

Abstract
Metastatic prostate tumors in the bone microenvironment stimulate bone resorption, resulting in release of growth factors from the bone matrix that play important roles in tumor growth and osteoclast induction. Transforming growth factor β (TGFβ) is one of the most abundantly stored cytokines in bone matrix, regulating diverse biological activities. Here we evaluate its involvement in prostate tumor growth in the bone microenvironment, comparing with tumor growth in the subcutaneous microenvironment as a control. Rat prostate tumors were transplanted onto the cranial bone and into the subcutis of F344 male rats. Tumor cell proliferation, apoptosis, and TGFβ signal transduction were compared between the tumor–bone interface and the tumor–subcutaneous interface. Effects of TGFβ on osteoclast differentiation were also evaluated in vitro. Inhibitory effects of TGFβ receptor 1 antisense oligonucleotide on TGFβ signaling, osteolysis, osteoblasts, and tumor growth were examined in vivo. Osteolytic changes were extensively observed at the tumor–bone interface, where the TGFβ level, TGFβ signal transduction, and tumor cell proliferation were higher than at the tumor–subcutaneous interface. In vitro treatment with receptor activator of nuclear factor‐κB ligand induced osteoclast differentiation of bone marrow stromal cells, and additional exposure to TGFβ exerted promotive effects on osteoclast induction. Intratumoral injection of TGFβ receptor 1 antisense oligonucleotide significantly reduced TGFβ signal transduction, osteolysis, induction of osteoclast and osteoblast, and tumor cell proliferation. Thus, we experimentally show that TGFβ derived from bone matrix promotes cell proliferation of rat prostate cancer and osteoclast activation‐associated osteolysis in the bone microenvironment. (Cancer Sci 2008; 99: 316–323)
Abbreviations:
- ASO
antisense oligonucleotide
- BrdU
bromodeoxyuridine
- ELISA
enzyme‐linked immunosorbent assay
- F344
Fisher 344
- IHC
immunohistochemical
- NF‐κB
nuclear factor‐κB
- NTB
non‐tumor–bone interface area
- NTS
non‐tumor–subcutaneous interface area
- PCNA
proliferating cell nuclear antigen
- PCR
polymerase chain reaction
- pSmad2
phosphorylated Smad2
- RANKL
receptor activator of nuclear factor‐κB ligand
- RCO
random control oligonucleotide
- TB‐interface
tumor–bone interface
- TGFβ
transforming growth factor β
- TS‐interface
tumor–subcutaneous interface
- TGFβR1‐ASO
transforming growth factor β receptor 1 antisense oligonucleotide
- TRAP
tartrate‐resistant acid phosphatase
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling
- VC
vehicle control.
Bone metastases are the most critical complications with advanced prostate cancer, resulting in severe pain, morbidity, and often mortality.( 1 , 2 ) Conventional therapies do not allow a curative outcome for prostate cancer bone metastasis,( 3 , 4 ) and elucidation of molecular mechanisms that regulate prostate tumor growth in the bone microenvironment is essential for development of new therapeutic approaches to prevent bone metastasis.
Once cancer cells reach the bone microenvironment, a reciprocal cellular interaction between tumor and bone stromal cells was observed.( 5 , 6 ) This interaction between the tumor and bone stromal cells has been commonly referred to as a “vicious cycle”, whereby tumor and stromal cells in the bone secrete several factors beneficial to each other.( 7 , 8 , 9 ) In the vicious cycle, metastatic prostate tumor cells in the bone microenvironment produce cytokines that stimulate osteoclastic bone resorption. This then results in the release of growth factors such as TGFβ,( 10 ) insulin‐like growth factors,( 10 ) and bone morphological proteins( 11 ) from the bone matrix.
In bone metastatic sites, prostate tumor cells, osteoblasts, and osteoclasts thus interact with each other.( 12 ) The activated osteoclasts expand the space in hard calcified bone tissue to assist tumor growth. Thus, osteolysis as well as tumor cell proliferation in the bone microenvironment plays critical roles in the growth of prostate cancer at bone metastasis sites.
TGFβ has been shown to regulate a diverse set of biological activities, including proliferation,( 13 ) apoptosis,( 14 ) differentiation,( 13 ) tumor cell motility, extracellular matrix deposition, and angiogenesis.( 15 , 16 ) It is one of the most abundantly stored cytokines in bone matrix,( 17 ) and bone‐derived TGFβ is known to stimulate bone absorption.( 18 , 19 ) However, its effects on tumor cell proliferation in the bone microenvironment have yet to be fully elucidated.
Previously, we established transplantable rat prostate carcinomas from tumors that were originally induced by 3,2′‐dimethyl‐4‐aminobiphenyl and testosterone propionate in F344 rats (PLS‐P).( 20 ) A 100% incidence of lung metastases was observed 12 weeks after subcutaneous transplantation in F344 male rats.( 21 ) Recently, we developed a syngeneic rat model that mimics human prostate cancer bone metastasis with respect to tumor stromal interaction.( 22 ) When the PLS‐P (moderately differentiated adenocarcinoma) was transplanted onto the surface of the calvaria, we observed osteolytic and osteoblastic changes at the tumor‐bone (TB) interface, mimicking the histopathological features of bone metastases of human prostate cancer.( 22 ) Thus, our model provides a good tool to examine prostate carcinoma behavior and growth in the bone microenvironment.
