

Abstract
We report that HSP105, identified by serological identification of antigens by recombinant expression cloning (SEREX), is overexpressed in a variety of human cancers, including colorectal, pancreatic, thyroid, esophageal, and breast carcinoma, but is not expressed in normal tissues except for the testis. The amino acid sequences and expression patterns of HSP105 are very similar in humans and mice. In this study, we set up a preclinical study to investigate the usefulness of a DNA vaccine producing mouse HSP105 whole protein for cancer immunotherapy in vivo using BALB/c and C57BL/6 mice, Colon26, a syngeneic endogenously HSP105‐expressing colorectal cancer cell line, and B16.F10, a melanoma cell line. The DNA vaccine was used to stimulate HSP105‐specific T‐cell responses. Fifty percent of mice immunized with the HSP105 DNA vaccine completely suppressed the growth of subcutaneous Colon26 or B16.F10 cells accompanied by massive infiltration of both CD4+ T cells and CD8+ T cells into tumors. In cell transfer or depletion experiments we proved that both CD4+ T cells and CD8+ T cells induced by these vaccines play critical roles in the activation of antitumor immunity. Evidence of autoimmune reactions was not present in surviving mice that had rejected tumor cell challenges. We found that HSP105 was highly immunogenic in mice and that the HSP105 DNA vaccination induced antitumor immunity without causing autoimmunity. Therefore, HSP105 is an ideal tumor antigen that could be useful for immunotherapy or the prevention of various human tumors that overexpress HSP105, including colorectal cancer and melanoma. (Cancer Sci 2005; 96: 695 – 705)
Abbreviations:
- C26 (C20)
Colon26 clone 20
- CRC
colorectal cancer
- CTL
cytotoxic T lymphocytes
- HE
hematoxylin and eosin
- HSP105
heat shock protein 105
- APC
antigen presenting cell
- mAb
monoclonal antibody
- MHC
major histocompatibility complex
- SEREX
serological identification of antigens by recombinant expression cloning
- TAA
tumor associated antigens.
Colorectal cancer (CRC) and melanoma are common and serious malignancies, for which surgery remains the main treatment, although the success of the treatment depends on the stage of the disease. Although adjuvant systemic chemotherapy or chemoradiation can confer a limited but significant survival advantage, novel and more effective therapies are needed. Identification of tumor associated antigens (TAA) expressed by CRC or melanomas remains one of the goals for designing novel immunological treatments for these tumors. Ideal targets for immunotherapy are gene products that are silenced in normal tissues except immune privilege tissue such as testis tissue, and that are overexpressed in cancer cells.
More than 2000 candidate TAA have been identified by using the serological identification of antigens by recombinant expression cloning (SEREX) method. We have also reported TAA identified by using this method.( 1 , 2 , 3 , 4 ) We earlier found that HSP105 (often called HSP110), as identified by SEREX was overexpressed specifically in a variety of human cancers, including colorectal, pancreatic, thyroid, esophageal, and breast carcinoma, but was not expressed in normal tissues except for testis tissue.( 1 , 5 ) We recently found that HSP105 was also overexpressed in melanoma (unpublished data). If HSP105 can induce strong antitumor immunity, it may be a potential candidate as a target antigen for cancer immunotherapy. In the present study, we set up a preclinical study to investigate the usefulness of a HSP105‐DNA vaccine, using BALB/c and C57BL/6 mice, the syngeneic endogenously HSP105‐expressing CRC cell line Colon26, and the melanoma cell line B16.F10. Using these models, we analyzed both the antitumor effects and side‐effects, including autoimmunity of the HSP105 DNA vaccination.
The pioneering studies of Srivastava and colleagues led to the proposal that several HSP, including HSP70, HSP90 and gp96, bind antigenic peptides and deliver these peptides (through receptor‐mediated endocytosis of the HSP) into the antigen‐processing pathway of the antigen presenting cell (APC) for presentation on major histocompatibility complex (MHC) class I molecules. This HSP‐involved pathway has been demonstrated to evoke potent antiviral and antitumor immune responses.( 6 ) However, many researchers have identified MHC class I‐presented peptide epitopes derived from HSP. HSP are rich sources of MHC‐bound peptides, and the expression of these peptides increases as a result of cellular stresses.( 7 )
Recently, Subjeck and colleagues tested a vaccine using the chaperoning properties of HSP110 as Srivastava and colleagues had done before them.( 8 , 9 ) They reported that HSP110 overexpression increases the immunogenicity of murine CT26 colon tumors.( 10 ) HSP110 cloned from CHO cells( 11 ) and HSP105 cloned from mice( 12 ) and humans( 13 ) are homologs. We show here that this HSP105 is highly immunogenic for stimulating tumor immunity against mouse CRC and melanoma. Furthermore, both CD4+ T cells and CD8+ T cells induced by the HSP105 DNA vaccination play critical roles in the activation of antitumor immunity. These findings indicate that HSP105 itself could be considered a valuable TAA for the immune‐based therapy of various tumors overexpressing HSP105, including CRC and melanoma.
Materials and Methods
Cell lines and mice
A subline of the BALB/c‐derived CRC cell line Colon26, C26 (C20),( 14 ) was provided by Dr Kyoichi Shimomura (Fujisawa Pharmaceutical Co., Japan). B16.F10 was kindly provided by the Cell Resource Center for Biomedical Research, Institute of Development, Aging, and Cancer, Tohoku University (Sendai, Japan). These cell lines were maintained in vitro in RPMI‐1640 medium supplemented with 10% fetal calf serum at 37°C in a 5% CO2 atmosphere. Female 7‐week‐old BALB/c mice (H‐2d) and C57BL/6 mice (H‐2b), purchased from Charles River Japan (Yokohama, Japan), were kept in the Center for Animal Resources and Development (CARD) of Kumamoto University, and handled in accordance with the animal care policy of Kumamoto University.
