCTGCTTGCTGATCCACATCTGC‐3′, with a product length of 838 bp.( 23 ) The PCR conditions were as follows: one cycle of denaturing at 94°C for 5 min, followed by 30 cycles of 94°C for 1 min, 60°C for 1 min and 72°C for 1 min, before a final extension at 72°C for 10 min. The PCR products were loaded onto 2% agarose gels and visualized with ethidium bromide under UV light. This experiment was carried out three times and representative data are shown.
Western blot. Whole‐cell protein extracts and nuclear protein extracts from tumor cells were prepared with RIPA Lysis Buffer (Santa Cruz) and a nuclear extract kit (Active Motif, Carlsbad, CA, USA), according to the manufacturer's instructions, respectively. Protein concentrations were determined using a Bio‐Rad assay kit (Bio‐Rad, Hercules, CA, USA). Lysates containing 100 µg of protein were mixed with loading‐buffer with 5%β‐mercaptoethanol, and heated for 5 min at 100°C. Samples were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred onto nitrocellulose membranes by semidry blotting. Membranes were incubated in blocking buffer (1 × TBS, 0.1% Tween 20, and 5% non‐fat dry milk) for 1 h at room temperature, followed by hybridization with antip‐STAT3[tyr‐705] antibody (Cell Signal, 1:1000 dilution), anti‐STAT3 antibody (Cell Signal, 1:1000 dilution), anti‐MMP‐2 antibody (Santa Cruz, 1:500 dilution), anti‐VEGF antibody (Santa Cruz, 1:500 dilution) or antiβ‐actin antibody (Labvision, Fremont, CA, USA, 1:100 dilution), at 4°C overnight. After three washes in TBS/0.1% Tween 20, the membranes were hybridizated with a horseradish peroxidase‐conjugated secondary antibody rabbit IgG (Santa Cruz, 1:5000 dilution) for 1 h at RT. After three washes in TBS/0.1% Tween 20, signals were detected by chemiluminescence using the Western Blotting Luminol Reagent (Santa Cruz). The same experiments were carried out three times and representative data are shown.
Electrophoretic mobility shift analysis. Nuclear protein extracts were prepared with a nuclear extract kit (Active Motif). Protein concentrations were determined using a Bio‐Rad assay kit (Bio‐Rad). STAT3 transcription factor activities were assessed by chemiluminescent electrophoretic mobility shift analysis (EMSA) Kit (Pierce, Rockford, IL, USA), according to the manufacturer's protocol. Briefly, nuclear protein extracts (10 µg) were incubated in a final volume of 20 µL of 10 × binding buffer, 50% Glycerol, 100 mM MgCl2, 1 µg/µL Poly (dIdC), 1% NP‐40 with the biotin end‐labeled or unlabeled double‐stranded STAT3 consensus‐binding motif 5′‐GATCCTTCTGGGAATTCCTAGATC‐3′ (Sangon) for 20 min at RT. The protein‐DNA complexes were electrophoresed on a 6% polyacrylamide gel in 0.5× TBE buffer and transferred onto Nylon membranes by semidry blotting. After cross‐linking transferred DNA to nylon membranes with UV‐light cross‐linker (Pharmacia), biotin‐labeled DNA was detected by chemiluminescence. This experiment was repeated three times and representative data are shown.
Statistical analysis. All data are expressed as mean ± standard deviation. Statistical significance of the in vitro data was determined by Student's t‐test (two‐tailed), while the significance of the differences between the median values of the in vivo data was determined using the two‐tailed Mann–Whitney test. Statistical significance was assigned if P < 0.05.
Results
STAT3 expression in SW1990 cells. STAT3 expressions in SW1990 cells and HeLa cells were determined by Western blot. STAT3 expression in SW1990 cells is almost as high as that in HeLa cells (a putative STAT3 overexpression tumor cell line), indicating that STAT3 was also up‐regulated in SW1990 cells (Fig. 1a). Immunocytochemistry showed that STAT3 was localized in the cytoplasm of SW1990 cells (Fig. 1b).