In order to elucidate the role of TGFβ on tumor growth and osteolysis in the bone microenvironment, we used our cranial bone–prostate cancer model. As a control, the same tumor was also transplanted into the subcutis. Tumor cell proliferation and osteoclast induction, TGFβ levels, and phosphorylation of Smad 2 as a marker of TGFβ signal transduction were then compared between the TB‐interface and the TS‐interface. To clarify the association between TGFβ and induction of osteoclast and osteoblast, effects of TGFβ on the osteclast differentiation from primary cultured bone marrow cells was also evaluated, along with effects of an antisense oligonucleotide for TGFβ receptor 1 on osteolysis and tumor growth in the bone microenvironment in vivo.
Materials and Methods
Animals. A total of 33 F344 male rats were obtained at 5 weeks of age from Charles River Japan (Atsugi, Japan) and maintained in plastic cages (three animals per cage) with hard wood chips as bedding in an air‐conditioned room at 22 ± 2°C and 55 ± 5% humidity with a 12/12 h L/D cycle. Food (Oriental MF; Oriental Yeast, Tokyo, Japan) and tap water were available ad libitum. The research was conducted according to the Guidelines for the Care and Use of Laboratory Animals of Nagoya City University Medical School and the experimental protocol was approved by the Institutional Animal Care and Use Committee (H17‐23).
Experimental procedure. PLS‐P (approximately 0.15 g) was transplanted onto the cranial bone and into the subcutis of the back of each of the F344 rats, as previously described.( 21 , 22 ) Tumor growth at the implanted sites and body weights were measured weekly. Four weeks after transplantation, 18 rats were killed, and cranial and subcutaneous tumors were removed and then divided in two, with one half used for histology (hematoxylin–eosin staining). The other half of each cranial tumor was separated into TB‐interface or non‐tumor‐bone interface (NTB) and the other half of each subcutaneous tumor was separated into TS‐interface or non‐tumor‐sub cutaneous interface (NTS). Pieces of the tumors and organs were then flash frozen for protein analysis or RNA extraction. For histological examination and immunohistochemistry, samples were fixed with paraformaldehyde for 48 h. The tissues were then transferred into a decalcification solution (15% ethylenediaminetetraacetic acid with glycerol, pH 7.4–7.5) for 6 days and subsequently paraffin embedded.
TRAP staining and immunohistochemistry. To detect activated osteoclasts in vitro and in vivo, TRAP assays were carried out according to manufacturer's instructions (Sigma‐Aldrich, St Louis, MO). TRAP‐positive multinucleated cells were counted under a light microscope. For the in vivo detection of pSmad2 and PCNA, pSmad2 (Cell Signaling Technology, Lake Placid, NY) and PCNA (DAKO, Denmark) antibodies were diluted at 1:50 and 1:200, respectively, in blocking solution. After exposure overnight at 4°C and washing, the slides were incubated with biotinylated species‐specific secondary antibodies diluted 1:500 for 60 min (Vector Laboratories), washed again and then incubated with avidin–peroxidase complex (ABC; Vector Laboratories) for 45 min. The sections were developed using diaminobenzidine tetrahydrochloride substrate (Sigma‐Aldrich), counterstained with hematoxylin, dehydrated, and permanently mounted. To examine DNA replication in PLS‐P in the two different microenvironments, levels of incorporation of BrdU were examined. All rats were injected intraperitoneally with 10 mg/kg BrdU (Sigma‐Aldrich) in saline 1 h before they were killed. Tumors were dissected and immunostained as described previously.( 23 ) To detect apoptotic cells in PLS‐P, formalin‐fixed paraffin‐embedded sections were analyzed by the TUNEL method using the in situ apoptosis detection TUNEL kit (Takara, Shiga, Japan) according to the manufacturer's recommended protocol. For quantitative analysis, immunostained sections were examined under a light microscope connected to an image analysis system, the Image Processor for Analytical Pathology (Sumika Technos Corp., Osaka, Japan). The total numbers of nuclei and those positive for pSmad2, BrdU, PCNA, and TUNEL were assessed at a magnification of ×600 for each lesion. Lesions examined were TB‐interface, NTB, whole cranial tumor, TS‐interface, NTS, and whole subcutaneous tumor. Approximately 8000 nuclei per selected area of tumor were counted.
ELISA and immunoblotting. Tumors and other tissues were homogenized in T‐PER (Pierce, Hercules, CA), and protein concentrations were determined with a bicinchoninic acid protein assay kit (Pierce). TGFβ levels in the lysates were measured using ELISA kits (R&D Systems, Minneapolis, MN) following the manufacturer's protocols. For Western blot analysis, aliquots of 20 µg of tissue lysates were boiled in sodium dodecyl sulfate sample buffer containing 0.5 mol/L β‐mercaptoethanol, separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes and immunoblotted. Separated proteins were probed overnight at 4°C with anti‐β‐actin antibody (Sigma‐Aldrich), anti‐pSmad2 antibody (Cell Signaling Tecnology), anti‐Smad2 antibody (Cell Signaling Technology), and anti‐TGFβ receptor1 antibody (sc‐398; Santa Cruz Biotechnolgy, Santa Cruz, CA). The blots were washed and incubated for 1 h with biotinylated antispecies secondary antibodies (Amersham Biosciences, Piscataway, NJ) then visualized with enhancement by chemiluminescence using the ECL Western Blotting Detection Reagent (Amersham Biosciences).