Histological and immunohistochemical analysis
Immunohistochemical detections of HSP105, CD8 and CD4 were carried out as described elsewhere.( 1 , 5 , 15 , 16 , 17 , 18 ) The primary antibody used in this study, rabbit polyclonal antihuman HSP105 was purchased from Santa Cruz (Santa Cruz, CA, USA). Hematoxylin and eosin (HE) staining and standard methods were used for histological analysis. We purchased Human Normal Organs and Cancer Multi Tissue Slide, BC4, from SuperBioChips Laboratories (Seoul, Korea) for immunohistochemical analysis.
Construction of a mouse HSP105 expression plasmid DNA
Plasmid pcDNA105, which expresses mouse HSP105 whole protein was generated as described elsewhere.( 12 ) To construct this plasmid, the mouse HSP105 full‐length cDNA derived from the pB105‐1 plasmid was subcloned into EcoRV–XbaI sites of the mammalian expression vector pcDNA3 (Invitrogen, Osaka, Japan). The pCAGGS expression vector was kindly provided by Dr Junichi Miyazaki (Osaka University, Japan) and this vector induces strong gene expression when injected into muscle.( 19 ) We constructed a pCAGGS‐HSP105 plasmid by inserting mouse HSP105 cDNA into the EcoRI site of the pCAGGS expression vector, which carries the CAG (cytomegalovirus immediate‐early enhancer/chicken β‐actin hybrid) promoter, and prepared the plasmid using a Qiagen EndoFree plasmid Mega kit (Qiagen GmbH, Hilden, Germany). We used the empty pCAGGS plasmid as a control.
DNA vaccination
We immunized mice twice by intramuscular injection into the anterior tibialis muscle. Booster immunization was carried out at 7 days after the primer immunization. The groups of mice were given the following vaccines: (i) saline group: given with 100 µL saline; (ii) control vector group: given 50 µg pCAGGS plasmids lacking inserts and diluted in 100 µL saline; (iii) HSP105 DNA vaccine group: given 50 µg of pCAGGS‐HSP105 plasmid diluted in 100 µL saline.
In vivo tumor challenge
Subcutaneous tumors were established by the injection of 3 × 104 C26 (C20) cells or 1 × 104 B16.F10 cells suspended in 100 µL Hanks’ Balanced Salt Solution (Gibco, Grand Island, NY, USA) medium into the right flank of BALB/c or C57BL/6 mice 7 days after the last vaccination. Tumor incidence and volume were assessed twice weekly using calipers until the mice died. Tumor area was calculated as a product of width and length. The results are presented as mean area of tumor ± SE; however, individual tumor area is presented for some experiments.
In vivo depletion of CD4+ T cells and CD8+ T cells
Each mouse was given a total of six intraperitoneal transfers (days −18, −15, −11, −8, −4, −1) of ascites (0.1 mL per mouse per transfer) from hybridoma‐bearing nude mice. The mAbs used were rat antimouse CD4 (clone GK1.5) and rat antimouse CD8 (clone 2.43). Normal rat IgG (Sigma, St. Louis, MO, USA; 200 µg per mouse per transfer) was used as a control. The depletion of T cell subsets by treatment with mAbs was confirmed by flow cytometric analysis of spleen cells, which showed a > 90% specific depletion.
Cell transfer in vivo
We purified CD8+ T cells, CD4+ T cells, and natural killer (NK) cells from spleen cells using the magnetic cell sorting system with antimouse CD8α (Ly‐2) mAb, antimouse CD4 (L3T4) mAb, antimouse NK (DX5) mAb, and these CD8+ T cells, CD4+ T cells, and NK cells were used for adoptive transfer into BALB/c mice. To investigate tumor growth in a homeostatic lymphocyte proliferation model, we intravenously injected 1.5 × 107 whole spleen cells or 3 × 106 CD8+ T cells, CD4+ T cells, NK cells, or CD8− CD4− NK− cells 3 days after sublethal irradiation (5 Gy). Subsequently, we subcutaneously inoculated BALB/c mice with C26 cells (3 × 104) 3 days after irradiated mice inoculated with cells.
Statistical analysis
We analyzed all data using the StatView statistical program for Macintosh (SAS, Cary, NC, USA) and evaluated statistical significance using the unpaired t‐test. The overall survival rate was calculated using the Kaplan–Meier method, and statistical significance was evaluated using Wilcoxon's test.
Results
Similar tissue and cancer‐specific expression of HSP105 in mice and humans
We have previously reported that HSP105 is overexpressed in a variety of human cancers, including colorectal, pancreatic, esophageal, thyroid, and breast cancer, whereas HSP105 is expressed at low levels in many normal tissues, except for testis tissue.( 1 , 5 ) In the present study, we carried out an immunohistochemical analysis of HSP105 using various human and mouse tissues (Fig. 1). Human HSP105 is overexpressed in almost all CRC cells, melanoma cells (unpublished data), and normal testis tissue, but there is no expression or only a low‐level expression of HSP105 in normal liver, brain, spleen, lung, and kidney tissue (Fig. 1a). Mouse HSP105 is also overexpressed in liver metastasis of the murine colorectal adenocarcinoma cell line C26 (C20), lung metastasis of the murine melanoma cell line B16.F10 and normal testis tissue, but there is no expression or only low‐level expression in normal liver, cerebrum, cerebellum, spleen, lung, and kidney tissue (Fig. 1b). Another group reported that HSP105/110 is expressed in neurons in the cerebrum and Purkinje cells in the cerebellum,( 20 ) we found the same pattern in the present study, but the level of expression in the neurons and Purkinje cells was much weaker than that in CRC and testis tissue (Fig. 1a,b). As a result, the expression levels of HSP105 protein in human colorectal, pancreatic, esophageal, thyroid, and breast cancers, melanoma, C26 tumors, and B16.F10 tumors were evidently much higher than those in all normal adult tissues, including brain, but not testis in both humans and mice. Because the expression pattern of HSP105 is very similar in humans and mice, we are able to analyze both the antitumor effects and side‐effects (including autoimmunity) of HSP105 vaccination using this mouse model of CRC and melanoma.
Figure 1.
