

Abstract
The rasH2 transgenic mice carry human c‐Ha‐ras proto‐oncogene, and are highly susceptible to chemical carcinogenesis. Previous studies showed that the mutation of c‐Ha‐ras induced by DMBA in the tumors of rasH2 were detected only in transgenes. To examine if the difference between the codons of the c‐Ha‐ras gene in human and mouse contributed to the tissue‐specific sensitivity to DMBA, we generated a line of transgenic mice, mras, carrying mouse c‐Ha‐ras genome with its own promoter. Western blot analysis showed that the protein expression of H‐RAS in the skin was increased in both rasH2 and mras compared with wild‐type. Chemical skin carcinogenesis was induced by DMBA and TPA. In rasH2 mice, the latency of tumor formation was shorter than wild‐type littermates. Both the number and the volume of skin tumors were increased in rasH2 than those of wild‐type. However, in mras mice, enhancement of tumor formation was not observed as compared with wild‐type. The mean number of tumors and the latency of tumor development was almost the same between mras and wild‐type littermates. Mutational analysis showed only A to T transversion in human c‐Ha‐ras transgenes at codon 61 but not in murine endogenous c‐Ha‐ras gene in the tumors of rasH2. In the tumors of wild‐type littermates and mras, A to T transversion in murine c‐Ha‐ras at codon 61 were detected. These results indicate that the differences in the codon of the c‐Ha‐ras gene between mouse and human might contribute to the tissue‐specific sensitivity of DMBA. (Cancer Sci 2006; 97: 842–847)
Abbreviations:
- DMBA
7,12‐dimethylbenz[a]anthracene
- GTP
guanosine triphosphate;
- MNU, N‐methyl‐N‐nitrosurea; PCR
polymerase chain reaction
- SPF, specific‐pathogen free; TPA
12‐O‐tetradecanoylphorbol‐13‐acetate;
Human cancers develop through a multistep process that involves the accumulation of genetic mutations.( 1 ) Based on work in experimental animal model systems, the carcinogenesis process can be divided both operationally and mechanistically into the initiation, promotion, and progression stages.( 2 , 3 ) The initiation stage is an irreversible event in which carcinogens damage DNA and induce mutations in critical genes in target stem cells. During the promotion stage, initiated cells undergo selective clonal expansion due to the acquisition of a proliferative advantage, and/or of the ability to evade growth inhibitory or apoptotic signals.( 3 ) This clonal expansion of initiated cells enhances the probability of additional genetic mutations that might lead to the development of malignant lesions.( 2 ) Malignant neoplastic cells possess an indefinite proliferative capability, thus being able to elude a commitment to terminal differentiation and postmitotic quiescence that normally regulates tissue homeostasis in an organism. In order to achieve a proliferative autonomy, malignant neoplastic cells have to either switch to an autocrine production of mitogenic factors or acquire activating mutations within the components of the signal transduction pathways that mediate mitogenic signaling. An example of this is the activating mutations of c‐Ha‐ras.( 4 ) The RAS proteins have essential roles in controlling the activity of several crucial signaling pathways that regulate normal cellular proliferation. The RAS proteins are members of a large superfamily of low‐molecular‐weight GTP‐binding proteins that regulates proliferation, differentiation, and cell survival. Three members of the RAS family – H‐RAS, K‐RAS and N‐RAS – are found to be activated by mutation in human tumors( 5 ) and are postulated to be involved in tumor development. Almost all RAS activation in tumors is accounted for by mutations at codons 12, 13 and 61.( 6 ) These mutations all compromise the GTPase activity of RAS, preventing GAP from promoting hydrolysis of GTP on RAS and therefore causing RAS to accumulate in the GTP‐bound, active form. The pivotal role of RAS proteins in the development of cancer prompted the study of carcinogenesis by using genetically engineered rodents in c‐Ha‐ras genes.