Rat bone marrow primary culture. Rat bone marrow cells were obtained from the tibiae of 4‐week‐old male F344 rats. Tibiae were removed aseptically, cut across the epiphyses, and the marrow was flushed out with α‐modification minimum essential medium (Sigma‐Aldrich) containing 10% fetal bovine serum (Sigma‐Aldrich) using an 18‐gauge needle. The cells were incubated for 3 h at 37°C in a humidified atmosphere containing 5% CO2 to separate the adherent stromal cell fraction from the non‐adherent osteoclast‐precursor cells as osteoclasts by the method reported previously.( 24 ) After incubation, the collected non‐adherent cells were cultured in α‐modification minimum essential medium containing 10% fetal bovine serum at 1.0 × 106 cells/mL/well in 24‐well plates (Corning, Corning, NY). The plates were incubated for 24 h, then 10 ng/mL soluble RANKL and various concentrations of TGFβ (0, 0.001, 0.01, 0.1, and 1 ng/mL) were added. Medium was changed every 3 days. After culturing for 1 week, cells were used for TRAP staining, and TRAP‐positive multinucleated cells were counted.
RNA extraction and real‐time PCR. Frozen tumor specimens were microdissected with careful separation of the TB‐interface, TS‐interface, NTB, and NTS. Total RNAs from each area were extracted using ISOGEN (Wako Pure Chemical, Tokyo, Japan). After DNase treatment, 1 µg of RNA was converted to cDNA with avian myoblastosis virus reverse transcriptase (Takara) in 20 µL reaction mixture. Two microliters of cDNA samples was amplified in 20 µL reaction mixtures using FastStart DNA Master SYBR Green I and a Light Cycler apparatus (Roche Diagnostics, Germany). Primers were used for TGFβ‐R1, 5′‐TCT‐GCT‐TCG‐TCT‐GCA‐TTG‐CA‐3′ and 5′‐CGG‐AAC‐CAT‐GAA‐CGC‐TCT‐TCT‐3′; for TGFβ‐R2, 5′‐ACC‐CTC‐CTT‐TTC‐CTC‐GAT‐CAA‐C‐3′ and 5′‐CCT‐GAC‐TGG‐AAG‐CAG‐CTT‐TCA‐T‐3′; and for glyceraldehyde‐3‐phosphate dehydrogenase, 5′‐AGC‐CTC‐GTC‐CCG‐TAG‐ACA‐AAA‐3′ and 5′‐GAT‐GAC‐AAG‐CTT‐CCC‐ATT‐CTC‐G‐3′.
Initial denaturation at 95°C for 10 min was followed by 40 cycles with denaturation at 95°C for 15 s, annealing at 60°C for 5 s, and elongation at 72°C for 30 s. The fluorescence intensity of the double‐stranded specific SYBR Green I, reflecting the amount of formed PCR product, was monitored at the end of each elongation step. Glyceraldehyde‐3‐phosphate dehydrogenase mRNA levels were used to normalize the sample cDNA content.
Synthesis and treatment of antisense oligonucleotide. Phosphorothioate ASOs used in this study were generated by Biognostik (Gottingen, Germany). A total of five ASOs, including two control ASOs were synthesized, the reverse complement of the target sequence containing no cross‐homology to other sequences on the GenBank database, and three ASOs for TGFβ receptor1 with different sequences. Five nanomoles of each ASO diluted with saline was injected intratumorally into five groups of three rats every 2 days from weeks 3 to 4. The saline vehicle was injected into the other group of three rats. Tumor growth at the implanted sites and body weights were measured weekly and 4 weeks after transplantation, rats were killed and processed as described above. We examined the extent of bone destruction in several sections from each rat using a ‘bone destruction index’, the ratio of the length of osteolysis to the length of the cranial bone beneath the transplanted tumor, as described previously.( 22 , 25 )
Statistical analysis. For in vivo data, statistical analysis was carried out using the Kruskal–Wallis and Bonferroni–Dunn's multiple comparison tests. In vitro data are presented as mean ± SD. The statistical significance of in vitro findings was analyzed using a two‐tailed Student's t‐test and Bonferroni–Dunn's multiple comparison tests. A value of P < 0.05 was considered significant.
Results
Histological differences between TB and TS‐interfaces. PLS‐P grew considerably both on the cranial bone (Fig. 1a) and in the back subcutis (Fig. 1b), with no significant differences in tumor volume (data not shown). Histological analysis of coronal sections of the cranial tumor revealed extensive bone defects with many multinucleated cells and nodular osteoid formations with the osteoblasts in response to the growth of moderately differentiated adenocarcinoma at the TB‐interface (Fig. 1c). In contrast, tumors at the TS‐interface showed signs of induction of microvessels and trace stromal cells (Fig. 1d). Multinucleated cells appeared in bone destruction sites (Fig. 1e) were identified as osteoclasts by strong positive staining for TRAP (Fig. 1f).
Figure 1.
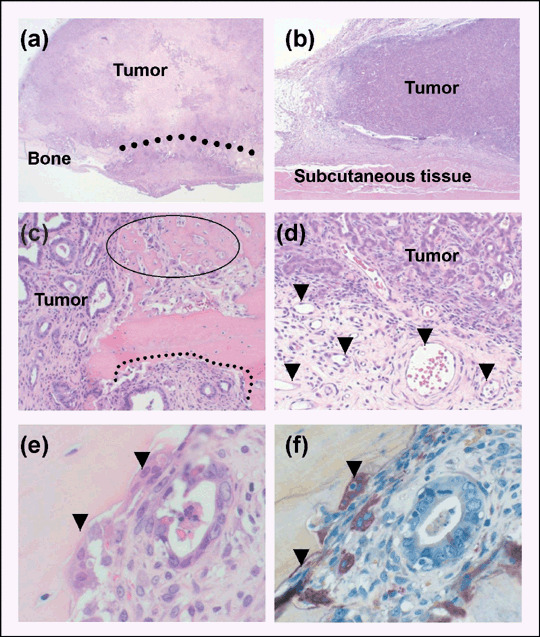
Histological differences between tumor–bone (TB) and tumor–subcutaneous (TS) interfaces in prostate tumors transplanted into the cranial surface and the subcutis of male rats. (a) Transplantation to the cranial surface destroyed bone. The dotted line indicates the bone destruction area (magnification: ×20). (b) Subcutaneous transplanted tumor (magnification: ×20). (c) Histology of the TB‐interface. The dotted line represents an osteolytic lesion. The circle indicates osteoid created by osteoblasts (magnification: ×100). (d) Histology of the TS‐interface. Arrowheads indicate microvessels (magnification: ×100). (e) High magnification of the TB‐interface. Arrowheads indicate multinucleated cells (magnification: ×400). (f) TRAP staining of a cranial transplanted tumor. Note tartrate‐resistant acid phosphatase‐positive multinucleated cells in the corresponding place as (e) (magnification: ×400).