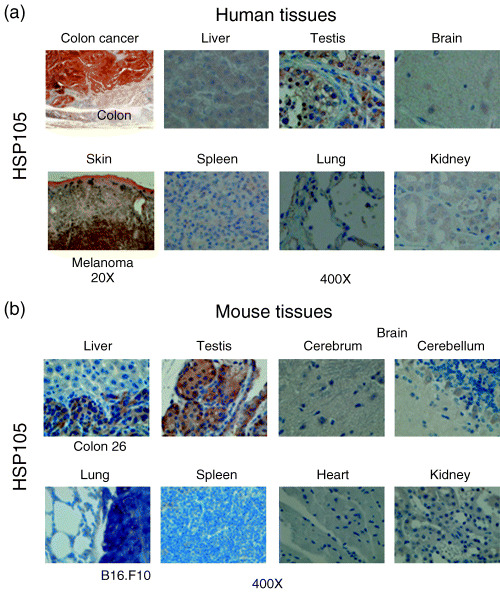
Expression of the HSP105 protein, a candidate for immunotherapy for CRC and melanoma, in human and mouse tissues and cells. Expression of HSP105 protein detected by immunohistochemical analysis in various (a) human and (b) mouse tissues. Objective magnification was 400× or 20×.
HSP105 DNA induced rejection of C26 and B16.F10 tumor challenge in mice
We investigated the effects of HSP105 DNA vaccination using a subcutaneously injected C26 (Fig. 2a–d) and B16.F10 (Fig. 2e–h) tumor model. Mice were divided into three groups: mice inoculated with (i) saline; (ii) pCAGGS, and (iii) pCAGGS‐HSP105. No mice died during the vaccination period.
Figure 2.
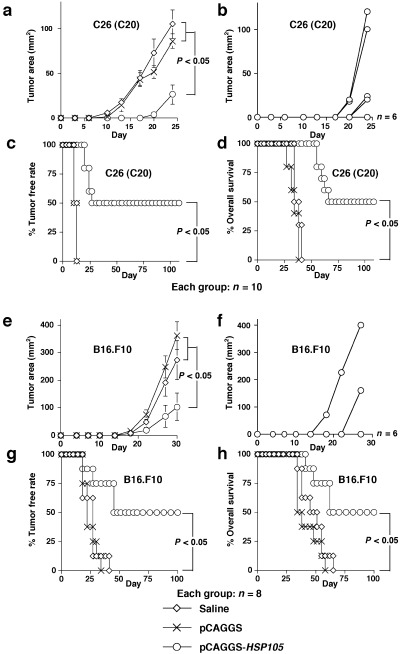
Vaccination with HSP105 DNA suppressed the growth of (a–d) C26 and (e–h) B16.F10 tumors in mice. Each group consisted of 10 (a–d) or eight (e–h) mice. (a,b,e,f) Suppression of the growth of HSP105‐expressing C26 (a,b) or B16.F10 (e,f) tumors inoculated subcutaneously into mice vaccinated with HSP105 DNA. The tumor area was calculated as the product of width and length. The result is presented as mean area of tumor ± SE, and we evaluated statistical significance using the unpaired t‐test (a,e). Growth curves of 10 and eight individual tumors in the mouse group treated with pCAGGS‐HSP105 are presented in (b) and (f), respectively. (c,d,g,h) Percentage tumor free rate (c,g) and percentage overall survival (d,h) were calculated using the Kaplan–Meier method, and the statistical significance of differences between groups was evaluated using Wilcoxon's test.
Subcutaneous inoculation of C26 cells (3 × 104) into the right flank was given 7 days after the last vaccination (Fig. 2a–d). In groups (i) and (ii), subcutaneous tumors appeared in some mice 10 days after inoculation. Measurement of tumor size was continued until 24 days after inoculation with the tumor cells, when one mouse died. The mean tumor size on day 24 in group (iii) mice (26.4 ± 10.8 mm2) was significantly smaller than that in the other two groups (105.0 ± 15.7, and 86.0 ± 8.3 mm2, respectively; P < 0.05; Fig. 2a). Six of the 10 mice (60%) in group (iii) did not have subcutaneous tumors on day 24 (Fig. 2b). All mice in groups (i) and (ii) had subcutaneous tumors within 13 days, and died within 41 days of inoculation with the tumor cells (Fig. 2c,d). Five of the 10 mice (50%) in group (iii) completely rejected the 3 × 104 C26 cells during the 108 days after the inoculation (Fig. 2c,d). A statistically significant difference in survival time was found between group (iii) and groups (i) and (ii) (P < 0.05).
Subcutaneous inoculation of B16.F10 cells (1 × 104) into the right flank was carried out 7 days after the last vaccination (Fig. 2e–h). Measurement of tumor size was continued until 30 days after inoculation with the tumor cells, when one mouse died. Mean tumor size on day 30 in group (iii) mice (103.9 ± 49.8 mm2) was significantly smaller than that in the other two groups (272.1 ± 69.7, and 361.6 ± 50.3 mm2, respectively; P < 0.05; Fig. 2e). Six of eight mice (75%) in group (iii) did not have subcutaneous tumors on day 30 (Fig. 2f). All mice in groups (i) and (ii) had subcutaneous tumors within 41 days, and died within 65 days of inoculation with the tumor cells (Fig. 2g,h). Four of eight mice (50%) in group (iii) completely rejected the 1 × 104 B16.F10 cells during the 100 days after the inoculation (Fig. 2g,h). A statistically significant difference in survival time was found between group (iii) and groups (i) and (ii) (P < 0.05). Therefore, the HSP105 DNA vaccine has the potential to prevent the growth of tumors expressing HSP105.
We also subcutaneously inoculated five surviving group (iii) mice that completely rejected the first challenges with C26 cells with further (3 × 104) C26 cells. These mice also rejected the second challenge with C26 cells, even at 108 days after the first challenge (data not shown). These results demonstrate that the effects of vaccination in group (iii) continued for a long time, and that the vaccination prevented the recurrence of HSP105‐expressing tumors.