The advantages of using genetically engineered mouse models for carcinogenicity studies are that the carcinogenic potential can be detected with smaller numbers of animals and the time to develop proliferative lesions is greatly reduced.( 7 ) Genetically modified animal models are also useful for elucidating the molecular and cellular processes that lead to cancer initiation, progression, and metastasis, and on the suitability to undergo therapeutic and chemopreventive trials.( 8 )
RasH2 mouse carries human c‐Ha‐ras proto‐oncogene with its own promoter region.( 9 ) RasH2 mouse spontaneously develops various tumors including hemangiosarcoma, lung adenoma, forestomach squamous cell papilomma and carcinoma, skin squamous cell papilloma and carcinoma, and lymphoma.( 10 ) As this transgene is under the control of its own promoter region, it is expressed in the whole body, allowing the study of carcinogenesis in different organs.( 11 ) RasH2 mice are highly susceptible to chemical carcinogenesis,( 12 , 13 , 14 , 15 ) and currently being evaluated to characterize their biological features by worldwide validation studies with a variety of chemicals.( 16 , 17 , 18 ) Mutational analysis of endogenous and transgenic c‐Ha‐ras genes shows that point mutations of the transgene at codons 12 and 61 have been implicated in the development of spontaneous tumors such as lung adenocarcinoma,( 9 , 19 ) skin and forestomach squamous cell carcinoma,( 19 , 20 ) skin papillomas,( 9 ) liver( 21 ) and splenic( 9 ) hemangiosarcomas, and of chemically induced tumors. In rasH2, a single dose of nitrosourea develops forestomach papillomas and uterine endometrial adenocarcinoma.( 19 , 22 , 23 ) Point mutation of c‐Ha‐ras is detected in tumors only at human transgenes and not at endogenous mouse genes, indicating that transgenic, but not endogenous, c‐Ha‐ras contributes to tumor development.( 9 , 24 ) In line with rasH2,( 25 ) Hras128 transgenic rats that integrated three copies of human c‐Ha‐ras proto‐oncogene with their own promoter regions,( 26 ) are sensitive to chemical carcinogenesis.( 11 , 27 ) In Hras128, treatment with DMBA followed by repeated applications of the tumor promoter TPA develops skin cancer.( 28 ) DMBA induces the mutation at codon 61 of the Ha‐ras gene. Consequent addition of TPA has a promoting effect which activates Stat3. The frequency of mutations in the transgene differed according to the induction agent used; the tumors induced by MNU have an extremely high incidence of point mutations at codon 12,( 9 , 11 , 24 , 29 ) but the tumors induced by urethane, vinyl carbamate,( 7 ) ethylnitrosourea have a high incidence of point mutations at codon 61.( 19 ) In Hras128, skin tumors induced by treatment of DMBA and TPA have mutations only at human c‐Ha‐ras gene at either codon 12 or 61.( 11 , 28 ) No mutations were detected in endogenous rat c‐Ha‐ras gene.( 11 , 28 ) Based on the fact that human c‐Ha‐ras transgenes are mainly modified by carcinogens, the rasH2 mouse is considered an adequate testing animal for the evaluation of environmental agents and their carcinogenic effects on humans.( 30 , 31 ) RasH2 mice have been widely used for short‐term carcinogene detection tests.( 24 , 30 , 31 , 32 ) Spontaneous and chemically induced tumors in rasH2 mice have been subjected to molecular analyses in order to clarify the mechanism underlying the observed enhancement of carcinogenesis.( 24 , 31 , 32 ) However, the mechanisms responsible for this enhanced carcinogenesis of rasH2 is not clear enough.( 33 )
Based on those lines of evidence, several factors might be considered to contribute to the enhanced carcinogenesis of c‐Ha‐ras transgenic mice: (i) multiple copies of transgenes are more susceptible to chemical modifications; (ii) human c‐Ha‐ras codons are more susceptible than murine codons to the induction of mutation by carcinogens; and (iii) the mutation rate is much higher for transgenes than for endogenous genes because of their respective stabilities. To further delineate the underlying mechanisms of increased tumor formation in human c‐Ha‐ras transgenic animals, we generated transgenic mice carrying the mouse c‐Ha‐ras genome with its own promoter, mras. The amino acid sequences of c‐Ha‐ras gene products are identical among mouse, rat and human, but the nucleotide sequences are different. We examine the chemically induced skin carcinogenesis in rasH2 and mras to see if differences in the codons of the c‐Ha‐ras gene contributed to the tissue‐specific sensitivity to DMBA.
Materials and Methods
Animals
We used a rasH2 transgenic mouse line generated from the mouse strain C57BL/6 J as previously described.( 29 ) To generate the mras mouse line, fertilized eggs were collected from sexually mature C57BL/6 J mice (CLEA, Tokyo, Japan) and microinjected with XbaI fragments of the mouse c‐Ha‐ras proto‐oncogene with its own promoter region from agarose gel. The genomic structure of transgenes for rasH2 and mras are shown in Figure 1. Subsequent matings between transgenic males and non‐transgenic C57BL/6 J female mice maintained the heterozygote transgenic mras strain. RasH2, mras and their wild‐type littermate mice were bred under SPF conditions. These mice were maintained in plastic cages with absorbent hardwood bedding in an experimental room controlled at a temperature of 23 ± 2°C and a relative humidity of 50 ± 10%, with a 12‐h light/dark cycle. The animals were allowed free access to a powdered basal diet CRF‐1 (Oriental Yeast, Tokyo, Japan) and to tap water. Heterozygotes for the transgene were identified by PCR using DNA samples from their tails.
Figure 1.