Figure 1.

Expression of signal transducers and activators of transcription 3 (STAT3) in SW1990 cells and HeLa cells. (a) STAT3 expression was determined by Western blot analysis. Detection of β‐actin was used as a loading control. HeLa cell line served as a positive control of STAT3 expression. (b) STAT3 distribution in SW1990 cells was examined by immunocytochemistry (400 ×), the buffy particle corresponded for STAT3 (shown by arrow).
Screening and selection of the most effective STAT3 specific shRNA expression vector targeting the STAT3 gene. To test the comparative inhibitory effects of STAT3 specific shRNA expression vector on STAT3 expression, SW1990 cells were tansfected transiently by the recombinant plasmids. The levels of STAT3 and p‐STAT3 protein were detected by Western blot 48 h after shRNA expression vector transfection. The results showed that the transfection of pRNAT‐Con did not reduce STAT3 and p‐STAT3 expression, while the transfection of pRNAT‐STAT3‐siRNA‐I, pRNAT‐STAT3‐siRNA‐II and pRNAT‐STAT3‐siRNA‐III significantly inhibited the STAT3 and p‐STAT3 expression in SW1990 cells. More specifically, the most obvious gene silencing effect was observed when pRNAT‐STAT3 siRNA‐II was applied, and in this case the expression of STAT3 mRNA and protein declined by about 78.5% and 71.8%, respectively (Fig. 2). Therefore, pRNAT‐STAT3‐siRNA‐II was chosen for stable transfection.
Figure 2.

Effects of signal transducers and activators of transcription 3 (STAT3) specific shRNA expression vector on STAT3 expression. (a) Reverse transcription‐polymerase chain reaction (RT‐PCR) analysis. The RNA samples (2 µg in each) extracted from SW1990 cells, SW1990 cells transfected with pRNAT‐Con, pRNAT‐STAT3‐siRNA‐I, pRNAT‐STAT3‐siRNA‐II and pRNAT‐STAT3‐siRNA‐III were subjected to RT‐PCR for STAT3 and β‐actin mRNAs as described in Materials and Methods. RT‐PCR for β‐actin was carried out in parallel to show an equal amount of total RNA in the sample. (b) Western blot analysis. Cellular whole and nuclear protein extracts (100 µg in each) were prepared from SW1990 cells, SW1990 cells transfected with pRNAT‐Con, pRNAT‐STAT3‐siRNA‐I, pRNAT‐STAT3‐siRNA‐II and pRNAT‐STAT3‐siRNA‐III. The expression of STAT3 protein was determined using Western blot analysis with an anti‐STAT3 antibody. The expression of p‐STAT3 protein was determined by hybridizing the same membrane filter with an antip‐STAT3 antibody. The levels of β‐actin expression were determined as a control for equivalent protein loading. Results shown are for one representative experiment of three.
Silencing of STAT3 expression by stable transfection of pRNAT‐STAT3‐siRNA‐II vector. To further evaluate the effects of STAT3 gene knockdown, SW1990 cells were stably transfected by pRNAT‐STAT3‐siRNA‐II vector. Stable transfectants containing the pRNAT‐STAT3‐siRNA‐II or pRNAT‐Con were selected in the presence of G418. Total and nuclear protein extracts were prepared from parental SW1990, pRNAT‐Con, and pRNAT‐STAT3‐siRNA‐II transfected cells, and the levels of STAT3 and p‐STAT3 protein expression in these cells were determined by Western blot. Densitometric measurements indicated that the levels of STAT3 mRNA expression in two of the representative transfectants containing pRNAT‐STAT3‐siRNA‐II (SW1990‐RNAi‐a and SW1990‐RNAi‐b) were decreased 96.8% and 92.5% as compared to parental SW1990 cells (Fig. 3a). Moreover, enforced expression of pRNAT‐STAT3‐siRNA‐II in SW1990 cells significantly suppressed STAT3 protein expression as determined using by Western blot analysis (Fig. 3b), and significantly suppressed STAT3 DNA binding activity as determined using EMSA (Fig. 3c). These results indicated that table transfection of pRNAT‐STAT3‐siRNA‐II vector silenced STAT3 expression.