TGFβ levels and signaling at TB and TS‐interfaces. TGFβ stored in the bone matrix has been shown to be released as a consequence of osteolysis.( 17 ) We compared TGFβ levels and TGFβ signaling between the TB and TS‐interfaces. ELISA assay revealed that the TGFβ level at the TB‐interface was higher than that at the TS‐interface (Fig. 2a). We also examined TGFβ levels in bone, prostate, lung, liver, kidney, and lymph node. The active form of TGFβ was the highest in bone (25 ng/g protein) and low in the others, including prostate (Fig. 2b). pSmad 2 was strongly positive in the nuclei of tumor cells at the TB‐interface (Fig. 2c) and weakly positive at the TS‐interface (Fig. 2d), whereas positive cells were rare at the NTB and NTS (data not shown). The positive cell index, evaluated by the Image Processor for Analytical Pathology (Sumika Technos Corp.), was higher at the TB than the TS‐interface (Fig. 2e). Western blot analysis revealed expression of pSmad2 also to be at the TB‐interface, correlating with total Smad2 expression (Fig. 2f).
Figure 2.
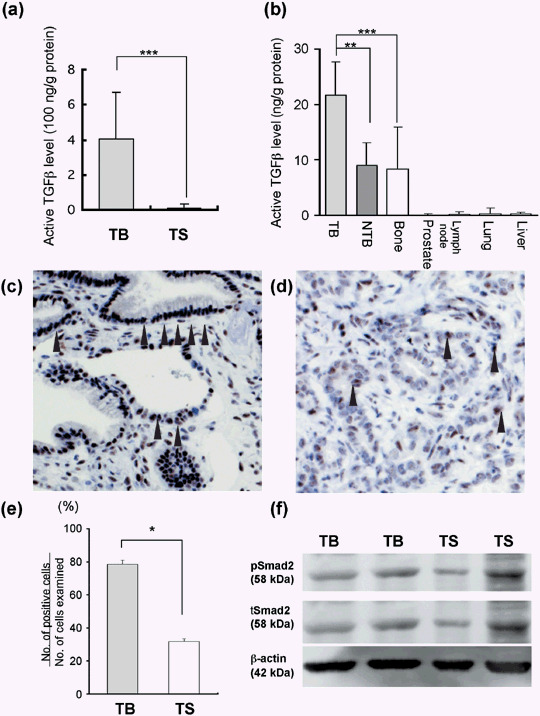
Transforming growth factor β (TGFβ) level and signaling at the tumor–bone (TB) and the tumor–subcutaneous (TS) interfaces in prostate tumors transplanted onto cranial bone and into the subcutis of male rats. (a) TGFβ level at the TB‐interface and the TS‐interface evaluated by enzyme‐linked immunosorbent assay. Data are mean ± SD from values for five samples. ***P < 0.001. (b) TGFβ level in several organs. (c,d) Immunostaining of phosphorylated Smad2 (pSmad2) at the TB‐interface (c) and the TS‐interface (d). The nuclei of pSmad2‐positive tumor cells are stained (arrowhead). (e) pSmad2‐positive tumor cell index evaluated by Image Processor for Analytical Pathology (Sumika Technos Corp., Osaka, Japan). *P < 0.05. (f) Western blot analysis of pSmad2 and total Smad2. Two different samples were loaded. NTB, non‐TB‐interface.
Tumor cell proliferation at TB and TS‐interfaces. BrdU‐positive cells were predominantly observed at the borders of the PLS‐P transplanted on the cranial bone (Fig. 3a) and in the subcutaneous area (Fig. 3b). At high magnification, positive cells were counted in three areas in both the cranial and subcutaneous tumors (TB‐interface (Fig. 3c), NTB, and whole cranial tumor; and TS‐interface (Fig. 3d), NTS, and whole subcutaneous tumor). The ratio of BrdU‐positive cells (labeling index) at the TB‐interface was significantly higher than that at the NTB, whole cranial tumor, TS‐interface (Fig. 3e), NTS, and whole subcutaneous tumor. Labeling indices at the NTB and NTS were almost the same as the TS‐interface value (Fig. 3e). TUNEL‐positive cells were also examined in the cranial and subcutaneous tumors, and the ratio of positive cells was not significantly different among the areas examined. (Fig. 3f).
Figure 3.