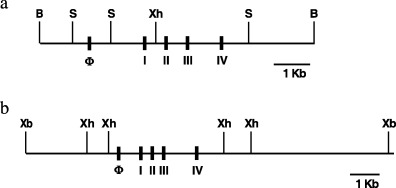
The genomic structure of transgenes for rasH2 mice (a) and mras mice (b). B, BamH I; S, Sac I; Xh, Xho I; Xb, Xba I.
Carcinogenesis study
Eight‐week‐old transgenic and wild‐type male littermates of the rasH2 and mras lines were initially treated with 25 µg of DMBA (Sigma‐Aldrich, St Louis, MO) dissolved in 0.1 mL of acetone painted onto shaved skin on the back. After 1 week, 0.2 µM of TPA (Sigma‐Aldrich) dissolved in 0.1 mL of acetone was applied twice per week for the following 20 weeks. Solutions were prepared every week, and supplied in light‐shaded bottles. Daily observations were made for mortality and clinical signs. The number and volume of skin tumors were recorded at the time of TPA application and at the time the animals were killed. All mice were killed 25 weeks after DMBA treatment by cervical translocation. Skin tissues were excised from the subcutaneous tissue with scissors, along with the fat pads. Skin tumors and normal skins without any macroscopic abnormalities, as negative controls, were cut in half. One piece of each tumor sample was fixed in formaldehyde then processed for paraffin embedding for histological examination. The remaining half‐pieces were immediately frozen in liquid nitrogen for DNA extraction, to investigate the mutation analysis. For histopathological examination, paraffin‐embedded back skin was sectioned to 4 mm thick, and stained with hematoxylin–eosin. All animals were handled in accordance with the guidelines established by the Institute of Medical Science, The University of Tokyo, and Teikyo University (Tokyo, Japan).
Expression of RAS protein in mice skin
Proteins were prepared from the skin of 6‐week‐old male mice as previously described.( 34 ) Proteins were separated by 12% sodium dodecylsulfate–polyacrylamide gel electrophoresis then transferred to an Immobilon membrane (Millipore, Billerica, MA, USA). Immunoblots were probed with polyclonal anti‐H‐ras (Santa Cruz Biotechnology, Santa Cruz, CA) antibody and visualized by enhanced chemiluminescence (ECL‐plus; Amersham Biosciences, Piscataway, NJ, USA).
Mutation analysis
DNA was extracted from tumor tissue by the proteinase K phenol–chloroform method. The analyses of mutations were focused on codons 12, 13 and 61 of the human and mouse c‐Ha‐ras gene because the known mutation pattern of this gene in chemically induced tumors in rasH2 suggest that there are hot spots for exposure to carcinogens in this model. Detection of mutations in the PCR method followed by oligonucleotide hybridization was carried out as described elsewhere.( 29 ) Oligonucleotides for use as primer for both human and murine c‐Ha‐ras genes or as probes for murine c‐Ha‐ras, were synthesized by Applied Biosystems 381 A synthesizer (Applied Biosystems, Foster City, CA, USA) using the manufacturer's protocols and reagents. The primer sequences were described in elsewhere.( 29 )
Results
Generation of mouse c‐Ha‐ras proto‐oncogene transgenic mice
Lines of the mras transgenic mice carried several copies of the mouse c‐Ha‐ras genes (data not shown). Mras mice grew normally and were fertile. The mras mice were alive after more than 30 months and appeared indistinguishable from their wild‐type littermates. They did not show any predisposition to spontaneous tumor formation.
Expression of H‐RAS protein in transgenic mice
We examined the expression of H‐RAS protein in the back skin of rasH2, mras, and wild‐type mice. Western blot analysis showed that the protein expression of H‐RAS was found increased in rasH2 and in mras skin compared with that of wild‐type (Fig. 2).
Figure 2.
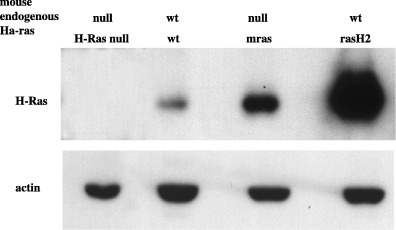
Elevated expression of H‐RAS protein in mras and rasH2 mouse dorsal skin.
To examine if the murine c‐Ha‐ras transgene in mras expressed the protein, we generated c‐Ha‐ras null mice with murine c‐Ha‐ras transgene by crossing mras mice with the mice with the targeted deletion of c‐Ha‐ras.( 35 ) Figure 2 shows that in c‐Ha‐ras null mice, no H‐RAS protein expression was detected by Western blot. The c‐Ha‐ras null mice with murine c‐Ha‐ras transgene expressed equivalent amounts of c‐Ha‐ras protein, which confirmed that the murine c‐Ha‐ras transgene was indeed expressed.