Figure 3.

Effects of stable transfection of pRNAT‐STAT3‐siRNA‐II vector on signal transducers and activators of transcription 3 (STAT3) expression. (a) Reverse transcription‐polymerase chain reaction (RT‐PCR) analysis. The RNA samples (2 µg in each) extracted from SW1990 cells, SW1990 cells transfected with a control vector (SW1990‐Con), and SW1990 cells transfected with STAT3‐RNAi (SW1990‐RNAi‐a and SW1990‐RNAi‐b) were subjected to RT‐PCR for STAT3 and β‐actin mRNAs as described in Materials and Methods. RT‐PCR for β‐actin was carried out in parallel to show an equal amount of total RNA in the sample. (b) Western blot analysis. Cellular whole and nuclear protein extracts (100 µg in each) were prepared from SW1990 cells, SW1990 cells transfected with a control vector (SW1990‐Con), and SW1990 cells transfected with STAT3‐RNAi (SW1990‐RNAi‐a and SW1990‐RNAi‐b). The expression of STAT3 protein was determined using Western blot analysis with an anti‐STAT3 antibody. The expression of p‐STAT3 protein was determined by hybridizing the same membrane filter with an antip‐STAT3 antibody. The levels of β‐actin expression were determined as a control for equivalent protein loading. (c) STAT3 DNA‐binding activity. Cell nuclear protein extracts (10 µg in each) were prepared from SW1990 cells, SW1990 cells transfected with a control vector (SW1990‐Con), and SW1990 cells transfected with STAT3‐RNAi (SW1990‐RNAi‐a and SW1990‐RNAi‐b) and subjected to electrophoretic mobility shift analysis (EMSA) using biotin end‐labeled oligonucleotide probes containing a consensus‐binding motif for STAT3. Results shown are for one representative experiment of three.
Inhibition of SW1990 cells invasion in vitro after STAT3 silencing. To evaluate the effect of STAT3 silencing on the invasiveness of SW1990 cells, an in vitro invasion system was set up as described in Materials and Methods. When compared with parental cells or cells transfected with pRNAT‐Con, pRNAT‐STAT3‐siRNA‐II transfected cells showed a substantial reduction in invasive capacity (Fig. 4a). Invasion of SW1990‐RNAi‐a and SW1990‐RNAi‐b cells was reduced to 27.3% and 28.3% from their respective parental cells (Fig. 4b).
Figure 4.
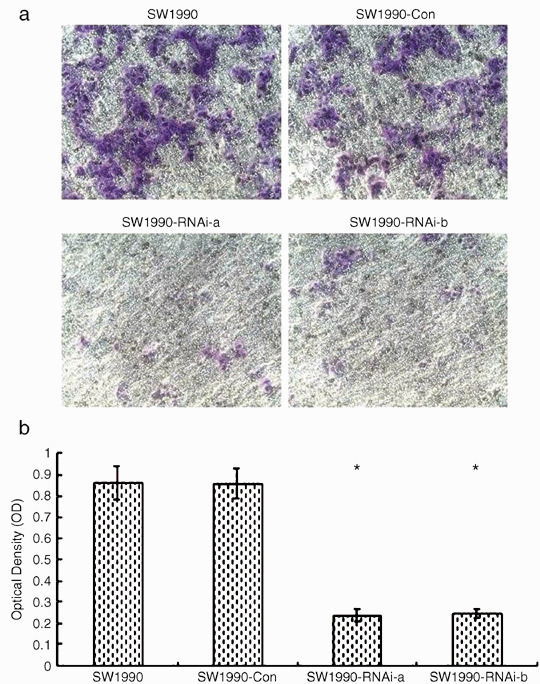
Effects of silence of signal transducers and activators of transcription 3 (STAT3) gene on SW1990 cell invasion in vitro. SW1990‐RNAi‐a, SW1990‐RNAi‐b, SW1990‐Con and the parental cell lines were seeded into the upper compartments of invasion chambers. Cells were allowed to invade for 48 h at 37°C. The tumor cells that invaded the ECMatrix and migrated through the polycarbonate membrane were stained by the staining solution and dissolved in 10% acetic acid. The dye/solution mixture was transferred onto a 96‐well plate for colorimetric reading of optical density (OD) at 560 nm. The number of migrated cells that penetrated through the ECMatrix‐coated filters was expressed as the OD at 560 nm. The standard deviation bars represent replicates within the assay. *P < 0.001 compared with that of parental SW1990 cells. Results shown are for one representative experiment of three.
