

Abstract
Current treatment modalities for cancer combine cytotoxic drugs against DNA and novel targeted drugs affecting signal transduction pathways, which are required for growth progression and metastasizing tumors. Classical chemotherapeutic regimens for gastro‐intestinal tumors include antimetabolites based on 5‐fluorouracil (5FU), the platinum analog oxaliplatin and the topoisomerase inhibitor irinotecan. The thymidine analog trifluorothymidine (TFT) has been shown to bypass resistance pathways for 5FU derivatives (S‐1, UFT, Xeloda) in model systems, while concurrent application with a thymidine phosphorylase inhibitor (TPI) increases the bioavailability of TFT, thereby potentiating the in vivo efficacy of TFT. The formulation TAS‐102 is given orally in a 1.0:0.5 molar ratio (TFT:TPI). The formulation is dual‐targeted due to the cytotoxic effect of TFT, which is enhanced by TPI, while TPI also exerts antiangiogenic effects by inhibiting thymidine phosphorylase (TP), also known as platelet‐derived endothelial cell growth factor. Evidence is accumulating from in vitro and in vivo preclinical studies that these properties favor further combinations with other cytotoxic agents currently being used in the treatment of gastro‐intestinal tumors. Also treatment with targeted agents will synergistically down‐regulate signal transduction pathways responsible for growth and progression of tumors. In this review, we summarize the available information on (clinical) pharmacology, mechanisms of action, pharmacodynamic and pharmacokinetic properties, early clinical trials and future directions of the new potent combination drug TAS‐102. (Cancer Sci 2007; 98: 779–789)
TAS‐102 is a promising novel combination drug, which combines several attractive features. One component is the antiangiogenic effect, which is also intended to enhance its cytotoxic effect. TAS‐102 contains the cytotoxic pyrimidine analog 5‐trifluoro‐2′‐deoxythymidine (TFT; trifluridine; 5‐trifluromethyl‐2′‐deoxyuridine; CF3dUrd; FTD; F 3TDR; F 3Thd) and the potent thymidine phosphorylase (TP) inhibitor (TPI), and is currently evaluated in phase I studies as an oral formulation in a molar ratio of 1.0:0.5 (Fig. 1).
Figure 1.
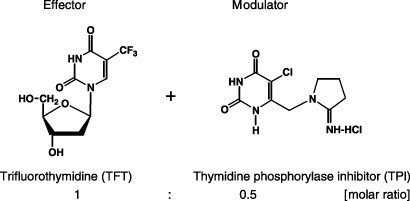
Chemical structure and functional mechanism of TAS‐102.
The thymidine analog trifluorothymidine was synthesized originally by Heidelberger et al.( 1 ) in 1964, who had already shown that TFT can be phosphorylated by thymidine kinase to its active monophosphate form in uninfected tissues mediating cytotoxicity.( 2 ) TFT has also been evaluated for its antitumor effects and was at least as effective against adenocarcinoma cells as compared to 5‐fluoro‐2′‐deoxyuridine (FdUrd).( 3 ) Based on these pharmacological data, a clinical study on TFT was carried out to evaluate its efficacy in patients with breast and colon carcinomas.( 4 ) Some of the administration schedules have been reported to produce a reduction of tumor size (eight out of 23 breast cancer and one out of six colon cancer). However, these clinical studies were unfortunately discontinued, because of a poor pharmacokinetic profile and side‐effects. TFT was also investigated as an antiviral agent and is registered as Viroptic,( 5 ) for use against herpes simplex virus (HSV) infections. The drug was previously approved by the Food and Drug Administration (FDA) in 1980 for the treatment of primary keratoconjonctivitis and epithelial keratitis.( 6 ) The mechanism of action of the individual components of TAS‐102, TFT and TPI, and their combined effects are discussed in view of its potential application as an anticancer formulation.