RasH2 but not mras showed enhanced chemical skin carcinogenesis after treatment with DMBA and TPA
Figure 3 shows the gross appearance of tumor formation in the back skin of mice 20 weeks after treatment initiation. On time‐sequence observation, rapid induction of grossly visible skin tumors was observed in rasH2 mice (Fig. 3). In rasH2 mice, the latency of tumor formation after treatment with DMBA was 3.5 weeks, but in wild‐type littermates it was 11 weeks (Fig. 4). Histologically the skin tumors were all diagnosed as squamous cell papilloma or squamous cell carcinoma (Fig. 5). By 25 weeks, on histopathological examination, all rasH2 mice developed squamous cell carcinomas (Fig. 5), whereas wild‐type mice needed more than 35 weeks to develop squamous cancer (data not shown). The decreased duration of latency in tumor development compared with wild‐type suggested that the rasH2 gene functioned as a tumor promoter. Both tumor numbers and volumes were clearly greater in rasH2 compared with those of wild‐type littermates. The mean number of tumors was 31.2 ± 11.2 in rasH2 (n = 5) and 3.8 ± 3.9 (n = 5) in wild‐type littermates. In mras mice, enhancement of tumor formation was not observed. The mean number of tumors was 4.6 ± 2.6 (n = 11) in mras and 3.0 ± 2.0 (n = 6) in wild‐type littermates. Latency of tumor development was almost the same in mras (10.5 weeks) and in wild‐type littermate (11.0 weeks).
Figure 3.
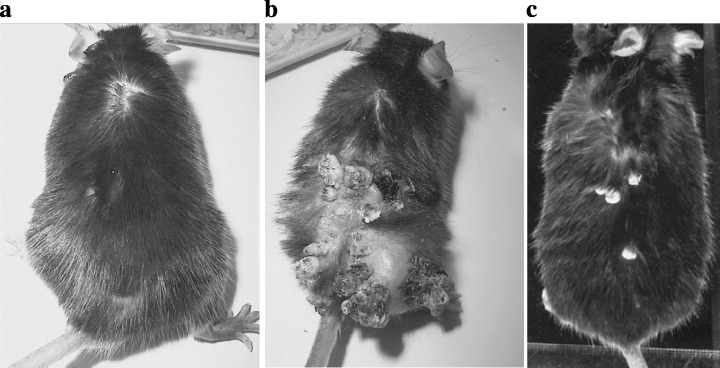
The gross appearance of tumor formation on the back skin of mice 25 weeks after the treatment with DMBA. (a) DMBA and TPA‐induced skin tiny papilloma in a control wild‐type mouse; (b) DMBA and TPA‐induced skin papilloma and squamous cell carcinoma in a rasH2 mouse; (c) DMBA and TPA‐induced skin papilloma in an mras mouse.
Figure 4.
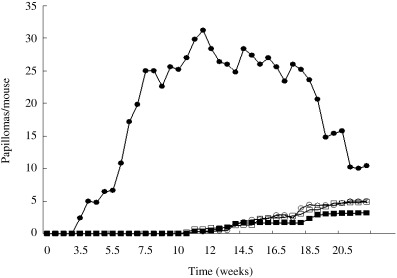
Average number of papillomas/mouse. Time dependence of skin tumor development on the back skin in male mice treated with DMBA and TPA. (•) rasH2 (transgenic mouse [Tg]); (○) rasH2 (wild‐type mouse [Wt]); (▪) mras (Tg); (□) mras (Wt).
Figure 5.
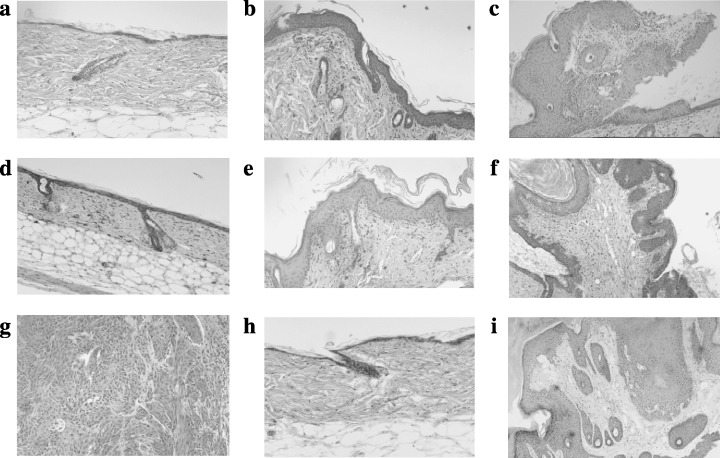
Histopathology of representative lesions of back skin tissue (hematoxylin–eosin stain). (a) Normal skin tissue in a control wild‐type mouse at 25 weeks; (b) DMBA and TPA‐induced skin hyperplasia in a wild‐type mouse at 25 weeks; (c) DMBA and TPA‐induced skin papilloma in a wild‐type mouse at 25 weeks; (d) normal skin tissue in a rasH2 mouse at 25 weeks; (e) DMBA and TPA‐induced skin hyperplasia in a rasH2 mouse at 25 weeks; (f) DMBA and TPA‐induced skin papilloma in a rasH2 mouse at 25 weeks; (g) DMBA and TPA‐induced skin squamous carcinoma in a rasH2 mouse at 25 weeks. Note the invasion into the deep subcutaneous and smooth muscle layers; (h) normal skin tissue in an mras mouse at 25 weeks; (i) DMBA and TPA‐induced skin papilloma in an mras mouse at 25 weeks.