Inhibition of SW1990 cells metastasis in vivo after STAT3 silencing. Parental SW1990 cells and SW1990 cells transfected with pRNAT‐STAT3‐siRNA‐II or pRNAT‐Con vector were used to evaluate experimental metastases. A total of 0.2 mL tumor cell suspension (5 × 105 cells/mouse) was injected into the lateral tail veins of nude mice. The mice were killed 21 days after tumor‐cell injection or when they had become moribund. As shown in Table 1, SW1990 and SW1990‐Con cells produced a large number of metastatic nodules in all injected mice. In contrast, the pRNAT‐STAT3‐siRNA‐II transfected cells did not produce metastatic nodules in any mice injected with these tumor cells. The lung metastasis nodus were proved by histopathological examination with H&E staining, which was shown in Figure 5(a,b). To determine whether STAT3, MMP‐2 and VEGF is expressed in the lung metastasis nodus of nude mice, we detected the level of STAT3, MMP‐2 and VEGF expression using immunohistochemistry analyses with anti‐STAT3, anti‐MMP‐2 and anti‐VEGF antibody. Immunohistochemistry analyses revealed that STAT3, MMP‐2 and VEGF were overexpressed in the lung metastasis nodus of nude mice (Fig. 5c).
Table 1.
Effects of silence of STAT3 gene on SW1990 cell metastasis in vivo
| Tumor cell lines | Experimental lung metastasis | |
|---|---|---|
| Incidence | Median (range) | |
| SW1990 | 6/6 | 55 (38–84) |
| SW1990‐Con | 6/6 | 61 (42–97) |
| SW1990‐RNAi‐a | 0/6 | All 0* |
| SW1990‐RNAi‐b | 0/6 | All 0* |
P < 0.001 compared with parental SW1990 cells
Figure 5.

Effects of silence of signal transducers and activators of transcription 3 (STAT3) gene on SW1990 cell metastasis in vivo. In total, 5 × 105 SW1990 cells, SW1990 cells transfected with a control vector (SW1990‐Con), and SW1990 cells transfected with STAT3‐RNAi (SW1990‐RNAi‐a and SW1990‐RNAi‐b) were injected i.v. into groups of nude mice (n = 6). The mice were killed on day 21 or when mice became moribund. The number of experimental lung metastases was determined with the aid of a dissecting microscope. Results were expressed as the median number and range of lung metastasis nodules. *P < 0.001 compared with that of parental SW1990 cells. Results shown here are for one representative experiment of three. (a) H&E staining of formalin‐fixed, paraffin‐embedded lung tissue of nude mice (100 ×). The metastasis nodus are indicated by arrows. (b) H&E staining of formalin‐fixed, paraffin‐embedded metastasis nodus of lung tissue of nude mice (400 ×). (c) In SW1990 cells injected nude mice, STAT3, matrix metalloproteinases‐2 (MMP‐2) and vascular endothelial growth factor (VEGF) distribution in metastasis nodus of lung tissue was examined by immunohistochemistry (400 ×), the buffy particle corresponded for STAT3, MMP‐2 and VEGF. Immunohistochemistry showed that STAT3, MMP‐2 and VEGF were distributed in cytoplasm.