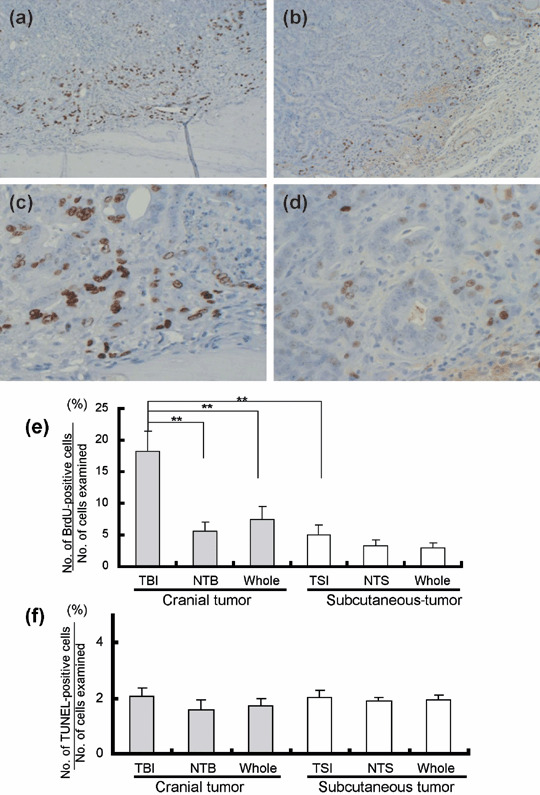
Tumor cell proliferation at the tumor–bone (TB) and the tumor–subcutaneous (TS) interfaces in prostate tumors transplanted onto cranial bone and into the subcutis of male rats. (a,b) Bromodeoxyuridine (BrdU)‐positive cells are predominantly located at the border of the tumor transplanted on the cranial bone (a) and in the subcutaneous area (b) (magnification: ×100 both plates). (c,d) High magnification of the TB‐interface (c) and the TS‐interface (d). Nuclei of BrdU‐positive cells are stained (magnification: ×400 both plates). (e) BrdU‐positive cell labeling indices for the TB‐interface, non‐TB‐interface (NTB), whole cranial tumor, TS‐interface, non‐TS‐interface (NTS), and whole subcutaneous tumor. (f) Terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate nick‐end labeling‐positive cell labeling indices for the TB‐interface, NTB, whole cranial tumor, TS‐interface, NTS, and whole subcutaneous tumor. **P < 0.01.
TGFβ promotes osteoclast differentiation in vitro. To examine the role of TGFβ in osteoclast differentiation, primary cultures of bone marrow cells obtained from F344 rats were incubated with an optimal concentration of soluble RANKL in the presence and absence of different concentrations of TGFβ for 7 days. TRAP stainig revealed that lower numbers of multinucleated cells were induced by medium alone treatment (Fig. 4a) than by RANKL treatment (Fig. 4b). Additional TGFβ significantly increased the number of osteoclasts (Fig. 4c,d) as compared with RANKL alone (Fig. 4e).
Figure 4.
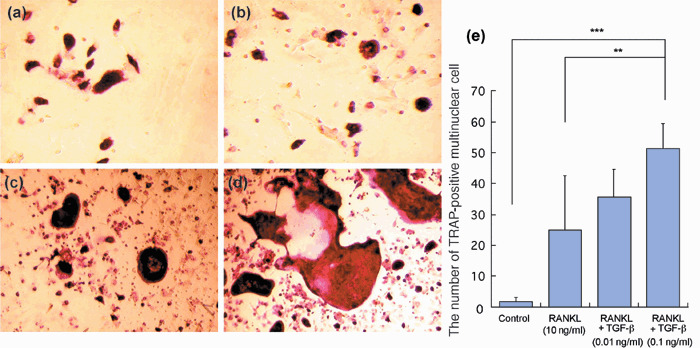
Transforming growth factor β (TGFβ) promotes osteoclast differentiation. Tartrate‐resistant acid phosphatase (TRAP) staining of cells from rat bone marrow cultured with α‐modification minimum essential medium alone (a), receptor activator of nuclear factor‐κB ligand (RANKL; 10 ng/mL) (b), RANKL + TGFβ (0.01 ng/mL) (c), and RANKL + TGFβ (0.1 ng/mL) (d) (magnification: ×200). Note the lower number of multinucleated cells induced by medium alone than with RANKL treatment. Additional TGFβ treatment is associated with an extra increment in osteoclasts. (e) Quantitative analysis of TRAP‐positive multinucleated cells. The numbers are averages of quadruplicate values. **P < 0.01; ***P < 0.001.
ASO study. We examined the silencing effect of TGFβ signaling on bone destruction and cell proliferation in vivo. Because cell proliferation, TGFβ levels, and indices of pSmad2 at the TS‐interface was similar to those at NTB, as shown in the 2, 3, we chose the NTB area as a control for the TB‐interface, instead of the TS‐interface. Western blot analysis showed that TGFβR1‐ASO treatment reduced TGFβR1 protein expression at the TB‐interface, but that vehicle control (VC) and random control oligonucleotide (RCO) did not (Fig. 5a). Quantitative reverse transcription–polymerase chain reaction analysis revealed similar findings for mRNA expression (Fig. 5a). Cranial tumor volume in VC‐ and RCO‐treated groups showed rapid increases, whereas growth in the TGFβR1‐ASO‐treated group was more gradual (Fig. 5b). TGFβR1‐ASO treatment did not significantly reduce tumor volume compared with VC (P = 0.17) and RCO (P = 0.08). TGFβR1‐ASO treatment did reduce the bone destruction ratio and osteoclast induction, whereas VC and RCO did not (Fig. 5c,d). TGFβR1‐ASO treatment also significantly reduced the induction of osteoblasts and osteoblastic lesions at the TB‐interface (Fig. 5e). Fewer PCNA‐positive cells were observed at the TB‐interface in TGFβR1‐ASO‐ than RCO‐treated groups (Fig. 5f). The index for pSmad2, reflecting the degree of endogenous TGFβ signal transduction, was also reduced by TGFβR1‐ASO, but not by VC or RCO (Fig. 5g).