Expression of HSP105 protein and infiltration of CD4+ T cells and CD8+ T cells in the injection sites
To observe HSP105 expression and infiltrating cells in muscles injected with the HSP105 DNA vaccine, we carried out intramuscular immunizations with pCAGGS DNA into the right anterior tibialis muscle, and with pCAGGS‐HSP105 DNA into the left anterior tibialis muscle of four mice. After 48 h, we killed the mice and evaluated the muscles by histological and immunohistochemical analysis (Fig. 3). In HE‐stained sections, there were some transverse sections of injection sites that included many cells in both the pCAGGS‐ and pCAGGS‐HSP105‐immunized muscles. But only in the transverse sections of the injection sites in pCAGGS‐HSP105‐immunized muscles could we observe many cells expressing HSP105 at a high level, and also a considerable number of both CD4+ T cells and CD8+ T cells. Although we did not immunohistochemically stain the dendritic cells in these traverse sections, we did find some dendritic cell‐like large cells.
Figure 3.
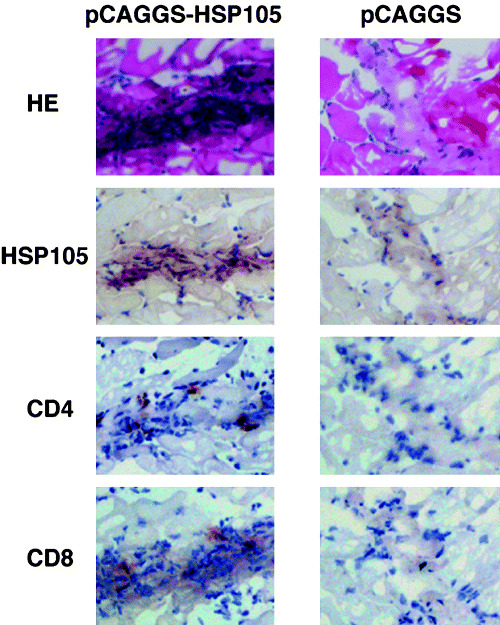
Expression of HSP105 protein and infiltration of CD4+ T cells and CD8+ T cells in the HSP105 DNA vaccine‐injected sites. To observe HSP105 expression and infiltrating cells in muscles injected with the HSP105 DNA vaccine, we carried out intramuscular immunizations with pCAGGS‐DNA into the right anterior tibialis muscle, and with pCAGGS‐HSP105 DNA into the left anterior tibialis muscle in four mice. After 48 h, we killed the mice and studied their muscle tissue by using HE staining and histological analysis, and immunohistochemical analysis of HSP105, CD4, and CD8. Representative results are shown. Objective magnification was 400×.
Infiltration of CD4+ T cells and CD8+ T cells into the C26 tumor after vaccination
To observe the antitumor effects of HSP105 DNA‐vaccination, we evaluated the tumor using immunohistochemical staining of CD8 and CD4. Figure 4a shows the tumor inoculation sites from two HSP105 DNA‐immunized mice, a saline‐inoculated mouse, and a pCAGGS‐immunized mouse that did not reject the tumor challenge. There were few lymphocytes in the tumors removed from both the saline‐inoculated mouse and the pCAGGS immunized mouse, but there were many CD4+ T cells and considerable numbers of CD8+ T cells making contact with the tumor cells and surrounding the tumors removed from the two HSP105 DNA‐immunized mice. These layers of CD4+ T cells surrounding the tumor were thick in the case of HSP105 DNA vaccinated mice. Furthermore, there were a considerable number of CD8+ T cells and CD4+ T cells that had infiltrated into the tumor (Fig. 4a).
Figure 4.
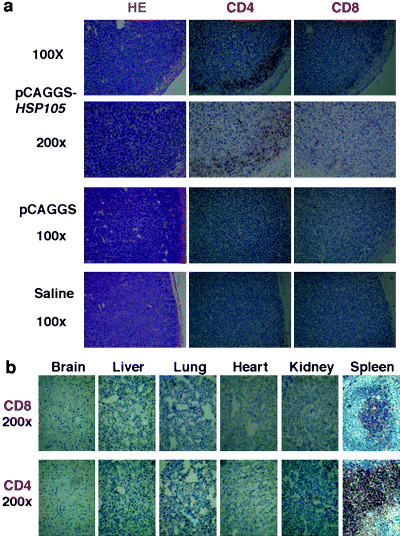
Vaccination with HSP105 DNA induced infiltration of both CD4+ T cells and CD8+ T cells into C26 tumors, but not into normal tissues. (a) Subcutaneous C26 tumors removed from two HSP105 DNA‐immunized mice, a saline‐inoculated mouse, and a pCAGGS‐immunized mouse that did not reject the tumor challenges were analyzed using immunohistochemical staining with anti‐CD4 mAb and anti‐CD8 mAb. (b) Normal tissues of mice vaccinated with HSP105 DNA were histologically and immunohistochemically examined. Objective magnification was 200×. The spleen was used as a positive control for staining of both CD4 and CD8.
Vaccination with HSP105 DNA did not induce damage of normal tissues
HSP105 expression in normal adult mice is limited to several tissues, and HSP105 expression levels in these tissues are lower than those in C26 (C20) tumor cells, which suggests a low risk of damage to normal tissue as a result of immune responses to the HSP105 antigen. To evaluate the risk of autoaggression by immunization against self‐HSP105, the tissues of mice immunized with HSP105 DNA were histologically examined. All mice were apparently healthy, and without abnormalities, suggesting autoimmunity for, for example, dermatitis, arthritis, or neurological disorders. The brain, liver, lung, heart, kidney, and spleen tissues of HSP105‐immunized mice were critically scrutinized and compared with those of normal mice. These tissues had normal structure and cellularity for each of the two groups examined, and pathological changes caused by immune response, such as infiltrations of CD8+ or CD4+ T cells, or tissue destruction and repair, were not present (Fig. 4b). Although CD4+ T cells and CD8+ T cells infiltrated into the C26 tumor (Fig. 4a), infiltration of CD4+ T cells or CD8+ T cells was not observed in any of the normal adult tissues examined (Fig. 4b). These results indicate that T cells stimulated with the HSP105 DNA vaccine do not recognize normal cells that express HSP105 at physiological levels.