To determine whether the inhibition of tumor metastases by SW1990‐RNAi cells in vivo was due to different growth rates in vitro, we compared their doubling time with that of control cells in cultures. The mean doubling time for parental and pRNAT‐Con‐transfected cells was 13.2 and 14.3 h, respectively, while the mean doubling time for STAT3‐RNAi‐transfected cells (SW1990‐RNAi‐a and b) was 13.5 and 14.6 h, respectively. Therefore, we found that the cell doubling time for SW1990 and SW1990‐Con cells was indistinguishable from that for the SW1990‐RNAi cells.
Inhibition of MMP‐2 and VEGF expression of SW1990 cells after STAT3 silencing. STAT3 activation contributes to oncogenesis through regulation of its target genes. To determine the effect of down‐regulation of STAT3 on metastasis‐related target gene expression, we assayed for the expression of MMP‐2 and VEGF by RT‐PCR, both of which are directly involved in tumor cell invasion and metastasis. As shown in Figure 6(a), the expression of MMP‐2 and VEGF mRNAs in SW1990 cells were significantly inhibited after STAT3 silencing. The densitometric analyses revealed MMP‐2 relative expression of SW1990‐RNAi‐a and SW1990‐RNAi‐b cells was reduced to 27% and 34% of that of parental SW1990 cells, respectively. VEGF relative expression of SW1990‐RNAi‐a and SW1990‐RNAi‐b cells was reduced to 45% and 49% of that of parental SW1990 cells, respectively. A similar inhibitory effect on protein levels is shown in Figure 6(b). These results demonstrated that the blockade of STAT3 signaling was able to inhibit SW1990 cell metastatic potentials by regulating MMP‐2 and VEGF.
Figure 6.
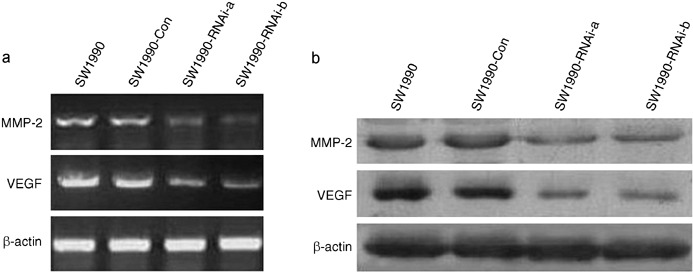
Effects of silence of signal transducers and activators of transcription 3 (STAT3) gene on the expression of matrix metalloproteinases‐2 (MMP‐2) and vascular endothelial growth factor (VEGF). (a) Reverse transcription‐polymerase chain reaction (RT‐PCR) analysis. The RNA samples (2 µg in each) extracted from SW1990 cells, SW1990 cells transfected with a control vector (SW1990‐Con), and SW1990 cells transfected with STAT3‐RNAi (SW1990‐RNAi‐a and SW1990‐RNAi‐b) were subjected to RT‐PCR for MMP‐2, VEGF, and β‐actin mRNAs as described in Materials and Methods. RT‐PCR for β‐actin was carried out in parallel to show an equal amount of total RNA in the sample. (b) Western blot analysis. Cellular whole protein extracts (100 µg in each) were prepared from SW1990 cells, SW1990 cells transfected with a control vector (SW1990‐Con), and SW1990 cells transfected with STAT3‐RNAi (SW1990‐RNAi‐a and SW1990‐RNAi‐b). The expression of MMP‐2 protein was determined using Western blot analysis with an anti‐MMP‐2 antibody. The expression of VEGF protein was determined by hybridizing the same membrane filter with an anti‐VEGF antibody. The levels of β‐actin expression were determined as a control for the equivalent protein loading. Results shown are for one representative experiment of three.
Discussion
Signal transducers and activators of transcription 3 is a key signal transduction protein that mediates signaling by numerous cytokines, peptide growth factors, and oncoproteins. There is much evidence that implicates the important roles for STAT3 in cell proliferation and survival in diverse human cancers, including pancreatic cancer. However, there are few data on its potential contribution to tumor invasion and metastasis. For a better understanding on the functions of STAT3 in the invasion and metastasis of pancreatic cancer, we have set up to investigate the effect and mechanism of the blockade of the STAT3 signaling pathway on the invasion and metastasis of human high metastatic pancreatic cancer cells SW1990.