Figure 5.

Antisense oligonucleotide study in prostate tumors transplanted onto cranial bone and into the subcutis of male rats. (a) Expression of transforming growth factor β receptor 1 (TGFβR1) at the tumor–bone (TB) interface treated with vehicle control (VC), TGFβR1 antisense oligonucleotide; TGFβR1‐ASO(1), TGFβR1‐ASO(2), TGFβR1‐ASO(3), random control oligonucleotide RCO(1) and RCO(2) shown by quantitative reverse transcription–polymerase chain reaction and Western blot analysis. Because TGFβR1‐ASO(1) exerted the most suppressive effect on TGFβR1 expression, we subsequently chose TGFβR1‐ASO(1) as ‘TGFβR1‐ASO’ and RCO(1) as ‘RCO’. (b) Tumor volume curves. Cranial tumor volume in VC and RCO groups shows a rapid increase, and the TGFβR1‐ASO group shows a gradual increase (n = 3 in each group). (c) Bone destruction at the TB‐interface. The dotted line indicates bone destruction. Quantitative analysis of the bone destruction index revealed that TGFβR1‐ASO significantly reduced bone destruction ratio (right). HE, hematoxylin–eosin. (Magnification: ×20). (d) Tartrate‐resistant acid phosphatase (TRAP) staining at the TB‐interface. Quantitative analysis indicated that TGFβR1‐ASO significantly reduced TRAP‐positive multinucleated cells (right) (magnification: ×100). (e) Osteoblasts at the TB‐interface. Many osteoblasts were observed in the woven bone at the TB‐interface in the RCO group (left) and less osteoblasts in the TGFβR1‐ASO group (middle). TGFβR1‐ASO treatment significantly reduced osteoblasts at the TB‐interface (right) (magnification: ×100). (f) Immunohistochemical staining of proliferating cell nuclear antigen (PCNA). Many PCNA‐positive cells were observed at the TB‐interface in the RCO group (left) and less numbers in the TGFβR1‐ASO group (middle). TGFβR1‐ASO treatment significantly suppressed PCNA index at the TB‐interface, but not at the non‐TB (NTB) interface (right) (magnification: ×100). (g) Immunohistochemical staining of phosphorylated Smad2 (pSmad2). Many positive cells were observed in the TB‐interface of RCO (left) and less in TGFβR1‐ASO (middle). The index of pSmad2‐positive cells at the TB‐interface revealed that TGFβR1‐ASO reduced TGFβ signaling, but that VC and RCO did not. No significant difference was observed at the NTB‐interface (right) (magnification: ×100). *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
In the present study, we compared prostate tumor growth at the TB‐interface with that at the TS‐interface to identify specific mechanisms for tumor growth in the bone microenvironment. Histological examination showed that prostate tumor cells were growing with the interaction among osteoclasts and osteoblasts at the TB‐interface, and tumor cells were growing with the induction of microvessels at the TS‐interface. To clarify the roles of the interaction at the TB‐interface, it is important to compare the behaviors of prostate tumor cells in bone microenvironments with an appropriate microenvironment as a control. Orthotopic transplantation appeared to be one appropriate control, but PLS‐P transplanted into the prostate grew rapidly and resulted in urinary retention, causing early death (unpublished data). Therefore, in the present study we chose the subcutaneous microenvironment for the control.
The difference in tumor volumes between cranial and subcutaneous tumors were not significant, although cell proliferation at the TB‐interface in cranial tumors was much higher than at the TS‐interface in subcutaneous tumors. Changes of tumor volumes correspond to cell proliferation in whole tumors, and the numbers of tumor cells examined at the TB and TS‐interfaces were approximately 10% of whole cranial and subcutaneous tumor cells. The reason for the discrepancy between the tumor volume and cell proliferation is that the TB and TS‐interfaces were such small lesions that the increase in cell proliferation at the TB and TS‐interfaces did not affect whole tumor volume, corresponding to whole tumor cell proliferation.
Interactions between tumor cells and osteoclasts has been shown to play important roles in the development of bone metastasis.( 5 , 26 , 27 ) Osteolysis by activated osteoclasts leads to the release of growth factors stored in the bone matrix.( 8 ) Because TGFβ is abundant in the bone matrix, the critical role of TGFβ has been indicated in the development of bone metastasis.( 22 ) In the present study, TGFβ levels evaluated by ELISA were higher in bone tissue than in prostate, lung, liver, or lymph nodes. Osteolytic changes were observed at the TB‐interface, where high TGFβ levels as well as TGFβ signal transduction were observed. Thus, the source of TGFβ was suggested to be derived from the bone matrix. Furthermore, intratumoral injection of TGFβR1‐ASO reduced TGFβ signal transduction at the TB‐interface. These results indicate that TGFβ is involved in the growth of prostate cancer in the bone microenvironment.