Somatic point mutational activation of the transgenes in tumors
Previous studies showed that the point mutations induced by DMBA in rasH2 were detected only in transgenes. We examined the somatic mutation of the human c‐Ha‐ras gene at codons 12 and 61 and the murine c‐Ha‐ras gene at codons 12, 13, and 61. Mutational analysis by PCR followed by oligonucleotide hybridization and direct sequencing showed that only A to T transversion in human c‐Ha‐ras transgenes at codon 61, but not in murine endogenous c‐Ha‐ras genes, were detected in the tumors of rasH2 (Fig. 6). No mutations were detected in untreated regions of skin and treated skin regions without tumor formations. However, in the tumors of wild‐type littermates and in mras mice, A to T transversion in murine c‐Ha‐ras transgenes were detected at codon 61 (Fig. 7).
Figure 6.
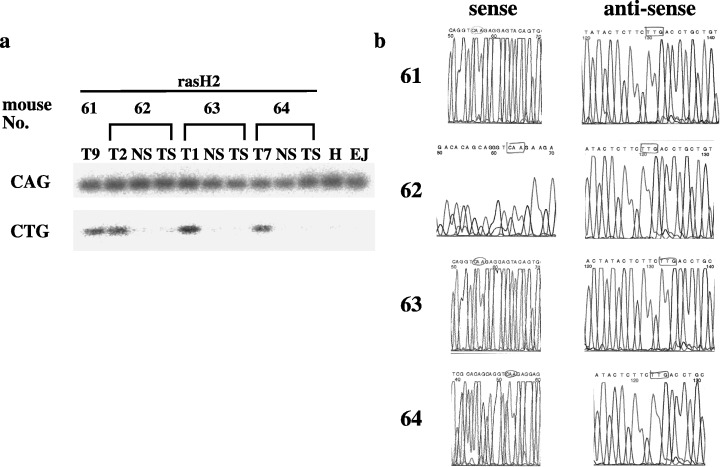
(a) Mutation analyses of human c‐Ha‐ras transgene at codon 61. A to T transversion at the second base of transgenic human c‐Ha‐ras codon 61 in papillomas detected by oligonucleotide hybridization. H, human genomic DNA; NS, skin of non‐treatment; T, tumor; TS, skin of DMBA and TPA treatment; (b) direct sequencing showed no mutation in papillomas of murine c‐Ha‐ras transgenes at codon 61.
Figure 7.
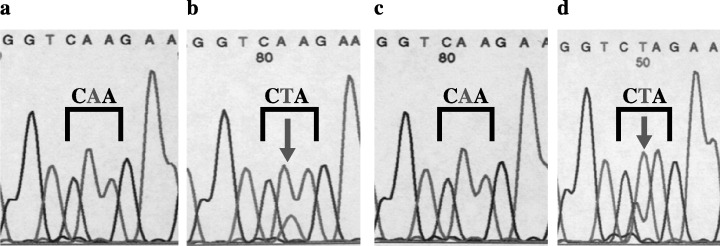
PCR‐direct sequencing of c‐Ha‐ras at codon 61 in mras mice. In the tumors of wild‐type littermates and in mras mice after DMBA and TPA treatment, A to T transversion in murine c‐Ha‐ras transgenes at codon 61 were detected (arrows). (a) Non‐neoplastic regions of mras; (b) papilloma in mras; (c) non‐neoplastic regions of wild‐type; (d) papilloma in wild‐type.
In summary, enhanced skin carcinogenesis induced by DMBA and TPA treatment was observed in rasH2 compared with wild‐type littermates. In tumors of rasH2, mutations of c‐Ha‐ras genes were detected only in human transgenes at codon 61, but not in endogenous mouse c‐Ha‐ras genes. On the contrary, no difference in the latency or incidence of tumor development was observed between mras and wild‐type littermates, although mras mice had increased expression of H‐RAS protein in the back skin compared with those of wild‐types.