Anti‐C26 tumor adoptive immunity elicited by injection with CD4+ T cells or CD8+ T cells from HSP105 DNA‐vaccinated mice
Antitumor responses could be augmented by homeostatic T cell proliferation in the periphery, involving the expansion of T cells recognizing MHC/tumor antigenic peptide ligands.( 21 , 22 , 23 ) To ascertain that the tumor rejections induced by HSP105 DNA vaccination were mediated through the activation of CD8+ T cells or CD4+ T cells, in a homeostatic lymphocyte proliferation model, we subcutaneously inoculated BALB/c mice with C26 cells (3 × 104) 6 days after sublethal irradiation (5 Gy). We intravenously injected 1.5 × 107 whole spleen cells or 3 × 106 CD8+ T cells, CD4+ T cells, NK cells, or CD8− CD4− NK− cells derived from each untreated or HSP105 DNA‐vaccinated mouse on day 3 before the tumor inoculation (Fig. 5a). Measurements of tumor size were continued for 22 days after inoculation with the tumor cells (Fig. 5b). Each group consisted of four mice. Inoculation with whole spleen cells or CD8+ T cells, CD4+ T cells, NK cells, or CD8− CD4− NK− cells derived from untreated mice, and with NK cells, or CD8− CD4− NK− cells derived from HSP105 DNA‐vaccinated mice did not cause the mice to reject challenges with C26 cells (3 × 104). Conversely, two of the four mice (50%) that were treated with whole spleen cells, CD8+ T cells, or CD4+ T cells derived from HSP105 DNA‐vaccinated mice completely rejected challenges with C26 cells (3 × 104; Fig. 5b–d). Thus, sublethally irradiated lymphopenic mice transfused with CD4+ T cells or CD8+ T cells derived from HSP105 DNA‐vaccinated mice displayed tumor growth inhibition. These results suggest that both CD4+ and CD8+ T cells play critical roles in antitumor immunity induced by immunization with the HSP105 DNA‐vaccine. The mice shown in Figure 5 were killed more than 100 days after lymphocyte transfer, respectively. All mice were apparently healthy and without abnormalities, suggesting autoimmunity for, for example, dermatitis, arthritis, or neurological disorders. The brain, liver, lung, heart, kidney, and spleen tissues of HSP105 DNA‐immunized mice were critically scrutinized and compared with those of normal mice. These tissues had normal structures and cellularity for each of the two groups examined, and pathological changes caused by immune response, such as CD8+ or CD4+ T lymphocyte infiltration or tissue destruction and repair, were not present, as shown in Figure 4b. These results indicate that T cells stimulated with HSP105 do not recognize normal cells that express HSP105 at physiological levels.
Figure 5.
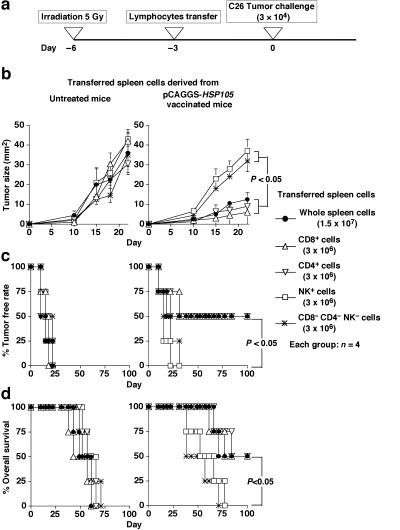
Injection of either CD4+ T cells or CD8+ T cells sensitized with HSP105 DNA vaccine into sublethally irradiated mice elicited effective antitumor adoptive immunity. (a) Experimental protocol; each group consisted of four mice. (b) Suppression of the growth of HSP105‐expressing C26 tumors inoculated subcutaneously into mice transferred with each group of spleen cells. Tumor area was calculated as the product of width and length. The result is presented as the mean area of tumor ± SE, and we evaluated the statistical significance using the unpaired t‐test. (c,d) Percentage tumor free rate (c) and percentage overall survival (d) were calculated using the Kaplan–Meier method, and the statistical significance of differences in survival time between groups was evaluated using Wilcoxon's test.
Involvement of both CD4+ T cells and CD8+ T cells in protection against B16.F10 induced by HSP105 DNA‐vaccination
To determine the role of CD4+ T cells and CD8+ T cells in the protection against B16.F10 tumor cells induced by HSP105 DNA‐vaccination, we depleted mice of CD4+ T cells or CD8+ T cells by treatment with anti‐CD4 or anti‐CD8 mAb in vivo. More than 90% of CD4+ T cells or CD8+ T cells were depleted (data not shown). During this procedure, mice were immunized with DNA vaccine and challenged with B16.F10 cells (Fig. 6a). Depletion of either CD4+ T cells or CD8+ T cells almost totally abrogated the protective immunity induced by immunization with HSP105 DNA vaccine (Fig. 6b–d). These results suggest that both CD4+ T cells and CD8+ T cells play critical roles in antitumor immunity induced by immunization with HSP105 DNA vaccine.
Figure 6.