Other oral fluoropyrimidine formulations
Several other oral fluoropyrimidine formulations are currently being developed or are already used in standard chemotherapeutic regimens (reviewed in De Bruin et al.( 7 ) and were developed to replace continuous infusions. They have at least equal efficacy compared with continuous infusions and improve the quality of life because of the oral route of administration. Ftorafur (Ft; 1‐(2‐tetrahydrofuryl)‐5‐fluorouracil, Tegafur or Futraful) is used in several combinations to improve its bioavailability. UFT consists of Ft combined with uracil in a 1:4 molar ratio, uracil being a natural substrate for dihydropyrimidine dehydrogenase (DPD) which prevents 5FU's breakdown by competition.( 7 ) S‐1, its successor follows a different strategy. This combination exists of Ft and two other compounds, 5‐chloro‐2,4‐dihydroxypyridine (CDHP) and potassium oxonate (OXO) (molar ratio Ft: CDHP: OXO; 1.0:0.4:1.0). CDHP and OXO have no antitumor activity and act as modulators of 5FU in the metabolism. CDHP functions as an inhibitor of DPD, thereby increasing the period of high 5FU in the circulation. CDHP is 200‐fold more potent than uracil in inhibiting DPD. OXO is added to limit the gastrointestinal toxicity of Ft. This toxic effect is the result of phosphoribosylation of 5FU to 5‐fluorouridine‐2′‐monophosphate (5‐FUMP) by orotate phosphoribosyl transferase (OPRT). OXO accumulates specifically in normal gastrointestinal tissues compared to tumors preventing activation of 5FU in normal mucosa, but not in the tumor.
A third rationally designed oral pro‐drug of 5FU is capecitabine (Xeloda), which is converted to 5FU in three steps, initially catalyzed by carboxyl esterase located almost exclusively in the liver, thereafter by cytidine deaminase expressed in the liver and various types of tumors and subsequently by platelet‐derived endothelial cell growth factor (PD‐ECGF)/TP. The design of capecitabine potentially had two advantages: enhanced activation at the tumor site and a decrease of drug concentration in the healthy tissue thereby decreasing systemic toxicity. The dose limiting toxicities were similar to those of i.v. administered fluoropyrimidines. Most common were the hand–foot syndrome and diarrhea. So, since capecitabine is at least as effective as the 5FU/LV standard,( 8 ) and it provides considerable benefits to the well being of the patient, it has become one of the most widely prescribed oral anticancer drugs.
Pharmacodynamics
TFT‐mediated cytotoxicity. The pharmacodynamic (PD) properties of TFT share some similarities with 5FU, but also with other deoxynucleoside analogs, particularly thymidine analogs, which are predominantly used as antiviral agents. The mechanism of action of TFT is depicted in Fig. 2. TFT is phosphorylated by thymidine kinase (TK) to its active monophosphate‐derivative TF‐TMP, which inhibits thymidylate synthase (TS) as was first shown by Reyes and Heidelberger.( 9 ) TS is one of the rate‐limiting enzymes in pyrimidine de novo deoxynucleotide synthesis and therefore it plays a central role in DNA synthesis as a target for chemotherapeutic approaches.( 10 ) TF‐TMP does not form a ternary complex and binds covalently to the active site of TS (tyrosine 146) thereby inhibiting its activity.( 11 , 12 ) TF‐TMP is a potent reversible inhibitor of TS with a Ki of 0.38 nM,( 9 ) and its activity remains inhibited when a constant influx of TFT is present; removal of the compound leads to a rapid recovery of TS activity.( 12 , 13 ) This is in contrast to the 5FU derivative 5‐fluoro‐2′‐deoxyuridine‐5′‐monophosphate (FdUMP), which inhibits TS after formation of a ternary complex with 5,10‐methylene‐tetrahydrofolate (CH2‐THF), thereby enhancing and prolonging this inhibition.( 14 ) CH2‐THF also serves as the cofactor for the reductive methylation of dUMP to dTMP (Fig. 2). Inhibition of TS leads to intracellular depletion of dTTP and subsequent dUMP accumulation, resulting in uracil misincorporation into the DNA leading to DNA damage.
Figure 2.
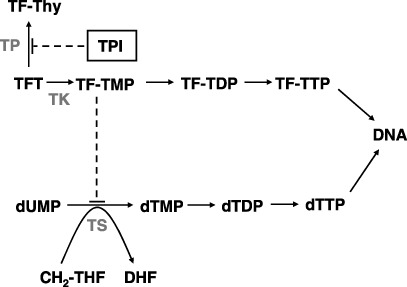
Mechanism of action of thymidine analog trifluorothymidine (TFT) and thymidine phosphorylase inhibitor (TPI).