Discussion
In this study, rasH2 mice demonstrated enhanced carcinogenesis in a chemically induced skin cancer model with DMBA as an initiator and TPA as a promoter. Tumors in rasH2 developed in shorter latency and in higher numbers and volumes compared with wild‐type littermates. In wild‐type mouse, codon 61 of the endogenous mouse c‐Ha‐ras gene was mutated by this chemically induced tumor model. However, tumors developed in rasH2 had mutations only at codon 61 of the human c‐Ha‐ras transgene from CAG (Gln) to CTG (Leu) without mutation at the mouse codon.( 24 ) This phenomenon has been observed in other models of chemically induced skin and mammary tumors in mice.( 36 ) The human c‐Ha‐ras transgene was used here as a probe to monitor the mutations because of its higher mutation rate compared to the rat endogenous gene.( 26 ) Most neoplasms in transgenic mice with human c‐Ha‐ras gene contain somatically activated transgenes, but no change in the endogenous gene.( 34 ) Also, in Tg.AC mice that bear a multicopy fusion transgene in which the mouse embryonic z‐globin promoter drives the expression of the v‐Ha‐ras oncogene,( 37 ) DMBA increases papillomagenesis by affecting the structure or expression of the endogenous c‐Ha‐ras gene.( 38 )
These lines of evidence suggest that the transgenes might be genetically unstable, and this very property of instability might be responsible for a higher incidence of mutation. Moreover, the locus and nature of the integration event of the transgene are critical. Why did the murine c‐Ha‐ras transgene with its own promoter fail to increase tumor formation, as in rasH2? The amino acid sequence of the c‐Ha‐ras gene is identical among mouse, rat and man. However, the base sequence is different in each. Our hypothesis is that this difference in the codon can contribute to tissue‐specific sensitivity to a particular carcinogen. It could be explained, in part, by the affinity of DMBA to codon 61. DMBA consistently produced A to T transversion at codon 61 of the c‐Ha‐ras gene, but little is known on the influence of the adjacent base on the efficiency of abduction. It might be possible that DMBA preferentially abducts human codon CAG to mouse codon CAA. Moreover, as rasH2 and mras both carry the genomic DNA fragments containing the c‐Ha‐ras gene, the difference of genomic structure between human and murine genomes affects the access of DMBA to the target in the genome. However, this does not fully explain why mras mice did not have enhanced tumorigenesis compared with wild‐type littermates, even though the protein expression of H‐RAS is higher in the transgenic mice. A previous study showed that the susceptibility of transgenes to a carcinogen was actually reduced in chemically induced mammary carcinogenesis in transgenic rats carrying multiple copies of a rat ras gene.( 39 ) If this finding is applied to mras, multiple copies of mouse c‐Ha‐ras transgenes might be less frequently mutated by DMBA. Another possibility is that the gene dosage of c‐Ha‐ras needs to exceed a certain threshold to show the enhanced carcinogenesis, as in rasH2. RasH2 carries five or six copies of the transgenes integrated into the mouse genome in a tandem array. Hras128, a transgenic rat line that showed enhanced carcinogenesis, also carries three copies of the human c‐Ha‐ras transgene. The number of integrated copy numbers of the transgene in mras might be less than that of rasH2. Further studies are required to find a plausible explanation for the difference in the induction of carcinogenesis between rasH2 and mras mice.
Acknowledgments
The authors appreciate K. Katsuki for her continuous support of this study.