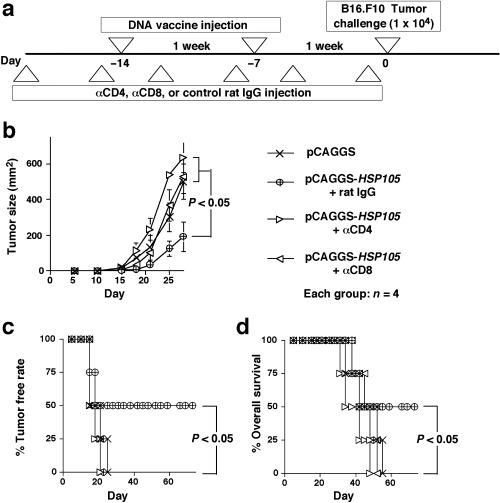
Involvement of both CD4+ T cells and CD8+ T cells in protection against B16.F10 induced by vaccination with HSP105 DNA. (a) Experimental protocol for in vivo depletion of CD4+ T cells and CD8+ T cells. Each group consisted of four mice. (b) Suppression of the growth of HSP105‐expressing B16.F10 tumors inoculated subcutaneously into mice vaccinated with HSP105 DNA. Tumor area was calculated as the product of width and length. Data are presented as mean area of tumor ± SE, and we evaluated the statistical significance using the unpaired t‐test. (c,d) Percentage tumor free rate (c) and percentage overall survival (d) were calculated using the Kaplan–Meier method, and the statistical significance of differences in survival time between groups was evaluated using Wilcoxon's test.
Discussion
Advances in molecular biology and tumor immunology have paved the way for identification of a large number of genes encoding TAA and antigenic peptides recognized by tumor‐reactive CTL, hence peptide‐based cancer immunotherapy has been the focus of much research.( 24 , 25 , 26 ) However, current clinical trials for peptide‐based immunotherapy have rarely resulted in tumor regression.( 27 ) The immunogenicity of these tumor antigenic peptides or the vaccination strategy may be sufficient to induce CTL responses but not to elicit CD4+ T cells.
DNA‐based immunization is potentially a powerful method for immunizing against microbial, viral, and tumor antigens through both humoral and cell‐mediated immune responses.( 28 ) The generation of T‐cell immunity involves local target cell transfection and protein antigen production, which is taken up by host APC, leading to cross‐presentation in draining lymph nodes; in addition, direct DNA transfection into APC in peripheral tissue has also been demonstrated.( 29 ) Compared with orthodox vaccines consisting of tumor proteins or viral components, DNA vaccination stimulates host immunity against transgene‐encoding proteins without the processes related to protein purification. In the present study, a DNA vaccine was used to activate HSP105‐specific tumor immunity.
Although the SEREX method facilitated the identification of tumor antigens that could be recognized by antibodies and CD4+ Th cells, few of their T cell epitopes have been determined.( 2 , 30 ) We previously reported that HSP105, identified by SEREX of pancreatic adenocarcinoma, was overexpressed specifically in a variety of human cancers, including pancreatic and colon adenocarcinoma.( 1 , 5 ) Other investigators identified HSP105 by SEREX using other cDNA libraries derived from tissues including colorectal cancer, melanoma, and normal testis. HSP105 are complexes associated with HSP70/HSC70,( 31 , 32 ) which negatively regulate HSP70/HSC70 chaperone activity.( 33 ) In addition, HSP105 protects neuronal cells against the apoptosis induced by various stresses.( 34 ) HSP105 consists of HSP105α and HSP105β. HSP105α is a constitutively expressed 105‐kDa HSP that is induced by a variety of stresses, whereas HSP105β is a 90‐kDa HSP that is specifically induced by heat shock at 42°C. HSP105β is a truncated form of HSP105α.( 12 ) We used in this study the mouse HSP105α DNA and protein. Recently, Subjeck and colleagues reported that recombinant HSP110 and cancer antigens such as Her2/neu or gp100 complexes are powerful cancer vaccines.( 8 , 9 , 35 ) Their HSP110( 11 ) and our HSP105α are in fact the same protein.
Although they noted that HSP110 did not have immunogenic properties, we emphasize in this study that HSP105 does have a strong immunogenic action. Although we did not identify the HSP105‐derived epitope peptides of CD8+ T‐cells or CD4+ T‐cells in this study, we did prove that HSP105 itself could induce both CD4+ T‐cells and CD8+ T‐cells to become reactive to tumor cells expressing HSP105. As shown in Figure 5, in a homeostatic lymphocyte proliferation model, we demonstrated that adoptive transfer of either CD4+ T cells or CD8+ T cells alone into sublethally irradiated mice was sufficient to reject C26 cells that do not express MHC class II molecules. To ascertain whether this is also true for B16.F10 that express both MHC class I and II molecules in the presence of interferon (IFN)‐γ, further investigation is needed. As shown in Figure 6, we demonstrated that both CD4+ T cells and CD8+ T cells were required for rejection of B16.F10 in the induction phase. In terms of the mechanism for the rejection of C26 tumors, we have other data relating to vaccination with HSP105 protein‐pulsed BM‐DC instead of HSP105 DNA vaccination. In those experiments, we also demonstrated that both CD4+ T cells and CD8+ T cells were required for rejection of not only B16.F10 but also C26 in the induction phase by depleting CD4+ T cells and CD8+ T cells using the in vivo administration of antibodies (unpublished data). Therefore, both HSP105‐specific CD4+ T cells and CD8+ T cells seem to be important for the rejection of HSP105‐expressing tumors in the induction phase, and either CD4+ T cells or CD8+ T cells can independently exert anti‐C26 tumor effects in the effector phase in a homeostatic lymphocyte proliferation model.
It has been reported that antigen‐specific CD4+ T‐cell help is required to activate memory CD8+ T cells to fully functional effector killer cells.( 36 ) The peptides derived from exogenous antigens acquired by endocytosis are typically presented on MHC class II molecules on the surface of APC, and activate CD4+ T cells. We observed in this study that CD4+ T cells specific to HSP105, in fact, have an important role in tumor rejection, even when tumors do not express MHC class II molecules, such as the C26 tumors used in this study. It was recently reported that tumor‐specific CD4+ T cells may have a pivotal role in preventing early tumorigenesis by secreting IFN‐γ and stimulating the classical macrophage‐activation pathway. This results in the inhibition of tumor cell growth, even when tumor cells themselves do not express MHC class II molecules.( 37 ) To better understand the mechanism of C26 tumor rejection by HSP105‐specific CD4+ T cells, further studies are needed. Furthermore, peptides derived from exogenous self‐antigen, HSP105, acquired by endocytosis are possibly presented by MHC class I molecules on the surface of APC by cross‐presentation to activate CD8+ T cells.