In addition to TFT‐mediated TS inhibition, Heidelberger originally showed that TFT itself can be incorporated into DNA.( 15 ) The triphosphate form of TFT (TF‐TTP) is incorporated into the DNA,( 16 , 17 ) eventually causing cell death due to DNA strand break formation,( 18 ) although the precise mechanism of TFT‐induced DNA damage remains to be elucidated. A relatively high dose of TFT appeared to have a similar cytotoxicity against 5FU‐resistant cells,( 18 ) unlike the effects of conventional 5FU analogs such as UFT or FdUrd, as well as its parent cells. These data demonstrated that excessive levels of TFT concentrations that are necessary to inhibit TS would be needed for DNA incorporation and require relatively high doses of TFT. Thus, a relatively short‐term and high concentration may cause more DNA damage than that of a low dose to show its similar cytotoxic effect. TFT was incorporated in a time‐dependent manner and not in a concentration‐dependent manner.( 17 ) TFT incorporation into DNA continued to increase during 8 h exposure of NUGC‐3 human gastric cancer cells to TFT at micromolar levels. Although intracellular TF‐TTP is rapidly eliminated from the tumor cells after removal of TFT from the culture medium, almost 80% of TFT, once incorporated into DNA, is not removed from the DNA for up to 24 h. These observations indicated that TFT incorporation into DNA is important. When tumor‐bearing mice were treated with TAS‐102 at a repeated dosing of 150 mg/kg per day a significantly higher incorporation of TFT into DNA of the tumor was found compared to single dosing.( 17 ) Altogether these in vitro and in vivo results demonstrated that when tumor cells were exposed for relatively repeated short‐term to high concentrations of TFT, the drug could induce most significant amounts of DNA damage due to enhanced DNA fragmentation resulting in most potent antitumor activity. This indicates that multiple daily dosing may result in better clinical benefits for TAS‐102‐treated cancer patients. However, it is not clear what type of DNA damage is induced by TFT, for example, does TFT cause a chain‐terminating effect and which DNA polymerase is involved?
TFT‐resistance. Most chemotherapeutic regimens in the treatment of gastrointestinal cancer patients are 5FU‐based. The presence of a fluoropyrimidine was actually necessary to obtain a maximal effect. To improve treatment it is important that an alternative chemotherapeutic regimen, such as TAS‐102, also exhibits antitumor effects against 5FU‐sensitive, and more favorably, against 5FU‐resistant tumor cells. Therefore a high bioavailability of TFT in vivo is required to increase its antitumor activity. At a high dose level of TFT alone (200 mg/kg per day) tumor growth inhibition rates were about 70% for both 5FU‐sensitive and ‐resistant NUGC‐3 gastric cancer cells (NUGC‐3/5FU) implanted s.c. in nude mice.( 18 ) Comparable tumor growth inhibition rates were seen for FdUrd‐resistant DLD‐1 colorectal cancer cells (DLD‐1/FdUrd) in mice treated with 150 mg/kg per day TFT. The 5FU‐resistant NUGC‐3 cells were characterized by decreased 5FU incorporation into the RNA and reduced activity of OPRT, a critical enzyme in the conversion of 5FU to its active metabolites,( 19 ) this is in contrast to the DLD‐1/FdUrd cells, which have increased TS levels.( 20 ) Therefore it can be concluded that TAS‐102, which contains TFT, has antitumor activity against 5FU‐resistant tumor cells with different mechanisms of acquired resistance. TAS‐102 itself was also effective in vivo against 5FU‐sensitive tumors from different tissue types, such as human pancreatic and oesophageal tumor cells.( 18 , 21 )