RNA interference represents a promising new experimental tool for the analysis of gene function and has rapidly progressed to become a key gene therapy technique in mammalian systems. Compared with traditional gene therapy methods, RNAi possesses the advantages of high specificity and high efficiency in down‐regulating gene expression, thus providing a new approach to the treatment of diseases including cancer.( 24 , 25 ) siRNA can be transfected into mammalian cells by various means. First, chemically synthesized siRNA can be introduced into cells.( 26 ) But variable transfection efficiency and the transient nature of silencing by exogenously delivered siRNA limit the applicability of chemically synthesized siRNA.( 27 ) To circumvent the disadvantage of chemically synthesized siRNA, short hairpin RNAs (shRNAs) are used. The transcribed shRNA from stably transfected plasmids is processed to siRNAs by cellular machinery in order to achieve stable RNAi. Studies from different cell types and against various genes have demonstrated the efficacy of endogenously expressed shRNA.( 28 , 29 , 30 )
In the present study, we used shRNAs targeting STAT3 to silence the expression of STAT3 in human high metastatic pancreatic cancer cells SW1990. We selected three shRNAs and cloned them into the expression plasmid pRNAT‐U6.1/Neo. Because this vector contains a U6 promoter upstream of the inserted DNA sequence, the shRNAs can be effectively expressed after being transfected into cancer cells. Our results demonstrated that recombinant plasmid pRNAT‐STAT3‐RNAi‐II were more effective in suppressing STAT3 expression in SW1990 cells. Therefore, we used recombinant plasmid pRNAT‐STAT3‐RNAi‐II to generate two SW1990 cell lines, SW1990‐RNAi‐a and SW1990‐RNAi‐b, which constitutively knocked down the expression of STAT3. Both SW1990‐RNAi‐a and SW1990‐RNAi‐b showed significant decreases in STAT3 expression. Attenuation of STAT3 changed the invasive and metastatic behavior of human SW1990 cells in both in vitro and in vivo settings. Invasion assay revealed that invasion ability of SW1990‐RNAi‐a and SW1990‐RNAi‐b cells was reduced to 27.3% and 28.3% of their parental SW1990 cells, respectively. Metastasis ability of SW1990‐RNAi‐a and SW1990‐RNAi‐b cells also significantly decreased compared to parental cells by using nude mice experimental metastasis analysis.
The inhibitory mechanism in the invasion and metastasis after STAT3 silence with RNAi is considered to be down‐regulation of genes related with proteolysis and angiogenesis. MMPs, which degrade the various ECM components, are believed to play key roles in the local invasiveness and metastasis in pancreatic cancer.( 31 , 32 , 33 ) Recently, some studies have found that STAT3 signaling directly regulates MMP‐2 expression, tumor invasion, and metastasis in the metastatic melanoma cells and proved MMP‐2 to be a target gene of STAT3.( 34 , 35 ) In our present study, the silence of STAT3 also reduced the mRNA and protein expression of MMP‐2 in SW1990 cells. Cancer cells also need to acquire angiogenesis phenotypes besides proteolysis for invasion and metastasis in vivo. VEGF, a potent angiogenic mitogen, is associated with poor survival and prognosis for pancreatic cancer. Furthermore, it is correlated with the increase in the microvessel density, local invasion, liver metastasis, and early recurrence after curative resection.( 36 , 37 , 38 ) A recent study has reported that VEGF is a downstream target gene of STAT3 and stable transfection of dominant‐negative STAT3 (DN‐STAT3) decreases VEGF expression of the pancreatic cancer cell line FG.( 39 ) In this work, we also found that the silence of STAT3 significantly decreased the mRNA and protein expression of VEGF in SW1990 cells.