References
- 1. Hahn WC, Weinberg RA. Modeling the molecular circuitry of cancer. Nat Rev Cancer 2002; 2: 331–41. [DOI] [PubMed] [Google Scholar]
- 2. Weinberg RA. Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Res 1989; 49: 3713–21. [PubMed] [Google Scholar]
- 3. DiGiovanni J. Multistage carcinogenesis in mouse skin. Pharmacol Ther 1992; 54: 63–128. [DOI] [PubMed] [Google Scholar]
- 4. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 5. Bos JL. Ras oncogenes in human cancer: a review. Cancer Res 1989; 49: 4682–9. [PubMed] [Google Scholar]
- 6. Lowy DR, Willumsen BM. Function and regulation of ras . Annu Rev Biochem 1993; 62: 851–91. [DOI] [PubMed] [Google Scholar]
- 7. Tomisawa M, Suemizu H, Ohnishi Y et al. Mutation analysis of vinyl carbamate or urethane induced lung tumors in rasH2 transgenic mice. Toxicol Lett 2003; 142: 111–7. [DOI] [PubMed] [Google Scholar]
- 8. Suzuki R, Kohno H, Suzui M et al. An animal model for the rapid induction of tongue neoplasms in human c‐Ha‐ras proto‐oncogene transgenic rats by 4‐nitroquinoline 1‐oxide: its potential use for preclinical chemoprevention studies. Carcinogenesis 2006; 27: 619–30. [DOI] [PubMed] [Google Scholar]
- 9. Saitoh A, Kimura M, Takahashi R et al. Most tumors in transgenic mice with human c‐Ha‐ras gene contained somatically activated transgenes. Oncogene 1990; 5: 1195–200. [PubMed] [Google Scholar]
- 10. Morton D, Alden CL, Roth AJ, Usui T. The Tg rasH2 mouse in cancer hazard identification. Toxicol Pathol 2002; 30: 139–46. [DOI] [PubMed] [Google Scholar]
- 11. Tsuda H, Fukamachi K, Ohshima Y et al. High susceptibility of human c‐Ha‐ras proto‐oncogene transgenic rats to carcinogenesis: a cancer‐prone animal model. Cancer Sci 2005; 96: 309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Okamura M, Moto M, Kashida Y, Machida N, Mitsumori K. Carcinogenic susceptibility to N‐bis(2‐hydroxypropyl)nitrosamine (DHPN) in rasH2 mice. Toxicol Pathol 2004; 32: 474–81. [DOI] [PubMed] [Google Scholar]
- 13. Imaoka M, Kashida Y, Watanabe T et al. Tumor promoting effect of phenolphthalein on development of lung tumors induced by N‐ethyl‐N‐nitrosourea in transgenic mice carrying human prototype c‐Ha‐ras gene. J Vet Med Sci 2002; 64: 489–93. [DOI] [PubMed] [Google Scholar]
- 14. Maronpot MM, Mitsumori K, Mann P et al. Interlaboratory comparison of the CB6F1‐Tg rasH2 rapid carcinogenicity testing model. Toxicology 2000; 146: 149–59. [DOI] [PubMed] [Google Scholar]
- 15. Yamamoto S, Hayashi Y, Mitsumori K, Nomura T. Rapid carcinogenicity testing system with transgenic mice harboring human prototype c‐HRAS gene. Lab Anim Sci 1997; 47: 121–6. [PubMed] [Google Scholar]
- 16. Gulezian D, Jacobson‐Kram D, McCullough CB et al. Use of transgenic animals for carcinogenicity testing: considerations and implications for risk assessment. Toxicol Pathol 2000; 28: 482–99. [DOI] [PubMed] [Google Scholar]
- 17. Storer RD. Current status and use of short/medium term models for carcinogenicity testing of pharmaceuticals – scientific perspective. Toxicol Lett 2000; 112–113: 557–66. [DOI] [PubMed] [Google Scholar]
- 18. Storer RD, French JE, Donehower LA et al. Transgenic tumor models for carcinogen identification: the heterozygous Trp53‐deficient and RasH2 mouse lines. Mutat Res 2003; 540: 165–76. [DOI] [PubMed] [Google Scholar]
- 19. Toyosawa K, Tanaka K, Imai T et al. Mutation and overexpression of the transgene in ethylnitrosourea‐induced tumors in mice carrying a human prototype c‐Ha‐ras gene. Toxicol Pathol 2003; 31: 491–5. [DOI] [PubMed] [Google Scholar]
- 20. Mitsumori K, Koizumi H, Nomura T, Yamamoto S. Pathological features of spontaneous and induced tumors in transgenic mice carrying a human prototype c‐Ha‐ras gene used for six‐month carcinogenicity studies. Toxicol Pathol 1998; 26: 520–31. [DOI] [PubMed] [Google Scholar]
- 21. Hayashi S, Mori I, Nonoyama T, Mitsumori K. Point mutations of the c‐H‐ras gene in spontaneous liver tumors of transgenic mice carrying the human c‐H‐ras gene. Toxicol Pathol 1998; 26: 556–61. [DOI] [PubMed] [Google Scholar]
- 22. Watanabe T, Kashida Y, Yasuhara K, Koujitani K, Hirose M, Mitsumori K. Rapid induction of uterine endometrial proliferative lesions in transgenic mice carrying a human prototype c‐Ha‐ras gene (rasH2 mice) given a single intraperitoneal injection of N‐ethyl‐N‐nitrosourea. Cancer Lett 2002; 188: 39–46. [DOI] [PubMed] [Google Scholar]