In an in vitro study 5FU‐resistant DLD‐1 cells (DLD‐1/5FU) did not show cross‐resistance to TFT or FdUrd, but DLD‐1 cells made resistant to TFT (DLD‐1/TFT) were cross‐resistant to FdUrd (about 90‐fold) and not to 5FU itself.( 20 ) Although colorectal cancer cells often show increased expression of TS as a resistance mechanism to TS inhibitors,( 14 ) the TS levels in DLD‐1/TFT cells remained unchanged, but TK activity was decreased significantly (37‐fold). Furthermore, H630 human colorectal carcinoma cells made resistant to TFT using either a long‐term continuous exposure schedule (H630‐cTFT) or short‐term repeated exposure schedule (H630‐4TFT), acquired different mechanisms of resistance (unpublished results). H630‐cTFT cells growing in medium containing 20 µM TFT had TS and TK levels comparable to the parental cells and showed a disturbed signal transduction (e.g. up‐regulated secretory phospholipase A2 expression, as shown with microarray analysis). The H630–4TFT cells exposed to TFT on a short‐term basis (250 µM TFT 4 h/week) had normal TS levels, but hardly had any TK activity, revealing the same TFT‐resistance mechanism as the DLD‐1/TFT‐resistant cells, which were exposed repeatedly on a long‐term basis (10 µM TFT 5 days/week). In both H630 TFT‐resistant cell lines, no change in TP levels was observed, although this was less relevant, because a high TP expression in colorectal cancer cells hardly influences the sensitivity to TFT.( 16 , 22 ) In agreement with the DLD‐1/TFT cells, both H630‐derived TFT‐resistant cell lines were not cross‐resistant to 5FU or the folate‐based direct TS inhibitor GW1843, but were cross‐resistant to FdUrd (about 160‐fold). In contrast to the DLD‐1/5FU, 5FU‐resistant H630 cells (H630‐R10) with increased TS levels were cross‐resistant to FdUrd and TFT, but this was dependent on exposure time.( 13 )
TPI increases bioavailability of TFT. The formulation TAS‐102 consisting of the cytotoxic TFT and the TP inhibitor TPI can be considered as a potential antiangiogenic therapy. In this formulation TPI has a dual effect, because it has an antiangiogenic effect and can enhance the bioavailability of TFT. Given as a single agent, TFT is rapidly degraded in the human body and follows first‐order kinetics with a short plasma half‐life of less than 20 min.( 23 ) In animal models it was demonstrated that the antitumor activity of TFT can be improved by increasing its bioavailability. For this purpose depot‐forms of TFT or TFT derivatives have been used that serve as pro‐drugs, which, besides increasing the bioavailability and antitumor activity of TFT, might decrease side‐effects and/or increase activation of the drugs. Takeda et al.( 24 ) explored the potential of several masked TFT derivatives as depot‐forms of TFT itself. Takeda already reported that TFT is more effective in divided dosing than as single administration, and the use of depot‐forms of TFT might even further increase its antitumor potency. Several acyl‐ and O‐alkylderivatives of TFT had significantly higher growth inhibition rates and therapeutic indices compared to TFT and might therefore be more useful than the parental compound, particularly 3′‐O‐benzyl‐TFT.( 25 , 26 ) Although these derivatives slowly release the drug in body fluids in order to achieve effective concentrations over an extended period of time, they might also affect healthy tissue thereby increasing the systemic side‐effects. Due to increased TK levels often observed in solid tumors,( 27 ) these agents may be an alternative for TFT.
The second approach to increase the bioavailability of TFT is to inhibit its degradation directly by use of a potent TP inhibitor. Initial mice studies already demonstrated that the TFT degradation product trifluorothymine (TF‐Thy) and its catabolite 5‐carboxyuracil were mainly formed in the liver, spleen and the intestines.( 2 ) In order to inhibit TP a variety of inhibitors have been developed. The commonly used 6‐amino‐5‐bromouracil (6A5BU),( 28 ) has been the benchmark pyrimidine‐based structure for the design of more potent inhibitors of TP for years. Freeman and colleagues synthesized a variety of TP inhibitors based on 6A5BU,( 29 , 30 ) and observed that 5‐halo‐2‐amino‐imidazolylmethyluracils were very potent inhibitors of Escherichia coli TP with enzyme IC50 of approximately 20 nM,( 30 ) which is at least 75‐fold more potent than 6A5BU. The corresponding 2‐nitro‐imidazolylmethyluracil pro‐drugs were found to be 1000‐fold less active than the parental compounds. However, because they are bioreductively activated they would give an advantage in enhanced activation in hypoxic areas in solid tumors. In addition, xanthine oxidase‐activated TP inhibitor pro‐drugs also belong to the possibilities.( 29 ) The first purine analog displaying inhibitory activity was 7‐deazaxanthine (7‐DX).( 31 )