In conclusion, the present study indicates that siRNA targeting of STAT3 mRNA via a plasmid based system effectively sustains knock‐down of the STAT3 gene expression in SW1990 cells. The impaired STAT3 expression results in reduced SW1990 cell invasiveness in vitro and metastasis in vivo through down‐regulation MMP‐2 and VEGF. Targeting STAT3 activation with RNAi may offer a novel strategy for preventing invasion and metastasis of pancreatic cancer.
References
- 1. Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer 2002; 2: 897–909. [DOI] [PubMed] [Google Scholar]
- 2. Li DH, Xie KP, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet 2004; 363: 1049–57. [DOI] [PubMed] [Google Scholar]
- 3. Neoptolemos JP, Cunningham D, Friess H et al. Adjuvant therapy in pancreatic cancer: historical and current perspectives. Ann Oncol 2003; 14: 675–92. [DOI] [PubMed] [Google Scholar]
- 4. Darnell JE Jr. STATs and gene regulation. Science 1997; 277: 1630–5. [DOI] [PubMed] [Google Scholar]
- 5. Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene 2000; 19: 2474–88. [DOI] [PubMed] [Google Scholar]
- 6. Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, Endo S, Johnson DE, Huang L, He Y, Kim JD. Constitutive activation of STAT3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci 2000; 97: 4227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dhir R, Ni Z, Lou W, Demiguel F, Grandis JR, Gao AC. STAT3 activation in prostatic carcinomas. Prostate 2002; 51: 241–6. [DOI] [PubMed] [Google Scholar]
- 8. Niu G, Bowman T, Huang M et al. Roles of activated Src and STAT3 signaling in melanoma tumor cell growth. Oncogene 2002; 21: 7001–10. [DOI] [PubMed] [Google Scholar]
- 9. Scholz A, Heinze S, Detjen KM et al. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003; 125: 891–905. [DOI] [PubMed] [Google Scholar]
- 10. Haura EB, Turkson J, Jove R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol 2005; 2: 315–24. [DOI] [PubMed] [Google Scholar]
- 11. Hsieh FC, Cheng G, Lin J. Evaluation of potential STAT3‐regulated genes in human breast cancer. Biochem Biophys Res Commun 2005; 335: 292–9. [DOI] [PubMed] [Google Scholar]
- 12. Kusaba T, Nakayama T, Yamazumi K et al. Expression of p‐STAT3 in human colorectal adenocarcinoma and adenoma; correlation with clinicopathological factors. J Clin Pathol 2005; 58: 833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suiqing C, Min Z, Lirong C. Overexpression of phosphorylated‐STAT3 correlated with the invasion and metastasis of cutaneous squamous cell carcinoma. J Dermatol 2005; 32: 354–60. [DOI] [PubMed] [Google Scholar]
- 14. Rivat C, De Wever O, Bruyneel E, Mareel M, Gespach C, Attoub S. Disruption of STAT3 signaling leads to tumor cell invasion through alterations of homotypic cell‐cell adhesion complexes. Oncogene 2004; 23: 3317–27. [DOI] [PubMed] [Google Scholar]
- 15. Hannon GJ. RNA interference. Nature 2002; 418: 244–51. [DOI] [PubMed] [Google Scholar]
- 16. Lee SH, Sinko PJ. siRNA_Getting the message out. Eur J Pharm Sci 2006; 27: 401–10. [DOI] [PubMed] [Google Scholar]
- 17. Hammond SM, Caudy AA, Hannon GJ. Post‐transcriptional gene silencing by double‐stranded RNA. Nat Rev Genet 2001; 2: 110–19. [DOI] [PubMed] [Google Scholar]
- 18. Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21‐ and 22‐nucleotide RNAs. Genes Dev 2001; 15: 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elbashir SM, Harborth J, Weber K, Tuschl T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 2002; 26: 199–213. [DOI] [PubMed] [Google Scholar]