Because HSP are present in all organisms, low levels of human HSP‐derived peptides serve as harbingers of autoimmune responses after CTL have been primed to respond to bacterial HSP‐derived peptides.( 38 ) However, because many cancers overexpress HSP, CTL‐based vaccines that elicit an anti‐HSP response might be effective against many different tumors.( 39 ) Indeed, in this study, HSP105 itself evoked T‐cell‐mediated tumor rejection without autoimmune reactions. In the present paper, all results shown in the figures were obtained using female mice, but we have carried out the same experiment using male mice. HSP105 DNA vaccination did not induce T‐cell infiltration or damage in testis tissue (in which HSP105 is highly expressed). Furthermore, HSP105 DNA vaccination was also able to induce antitumor immunity in male mice (data not shown), indicating that male mice did not acquire immunological tolerance to HSP105 expressed in testis tissue.
To substantiate the specificity for HSP105, we searched for mouse cancer cell lines derived from BALB/c mice and C57BL/6 mice that do not express HSP105. However, all cancer cell lines we examined strongly expressed HSP105. BALB/3T3 fibroblasts expressed HSP105 relatively weakly, but these cells unfortunately did not form tumors in mice. Further investigations are needed to clarify whether HSP105 DNA vaccination affects the growth of some tumors that do not express HSP105.
We showed in this study that HSP105 DNA vaccination can prime T cells to be reactive to tumor cells expressing HSP105 in vivo, and that growth of C26 and B16.F10 cells expressing HSP105 was prevented without inducing autoimmune destruction in murine subcutaneous CRC and melanoma models. We believe that HSP105 DNA vaccination is a novel strategy for the prevention of CRC and melanoma in patients treated surgically who are at high risk of recurrence of CRC or melanoma. Whether or not HSP105 is an ideal target for immunotherapy in human cancers will continue to be investigated in our laboratory.
Acknowledgments
We thank Drs Hidetake Matsuyoshi, Shinya Hirata, Yoshiaki Ikuta, and Daiki Fukuma (Department of Immunogenetics, Graduate School of Medical Sciences, Kumamoto University) for technical assistance, Dr Junichi Miyazaki, Osaka University, for providing the pCAGGS vector, and Dr Kyoichi Shimomura (Fujisawa Pharmaceutical Co.), for providing the cell line. This work was supported in part by Grants‐in‐Aid (no. 12213111 to Y. Nishmura, and no. 14770142 to T. Nakatsura) from the Ministry of Education, Science, Technology, Sports and Culture, Japan.
M. Miyazaki and T. Nakatsura contributed equally to this work.
References
- 1. Nakatsura T, Senju S, Yamada K, Jotsuka T, Ogawa M, Nishimura Y. Gene cloning of immunogenic antigens overexpressed in pancreatic cancer. Biochem Biophys Res Commun 2001; 281: 936–44. [DOI] [PubMed] [Google Scholar]
- 2. Nakatsura T, Senju S, Ito M, Nishimura Y, Itoh K. Cellular and humoral immune responses to a human pancreatic cancer antigen, coactosin‐like protein, originally defined by the SEREX method. Eur J Immunol 2002; 32: 826–36. [DOI] [PubMed] [Google Scholar]
- 3. Monji M, Senju S, Nakatsura T et al. Head and neck cancer antigens recognized by the humoral immune system. Biochem Biophys Res Commun 2002; 294: 734–41. [DOI] [PubMed] [Google Scholar]
- 4. Monji M, Nakatsura T, Senju S et al. Identification of a novel human cancer/testis antigen, KM‐HN‐1, recognized by cellular and humoral immune responses. Clin Cancer Res 2004; 10: 6047–57. [DOI] [PubMed] [Google Scholar]
- 5. Kai M, Nakatsura T, Egami H, Senju S, Nishimura Y, Ogawa M. Heat shock protein 105 is overexpressed in a variety of human tumors. Oncol Rep 2003; 10: 1777–82. [PubMed] [Google Scholar]
- 6. Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol 2002; 20: 395–425. [DOI] [PubMed] [Google Scholar]
- 7. Hickman‐Miller HD, Hildebrand WH. The immune response under stress. The role of HSP‐derived peptides. Trends Immunol 2004; 25: 427–33. [DOI] [PubMed] [Google Scholar]
- 8. Manjili MH, Henderson R, Wang XY et al. Development of a recombinant HSP110‐HER‐2/neu vaccine using the chaperoning properties of HSP110. Cancer Res 2002; 62: 1737–42. [PubMed] [Google Scholar]
- 9. Wang XY, Chen X, Manjili MH, Repasky E, Henderson R, Subjeck JR. Targeted immunotherapy using reconstituted chaperone complexes of heat shock protein 110 and melanoma‐associated antigen gp100. Cancer Res 2003; 63: 2553–60. [PubMed] [Google Scholar]
- 10. Wang XY, Li Y, Manjili MH, Repasky EA, Pardoll DM, Subjeck JR. Hsp110 over‐expression increases the immunogenicity of the murine CT26 colon tumor. Cancer Immunol Immunother 2002; 51: 311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee‐Yoon D, Easton D, Murawski M, Burd R, Subjeck JR. Identification of a major subfamily of large hsp70‐like proteins through the cloning of the mammalian 110‐kDa heat shock protein. J Biol Chem 1995; 270: 15725–33. [DOI] [PubMed] [Google Scholar]
- 12. Yasuda K, Nakai A, Hatayama T, Nagata K. Cloning and expression of murine high molecular mass heat shock proteins, HSP105. J Biol Chem 1995; 270: 29718–23. [DOI] [PubMed] [Google Scholar]
- 13. Ishihara K, Yasuda K, Hatayama T. Molecular cloning, expression and localization of human 105 kDa heat shock protein, hsp105. Biochim Biophys Acta 1999; 1444: 138–42. [DOI] [PubMed] [Google Scholar]