Yano et al. described the synthesis and evaluation of novel orally active 6‐methylene‐bridged uracil‐derived inhibitors of human TP,( 32 , 33 ) and identified several compounds with IC50 values around 1 µM for human TP.( 32 ) These compounds were selected for further evaluation because of their inhibitory potency and their pharmacokinetic profile was optimized to inhibit TP.( 33 ) The most potent human TP inhibitor of the whole series that was synthesized was the 5‐chloro‐6‐(2‐iminopyrrolidin‐1‐yl)methyl‐2,4(1H,3H)‐pyrimidine dione hydrochloride (TPI) with an IC50 of 35 nM for TP, but was a poor inhibitor of uridine phosphorylase (UP) (IC50 > 100 µM),( 34 ) a pyrimidine phosphorylase also known to cleave thymidine (TdR) and other pyrimidine nucleosides. The Ki value for TPI using recombinant human TP was 17 nM,( 34 ) TPI was a potent inhibitor of TFT phosphorolysis in extracts from human liver, small intestine and tumor tissues, and at equimolar doses TPI increased Cmax and area under the plasma concentration time curve (AUC) of orally administered TFT (10 mg/kg) to monkeys at least 70‐fold (3, 4, respectively). Initial mice experiments showed that at equimolar doses TPI significantly potentiated TFT‐induced growth inhibition of xenografted AZ‐521 human gastric cancers in nude mice.( 34 ) Comparable antitumor activity was shown against DLD‐1 colorectal cancer xenografts, which reached a maximum tumor growth inhibition rate of 70% (Fig. 5).( 35 ) TPI augments TFT cytotoxicity maximally in vivo due to elevation of drug exposure of tumor cells to TFT. The optimal molar drug ratio to reach maximal TFT plasma levels in monkeys was 2:1 (TFT:TPI) (Fig. 6), which was subsequently developed as TAS‐102.( 35 ) In vitro studies have shown that despite high TFT phosphorolysis, high TP‐expressing colorectal Colo320TP1 and non‐small cell lung H460TP2 cancer cells are not more resistant to TFT, while the addition of TPI did not increase TFT sensitivity.( 16 , 22 ) A moderate almost twofold increase of the formation of active TFT metabolites was found, although it was not related to increased incorporation of TFT into DNA.( 16 ) In addition, favorable enzyme properties possibly allow the TFT activation pathway to be maximally saturated, which is hardly influenced by TFT degradation by TP. TPI only affected TFT‐mediated cytotoxicity at very short exposure in cells expressing very high TP,( 16 ) with apparently maximum incorporation of TFT into DNA. This means that the mode of in vitro cytotoxicity exerted by TFT is also dependent on exposure times used. The effect of TPI was not dependent on the activity of TP, since all models displayed different activities of TP.
Figure 3.
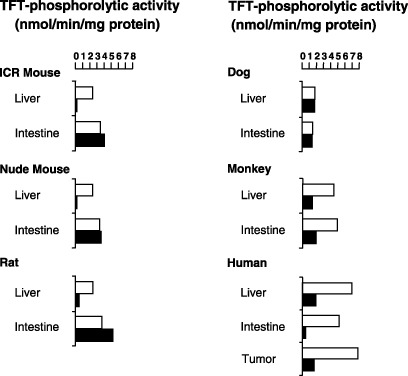
Inhibitory effect of thymidine phosphorylase inhibitor (TPI) on thymidine analog trifluorothymidine (TFT)‐phosphorolytic activity in crude extracts from various mammalian tissues. Radio labeled TFT was incubated with enzyme extracts from rodent, dog, monkey and human tissues in the presence (black bars) and absence (white bars) of 1 µM TPI. Values are means of duplicate determinations. Data were adapted from Fukushima et al. (2000; with permission).( 34 )
Figure 4.
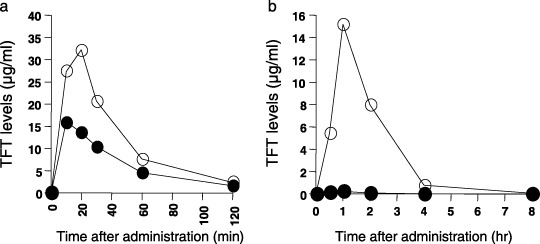
Plasma thymidine analog trifluorothymidine (TFT) levels in mice (a) and monkeys (b) following oral administration in the presence and absence of thymidine phosphorylase inhibitor (TPI). TFT (black circles) was administered in doses of 50 mg/kg (normal mice) or 10 mg/kg (normal monkeys), or in combination with an equimolar amount of TPI (white circles). Values are means of several experiments (n = 6 mice; n = 3 monkeys). Data were adapted from Fukushima et al. (2000; with permission).( 34 )
Figure 5.