Previous studies have shown that suppression of TGFβ signaling reduced bone metastasis of breast cancer and tumor growth in vivo.( 28 , 29 , 30 ) In the present study, TGFβ expression and tumor cell proliferation was higher at the TB‐interface than at the TS‐interface, and higher cell proliferation at the TB‐interface was well‐correlated with higher TGFβ levels. Treatment with TGFβR1‐ASO reduced TGFβ signal transduction and cell proliferation at the TB‐interface. Taken together, these results indicate that TGFβ might promote proliferation of tumor cells in the bone microenvironment. However, further studies are necessary as TGFβ has also been shown to suppress tumor cell proliferation in vitro.( 31 , 32 , 33 , 34 )
In the present study, IHC stainings showed that pSmad2 was strongly positive at the TB‐interface and weakly positive at the TS‐interface. However, Western blot analysis showed that the levels of phosphorylation of Smad2 were comparable between the two interfaces. The TB‐interface evaluated by IHC was clearly defined microscopically, and the protein samples for Western blotting were derived from the specimens dissected macroscopically at the time the animals were killed. Therefore, the protein samples at the TB‐interface might include the microscopically‐defined TB‐interface and the surrounding tissues that are weakly positive for pSmad2. This might be one of the reasons for the discrepancy in the pSmad2 expression between IHC and Western blots. However, further study is necessary to confirm pSmad2 expression at the TB‐interface.
Osteolysis is essential for tumor growth in hard calcified bone tissue and is associated with release of several cytokines from the bone matrix. RANKL has been shown to be involved in the induction of osteoclasts that are responsible for bone absorption.( 35 , 36 ) TGFβ has been shown to induce suppressors of cytokine signaling that enhance RANKL‐induced osteoclasts differentiation in mononuclear phagocyte precursors.( 37 ) Thus, TGFβ might promote RANKL‐induced osteoclast differentiation through suppressors of cytokine signaling. In the present study, TGFβ exerted a synergistic promotive effect on osteoclast induction with RANKL in vitro. In addition, we previously found high RANKL expression at the TB‐interface.( 22 ) The present study indicated that TGFβ levels at the TB‐interface were correlated with the degree of osteolysis. Combined, the data suggest that TGFβ derived from the bone matrix promotes osteolysis through synergistic induction of osteoclasts with RANKL.
In conclusion, we have shown that TGFβ derived from the bone matrix promoted the cell proliferation of prostate cancer in the bone microenvironment and expanded osteolysis through the induction of osteoclasts in combination with RANKL. TGFβ targeted therapy could therefore prove beneficial for patients of prostate cancer with bone metastasis.
Acknowledgments
This work was supported in part by a research grant from the Princess Takamatsu Cancer Research Fund, a grant‐in‐aid for Scientific Research (C) from the Japan Society for Promotion of Science, a grant‐in‐aid for the 2nd Term Comprehensive 10‐year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan, a grant‐in‐aid for Research in Nagoya City University, and a grant from the Society for Promotion of Pathology of Nagoya, Japan.
References
- 1. Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin 2007; 57: 43–66. [DOI] [PubMed] [Google Scholar]
- 2. Roodman GD. Mechanisms of bone metastasis. N Engl J Med 2004; 350: 1655–64. [DOI] [PubMed] [Google Scholar]
- 3. Petrylak DP, Tangen CM, Hussain MH et al . Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004; 351: 1513–20. [DOI] [PubMed] [Google Scholar]
- 4. Tannock IF, De Wit R, Berry WR et al . Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004; 351: 1502–12. [DOI] [PubMed] [Google Scholar]
- 5. Mundy GR. Mechanisms of bone metastasis. Cancer 1997; 80: 1546–56. [DOI] [PubMed] [Google Scholar]
- 6. Cher ML, Towler DA, Rafii S et al . Cancer interaction with the bone microenvironment: a workshop of the National Institutes of Health Tumor Microenvironment Study Section. Am J Pathol 2006; 168: 1405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2002; 2: 584–93. [DOI] [PubMed] [Google Scholar]
- 8. Chirgwin JM, Guise TA. Molecular mechanisms of tumor–bone interactions in osteolytic metastases. Crit Rev Eukaryot Gene Expr 2000; 10: 159–78. [PubMed] [Google Scholar]
- 9. Kakonen SM, Mundy GR. Mechanisms of osteolytic bone metastases in breast carcinoma. Cancer 2003; 97: 834–9. [DOI] [PubMed] [Google Scholar]
- 10. Finkelman RD, Mohan S, Jennings JC, Taylor AK, Jepsen S, Baylink DJ. Quantitation of growth factors IGF‐I, SGF/IGF‐II, and TGF‐beta in human dentin. J Bone Miner Res 1990; 5: 717–23. [DOI] [PubMed] [Google Scholar]
- 11. Solheim E. Growth factors in bone. Int Orthop 1998; 22: 410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chung LW. Prostate carcinoma bone–stroma interaction and its biologic and therapeutic implications. Cancer 2003; 97: 772–8. [DOI] [PubMed] [Google Scholar]
- 13. Huang X, Lee C. Regulation of stromal proliferation, growth arrest, differentiation and apoptosis in benign prostatic hyperplasia by TGF‐beta. Front Biosci 2003; 8: s740–9. [DOI] [PubMed] [Google Scholar]