- 23. Watanabe T, Kashida Y, Ueda M et al. Inhibition by ethinylestradiol of N‐ethyl‐N‐nitrosourea‐initiated uterine carcinogenesis in transgenic mice carrying a human prototype C‐Ha‐ras gene (rasH2 mice). Toxicol Pathol 2003; 31: 496–505. [DOI] [PubMed] [Google Scholar]
- 24. Maruyama C, Tomisawa M, Wakana S et al. Overexpression of human H‐ras transgene is responsible for tumors induced by chemical carcinogens in mice. Oncol Rep 2001; 8: 233–7. [DOI] [PubMed] [Google Scholar]
- 25. Suemizu H, Muguruma K, Maruyama C et al. Transgene stability and features of rasH2 mice as an animal model for short‐term carcinogenicity testing. Mol Carcinog 2002; 34: 1–9. [DOI] [PubMed] [Google Scholar]
- 26. Asamoto M, Ochiya T, Toriyama‐Baba H et al. Transgenic rats carrying human c‐Ha‐ras proto‐oncogenes are highly susceptible to N‐methyl‐N‐nitrosourea mammary carcinogenesis. Carcinogenesis 2000; 21: 243–9. [DOI] [PubMed] [Google Scholar]
- 27. Han BS, Fukamachi K, Takasuka N et al. Inhibitory effects of 17beta‐estradiol and 4‐n‐octylphenol on 7,12‐dimethylbenz[a]anthracene‐induced mammary tumor development in human c‐Ha‐ras proto‐oncogene transgenic rats. Carcinogenesis 2002; 23: 1209–15. [DOI] [PubMed] [Google Scholar]
- 28. Park CB, Fukamachi K, Takasuka C et al. Rapid induction of skin and mammary tumors in human c‐Ha‐ras proto‐oncogene transgenic rats by treatment with 7,12‐dimethylbenz[a]anthracene followed by 12‐O‐tetradecanoylphorbol 13‐acetate. Cancer Sci 2004; 95: 205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ando K, Saitoh A, Hino O, Takahashi R, Kimura M, Katsuki M. Chemically induced forestomach papillomas in transgenic mice carry mutant human c‐Ha‐ras transgenes. Cancer Res 1992; 52: 978–82. [PubMed] [Google Scholar]
- 30. Yamamoto S, Mitsumori K, Kodama Y et al. Rapid induction of more malignant tumors by various genotoxic carcinogens in transgenic mice harboring a human prototype c‐Ha‐ras gene than in control non‐transgenic mice. Carcinogenesis 1996; 17: 2455–61. [DOI] [PubMed] [Google Scholar]
- 31. Mitsumori K, Wakana S, Yamamoto S et al. Susceptibility of transgenic mice carrying human prototype c‐Ha‐ras gene in a short‐term carcinogenicity study of vinyl carbamate and ras gene analyses of the induced tumors. Mol Carcinog 1997; 20: 298–307. [DOI] [PubMed] [Google Scholar]
- 32. Mori I, Yasuhara K, Hayashi S, Nonoyama T, Nomura T, Mitsumori K. Carcinogen dose‐dependent variation in the transgene mutation spectrum in urethane‐induced lung tumors in transgenic mice carrying the human prototype c‐Ha‐ras gene. Cancer Lett 2000; 153: 199–209. [DOI] [PubMed] [Google Scholar]
- 33. Mitsumori K. Possible mechanism on enhanced carcinogenesis of genotoxic carcinogens and unsolved mechanisms on lesser carcinogenic susceptibility to some carcinogens in rasH2 mice. J Toxicol Sci 2003; 28: 371–83. [DOI] [PubMed] [Google Scholar]
- 34. Ise K, Nakamura K, Nakao K et al. Targeted deletion of the H‐ras gene decreases tumor formation in mouse skin carcinogenesis. Oncogene 2000; 19: 2951–6. [DOI] [PubMed] [Google Scholar]
- 35. Manabe T, Aiba A, Yamada A et al. Regulation of long‐term potentiation by H‐Ras through NMDA receptor phosphorylation. J Neurosci 2000; 20: 2504–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsuda H, Asamoto M, Ochiya T et al. High susceptibility of transgenic rats carrying the human c‐Ha‐ras proto‐oncogene to chemically‐induced mammary carcinogenesis. Mutat Res 2001; 477: 173–82. [DOI] [PubMed] [Google Scholar]
- 37. Leder A, Kuo A, Cardiff RD et al. v‐Ha‐ras transgene abrogates the initiation step in mouse skin tumorigenesis: effects of phorbol esters and retinoic acid. Proc Natl Acad Sci USA 1990; 87: 9178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Owens DM, Spalding JW, Tennant RW et al. Genetic alterations cooperate with v‐Ha‐ras to accelerate multistage carcinogenesis in TG.AC transgenic mouse skin. Cancer Res 1995; 55: 3171–8. [PubMed] [Google Scholar]
- 39. Thompson TA, Haag JD, Lindstrom MJ et al. Decreased susceptibility to NMU‐induced mammary carcinogenesis in transgenic rats carrying multiple copies of a rat ras gene driven by the rat Harvey ras promoter. Oncogene 2002; 21: 2797–804. [DOI] [PubMed] [Google Scholar]