- 20. Huang S, Jean D, Luca M, Tainsky M, Bar‐Eli M. Loss of AP‐2 results in downregulation of c‐KIT and enhancement of melanoma tumorigenicity and metastasis. EMBO J 1998; 17: 4358–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uchima Y, Sawada T, Nishihara T, Maeda K, Ohira M, Hirakawa K. Inhibition and mechanism of action of a protease inhibitor in human pancreatic cancer cells. Pancreas 2004; 29: 123–31. [DOI] [PubMed] [Google Scholar]
- 22. Brown KJ, Maynes SF, Bezos A, Maguire DJ, Ford MD, Parish CR. A novel in vitro assay for human angiogenesis. Lab Invest 1996; 75: 539–55. [PubMed] [Google Scholar]
- 23. Zhu Z, Yao J, Wang F, Xu Q. TNF‐alpha and the phenotypic transformation of human peritoneal mesothelial cell. Chin Med J (Engl) 2002; 115: 513–17. [PubMed] [Google Scholar]
- 24. Leung RK, Whittaker PA. RNA interference: from gene silencing to gene‐specific therapeutics. Pharmacol Ther 2005; 107: 222–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uprichard SL. The therapeutic potential of RNA interference. FEBS Lett 2005; 579: 5996–6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21‐and 22‐nucleotide RNAs. Genes Dev 2001; 15: 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat Rev Genet 2002; 3: 737–47. [DOI] [PubMed] [Google Scholar]
- 28. Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence‐specific silencing in mammalian cells. Genes Dev 2002; 16: 948–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paddison PJ, Caudy AA, Hannon GJ. Stable suppression of gene expression by RNAi in mammalian cells. Proc Natl Acad Sci USA 2002; 99: 1443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002; 19: 550–3. [DOI] [PubMed] [Google Scholar]
- 31. Bloomstom M, Zeros EE, Rosemurgy AS 2nd. Matrix metalloproteinases and their role in pancreatic cancer: a review of preclinical studies and clinical trials. Ann Surg Oncol 2002; 9: 668–74. [DOI] [PubMed] [Google Scholar]
- 32. Matsuyama Y, Takao S, Aikou T. Comparison of matrix metalloproteinase expression between primary tumors with or without liver metastasis in pancreatic and colorectal carcinomas. J Surg Oncol 2002; 80: 105–10. [DOI] [PubMed] [Google Scholar]
- 33. Tan X, Egami H, Ishikawa S et al. Involvement of matrix metalloproteinase‐7 in invasion‐metastasis through induction of cell dissociation in pancreatic cancer. Int J Oncol 2005; 26: 1283–9. [PubMed] [Google Scholar]
- 34. Xie TX, Wei D, Liu M et al. STAT3 activation regulates the expression of matrix metalloproteinase‐2 and tumor invasion and metastasis. Oncogene 2004; 23: 3550–60. [DOI] [PubMed] [Google Scholar]
- 35. Xie TX, Huang FJ, Aldape KD et al. Activation of STAT3 in human melanoma promotes brain metastasis. Cancer Res 2006; 66: 3188–96. [DOI] [PubMed] [Google Scholar]
- 36. Itakura J, Ishiwata T, Friess H et al. Enhanced expression of vascular endothelial growth factor in human pancreatic cancer correlates with local disease progression. Clin Cancer Res 1997; 3: 1309–16. [PubMed] [Google Scholar]
- 37. Seo Y, Baba H, Fukuda T, Takashima M, Sugimachi K. High expression of vascular endothelial growth factor is associated with liver metastasis and a poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer 2000; 88: 2239–45. [DOI] [PubMed] [Google Scholar]
- 38. Niedergethmann M, Hildenbrand R, Wostbrock B et al. High expression of vascular endothelial growth factor predicts early recurrence and poor prognosis after curative resection for ductal adenocarcinoma of the pancreas. Pancreas 2002; 25: 122–9. [DOI] [PubMed] [Google Scholar]
- 39. Wei D, Le X, Zheng L et al. STAT3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003; 22: 319–29. [DOI] [PubMed] [Google Scholar]