- 14. Edlund S, Bu S, Schuster N et al . Transforming growth factor‐β 1 (TGF‐β)‐induced apoptosis of prostate cancer cells involves Smad7‐dependent activation of p38 by TGF‐β‐activated kinase 1 and mitogen‐activated protein kinase kinase 3. Mol Biol Cell 2003; 14: 529–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang EY, Moses HL. Transforming growth factor β 1‐induced changes in cell migration, proliferation, and angiogenesis in the chicken chorioallantoic membrane. J Cell Biol 1990; 111: 731–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nicolas FJ, Hill CS. Attenuation of the TGF‐β–Smad signaling pathway in pancreatic tumor cells confers resistance to TGF‐β‐induced growth arrest. Oncogene 2003; 22: 3698–711. [DOI] [PubMed] [Google Scholar]
- 17. Delany AM, Canalis E. Growth Factors and Cytokines in Health and Disease. Greenwich CT: JAI Press; 1997. [Google Scholar]
- 18. Kakonen SM, Selander KS, Chirgwin JM et al . Transforming growth factor‐β stimulates parathyroid hormone‐related protein and osteolytic metastases via Smad and mitogen‐activated protein kinase signaling pathways. J Biol Chem 2002; 277: 24571–8. [DOI] [PubMed] [Google Scholar]
- 19. Yin JJ, Selander K, Chirgwin JM et al . TGF‐β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 1999; 103: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kato K, Takahashi S, Mori T et al . Establishment of transplantable rat prostate carcinomas from primary lesions induced by 3,2′‐dimethyl‐4‐aminobiphenyl and testosterone. J Toxicol Pathol 1998; 11: 27–32. [Google Scholar]
- 21. Zhao L, Futakuchi M, Suzuki S et al . Kinetics of marked development of lung metastasis of rat prostatic carcinomas transplanted in syngeneic rats. Clin Exp Metastasis 2005; 22: 309–18. [DOI] [PubMed] [Google Scholar]
- 22. Lynch CC, Hikosaka A, Acuff HB et al . MMP‐7 promotes prostate cancer‐induced osteolysis via the solubilization of RANKL. Cancer Cell 2005; 7: 485–96. [DOI] [PubMed] [Google Scholar]
- 23. Ogawa K, Kimoto N, Asamoto M, Nakanishi M, Takahashi S, Shirai T. Aberrant expression of p27 (Kip1) is associated with malignant transformation of the rat urinary bladder epithelium. Carcinogenesis 2000; 21: 117–21. [DOI] [PubMed] [Google Scholar]
- 24. Grano M, Mori G, Minielli V et al . Rat hindlimb unloading by tail suspension reduces osteoblast differentiation, induces IL‐6 secretion, and increases bone resorption in ex vivo cultures. Calcif Tissue Int 2002; 70: 176–85. [DOI] [PubMed] [Google Scholar]
- 25. Hikosaka A, Futakuchi M, Ogiso T, Suzuki S, Kohri K, Shirai T. Lack of prophylactic effect of incadronate on skeletal lesions associated with implants of prostate cancer. Eur Urol 2006; 49: 176–82. [DOI] [PubMed] [Google Scholar]
- 26. Lipton A. Future treatment of bone metastases. Clin Cancer Res 2006; 12: 6305s–8s. [DOI] [PubMed] [Google Scholar]
- 27. Kim SJ, Uehara H, Yazici S et al . Modulation of bone microenvironment with zoledronate enhances the therapeutic effects of STI571 and paclitaxel against experimental bone metastasis of human prostate cancer. Cancer Res 2005; 65: 3707–15. [DOI] [PubMed] [Google Scholar]
- 28. Ehata S, Hanyu A, Fujime M et al . Ki26894, a novel transforming growth factor‐β type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci 2007; 98: 127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bandyopadhyay A, Agyin JK, Wang L et al . Inhibition of pulmonary and skeletal metastasis by a transforming growth factor‐β type I receptor kinase inhibitor. Cancer Res 2006; 66: 6714–21. [DOI] [PubMed] [Google Scholar]
- 30. Hiraga T, Myoui A, Choi ME, Yoshikawa H, Yoneda T. Stimulation of cyclooxygenase‐2 expression by bone‐derived transforming growth factor‐β enhances bone metastases in breast cancer. Cancer Res 2006; 66: 2067–73. [DOI] [PubMed] [Google Scholar]
- 31. Guo Y, Kyprianou N. Restoration of transforming growth factor β signaling pathway in human prostate cancer cells suppresses tumorigenicity via induction of caspase‐1‐mediated apoptosis. Cancer Res 1999; 59: 1366–71. [PubMed] [Google Scholar]
- 32. Schniewind B, Groth S, Sebens Muerkoster S et al . Dissecting the role of TGF‐β type I receptor/ALK5 in pancreatic ductal adenocarcinoma: Smad activation is crucial for both the tumor suppressive and prometastatic function. Oncogene 2007; 26: 4850–62. [DOI] [PubMed] [Google Scholar]
- 33. Rowland‐Goldsmith MA, Maruyama H, Kusama T, Ralli S, Korc M. Soluble type II transforming growth factor‐β (TGF‐β) receptor inhibits TGF‐β signaling in COLO‐357 pancreatic cancer cells in vitro and attenuates tumor formation. Clin Cancer Res 2001; 7: 2931–40. [PubMed] [Google Scholar]
- 34. Tuxhorn JA, McAlhany SJ, Yang F, Dang TD, Rowley DR. Inhibition of transforming growth factor‐β activity decreases angiogenesis in a human prostate cancer‐reactive stroma xenograft model. Cancer Res 2002; 62: 6021–5. [PubMed] [Google Scholar]
- 35. Udagawa N, Takahashi N, Jimi E et al . Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony‐stimulating factor: receptor activator of NF‐kappa B ligand. Bone 1999; 25: 517–23. [DOI] [PubMed] [Google Scholar]
- 36. Ohshiba T, Miyaura C, Inada M, Ito A. Role of RANKL‐induced osteoclast formation and MMP‐dependent matrix degradation in bone destruction by breast cancer metastasis. Br J Cancer 2003; 88: 1318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fox SW, Haque SJ, Lovibond AC, Chambers TJ. The possible role of TGF‐β‐induced suppressors of cytokine signaling expression in osteoclast/macrophage lineage commitment in vitro . J Immunol 2003; 170: 3679–87. [DOI] [PubMed] [Google Scholar]