- 14. Hattori K, Matsushita R, Kimura K, Abe Y, Nakashima E. Synergistic effect of indomethacin with adriamycin and cisplatin on tumor growth. Biol Pharm Bull 2001; 24: 1214–7. [DOI] [PubMed] [Google Scholar]
- 15. Nakatsura T, Kageshita T, Ito S et al. Identification of glypican‐3 as a novel tumor marker for melanoma. Clin Cancer Res 2004; 10: 6612–21. [DOI] [PubMed] [Google Scholar]
- 16. Yoshitake Y, Nakatsura T, Monji M et al. Proliferation potential‐related protein, an ideal esophageal cancer antigen for immunotherapy, identified using complementary DNA microarray analysis. Clin Cancer Res 2004; 10: 6437–48. [DOI] [PubMed] [Google Scholar]
- 17. Nakatsura T, Komori H, Kubo T et al. Mouse homologue of a novel human oncofetal antigen, glypican‐3, evokes T cell‐mediated tumor rejection without autoimmune reactions in mice. Clin Cancer Res 2004; 10: 8630–40. [DOI] [PubMed] [Google Scholar]
- 18. Matsuyoshi H, Senju S, Hirata S, Yoshitake Y, Uemura Y, Nishimura Y. Enhanced priming of antigen‐specific CTLs in vivo by embryonic stem cell‐derived dendritic cells expressing chemokine along with antigenic protein: application to antitumor vaccination. J Immunol 2004; 172: 776–86. [DOI] [PubMed] [Google Scholar]
- 19. Maruyama H, Higuchi N, Nishikawa Y et al. High‐level expression of naked DNA delivered to rat liver via tail vein injection. J Gene Med 2002; 4: 333–41. [DOI] [PubMed] [Google Scholar]
- 20. Hylander BL, Chen X, Graf PC, Subjeck JR. The distribution and localization of hsp110 in brain. Brain Res 2000; 869: 49–55. [DOI] [PubMed] [Google Scholar]
- 21. Dummer W, Niethammer AG, Baccala R et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest 2002; 110: 185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maine GN, Mule JJ. Making room for T cells. J Clin Invest 2002; 110: 157–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dudley ME, Wunderlich JR, Robbins PF et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298: 850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van Der Bruggen P, Traversari C, Chomez P et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991; 254: 1643–7. [DOI] [PubMed] [Google Scholar]
- 25. Kawakami Y, Eliyahu S, Delgado CH et al. Identification of a human melanoma antigen recognized by tumor‐infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA 1994; 91: 6458–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen YT, Gure AO, Tsang S et al. Identification of multiple cancer/testis antigens by allogeneic antibody screening of a melanoma cell line library. Proc Natl Acad Sci USA 1998; 95: 6919–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rosenberg SA, Yang JC, Schwartzentruber DJ et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 1998; 4: 321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kumar V, Sercarz E. Genetic vaccination: the advantages of going naked. Nat Med 1996; 2: 857–9. [DOI] [PubMed] [Google Scholar]
- 29. Leitner WW, Ying H, Restifo NP. DNA and RNA‐based vaccines: principles, progress and prospects. Vaccine 1999; 18: 765–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jager E, Chen YT, Drijfhout JW et al. Simultaneous humoral and cellular immune response against cancer‐testis antigen NY‐ESO‐1: definition of human histocompatibility leukocyte antigen (HLA)‐A2‐binding peptide epitopes. J Exp Med 1998; 187: 265–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wakatsuki T, Hatayama T. Characteristic expression of 105‐kDa heat shock protein (HSP105) in various tissues of nonstressed and heat‐stressed rats. Biol Pharm Bull 1998; 21: 905–10. [DOI] [PubMed] [Google Scholar]
- 32. Hatayama T, Yasuda K. Association of HSP105 with HSC70 in high molecular mass complexes in mouse FM3A cells. Biochem Biophys Res Commun 1998; 248: 395–401. [DOI] [PubMed] [Google Scholar]
- 33. Yamagishi N, Nishihori H, Ishihara K, Ohtsuka K, Hatayama T. Modulation of the chaperone activities of Hsc70/Hsp40 by Hsp105alpha and Hsp105beta. Biochem Biophys Res Commun 2000; 272: 850–5. [DOI] [PubMed] [Google Scholar]
- 34. Hatayama T, Yamagishi N, Minobe E, Sakai K. Role of hsp105 in protection against stress‐induced apoptosis in neuronal PC12 cells. Biochem Biophys Res Commun 2001; 288: 528–34. [DOI] [PubMed] [Google Scholar]
- 35. Manjili MH, Wang XY, Chen X et al. HSP110‐HER2/neu chaperone complex vaccine induces protective immunity against spontaneous mammary tumors in HER‐2/neu transgenic mice. J Immunol 2003; 171: 4054–61. [DOI] [PubMed] [Google Scholar]
- 36. Gao FG, Khammanivong V, Liu WJ, Leggatt GR, Frazer IH, Fernando GJ. Antigen‐specific CD4+ T‐cell help is required to activate a memory CD8+ T cell to a fully functional tumor killer cell. Cancer Res 2002; 62: 6438–41. [PubMed] [Google Scholar]
- 37. Corthay A, Skovseth DK, Lundin KU et al. Primary antitumor immune response mediated by CD4+ T cells. Immunity 2005; 22: 371–83. [DOI] [PubMed] [Google Scholar]
- 38. Zugel U, Schoel B, Yamamoto S, Hengel H, Morein B, Kaufmann SH. Cross recognition by CD8 T cell receptor alpha beta cytotoxic T lymphocytes of peptides in the self and the mycobacterial hsp60 which share intermediate sequence homology. Eur J Immunol 1995; 25: 451–8. [DOI] [PubMed] [Google Scholar]
- 39. Faure O, Graff‐Dubois S, Bretaudeau L et al. Inducible Hsp70 as target of anticancer immunotherapy: Identification of HLA‐A*0201‐restricted epitopes. Int J Cancer 2004; 108: 863–70. [DOI] [PubMed] [Google Scholar]