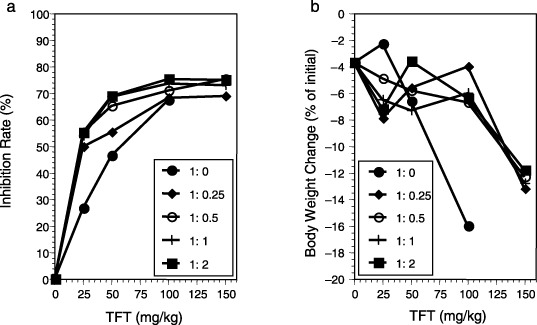
Antitumor activity (a) and body weight change (b) as a result of different treatment ratios of thymidine analog trifluorothymidine : thymidine phosphorylase inhibitor (TFT : TPI) in human DLD‐1 colorectal cancer xenografts in nude mice. Data were adapted from Emura et al. (2005; with permission).( 35 )
Figure 6.
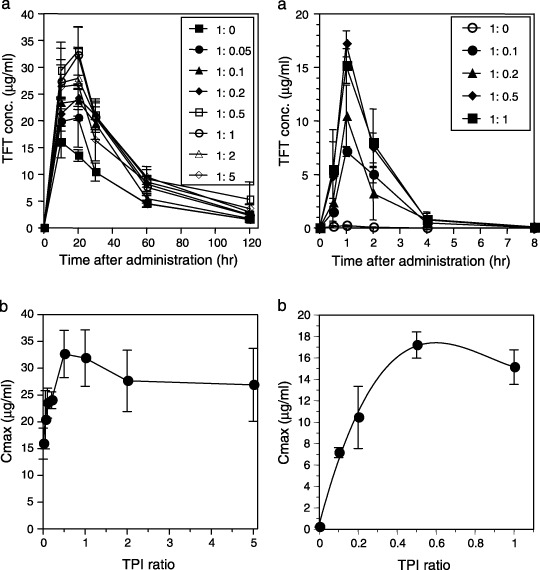
Plasma thymidine analog trifluorothymidine (TFT) levels in ICR mice (left) and monkeys (right) following oral administration in the presence and absence of thymidine phosphorylase inhibitor (TPI) at different molar ratios. TFT was administered in doses of 50 mg/kg (mice) or 10 mg/kg (monkeys). TFT plasma levels over time (a) and relationship Cmax and various TPI : TFT ratios (b) were determined. Data were adapted from Emura et al. (2005; with permission).( 35 )
Tumor angiogenesis. To improve cancer therapy there is an obvious need to develop novel treatment options. Angiogenesis is an important process in stimulating tumor growth and metastasis of many cancers. Targeting angiogenesis in addition to current chemotherapy is an important new development.( 36 ) Vascular endothelial growth factor (VEGF) and its receptors (VEGF‐R) are often elevated in various malignancies and therefore are excellent chemotherapeutic targets in the treatment of colorectal tumors and other solid tumors.( 37 ) Most antiangiogenic drugs target growth factors (and their receptors) or inhibit endothelial cell proliferation or/and signal transduction. VEGF, but also fibroblast growth factor (FGF), endothelial cell growth factor (ECGF) and platelet‐derived growth factor (PDGF) and their tyrosine kinase receptors are major regulators of angiogenesis.( 38 , 39 ) Promising new antiangiogenic drugs are under investigation.
Next to the mentioned factors, PD‐ECGF plays a role in tumor angiogenesis.( 40 ) PD‐ECGF was also identified as TP,( 41 , 42 ) and gliostatin.( 40 ) PD‐ECGF/TP is a potent pro‐angiogenic factor that promotes tumor growth.( 43 , 44 ) In addition to VEGF, increased PD‐ECGF/TP expression is seen in colorectal tumor tissue compared to normal tissue,( 45 ) and high PD‐ECGF/TP levels are a prognostic factor for poor survival in colorectal( 46 ) and gastric cancers (reviewed by De Bruin et al. 7 ). Using staining techniques PD‐ECGF/TP is often overexpressed in tumor infiltrating cells (mainly macrophages) and colon cancer cells themselves.( 7 , 47 , 48 ) Takahashi et al.( 49 ) previously reported that high blood vessel density is well correlated with increased PD‐ECGF/TP expression and is associated with metastasis formation in human colorectal cancer,( 48 ) and gastric cancer. High PD‐ECGF/TP expression is often associated with high vessel counts in colorectal tumors expressing low VEGF, in contrast to high VEGF expressing tumors with high vessel density.( 50 ) Therefore PD‐ECGF expression in human colorectal tumors by infiltrating cells provides an additional alternate mechanism for tumor neovascularization.
Both VEGF and PD‐ECGF/TP are important in human angiogenesis induced by both tumor and normal cells and therefore these factors and their receptors are good targets for antiangiogenic therapy.( 7 ) Although there are several other factors/parameters that regulate angiogenesis, VEGF and PD‐ECGF/TP expression levels and counting microvessels (angiogenic index) within the tumors will have a prognostic value. For PD‐ECGF/TP Takebayashi et al.( 46 ) already postulated that PD‐ECGF/TP inhibition might improve prognosis for some colorectal cancer patients. Tumor growth and metastasis are dependent on a sufficient blood supply and therefore, inhibition of tumor‐induced angiogenesis in combination with classic cytotoxic chemotherapy is a strategy to improve survival for patients with solid tumors. Several (pre)clinical studies have already shown enhanced tumor regression by combining an antiangiogenic agent with cytotoxic agents.( 51 , 52 )
The downstream mediator of PD‐ECGF/TP 2‐deoxy‐D‐ribose promotes angiogenesis by enhancing chemotaxis of vascular endothelial cells,( 53 ) and may confer resistance to apoptosis induced by hypoxia.( 54 ) PD‐ECGF/TP enzymatic activity is needed for angiogenesis, tumor growth, invasion and metastasis, which is all related to the often up‐regulated PD‐ECGF/TP in tumor stroma. The potential of TPI to block these processes was investigated in vivo.( 35 , 55 , 56 ) TPI significantly inhibited PD‐ECGF/TP‐induced neovascularization in a dose‐dependent manner in mice models. Furthermore, KB human epidermoid carcinoma expressing high PD‐ECGF/TP (KB/TP) in the mouse dorsal air sac assay, had a higher level of blood vessel formation than low PD‐ECGF/TP expressing KB cells, while TPI could suppress this process.( 56 ) Fig. 7 shows that TPI strongly inhibits KB/TP‐induced formation of new blood vessels. Additionally, TPI alone decreased the growth rate of KB/TP cells (Fig. 8) and increased the apoptotic index significantly, indicating the antitumor activity of TPI as a single agent and its important role in tumor growth.
Figure 7.
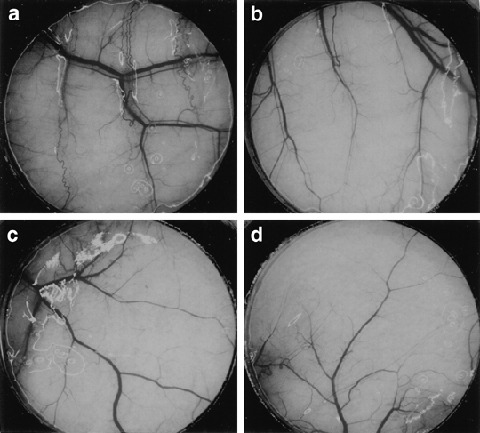
The effect of thymidine phosphorylase inhibitor (TPI) on TP‐induced angiogenesis. Mice implanted with a chamber containing human epidermoid carcinoma cells transfected with the PD‐ECGF/TP gene (KB/TP) (A and B) and human epidermoid carcinoma cells transfected with empty control vector (KB/CV) (c, d) cells were treated with vehicle (a, c) or 50 mg/kg per day TPI (b, d). Magnification: 20 ×. Data were adapted from Matsushita et al. (1999; with permission).( 56 )
Figure 8.
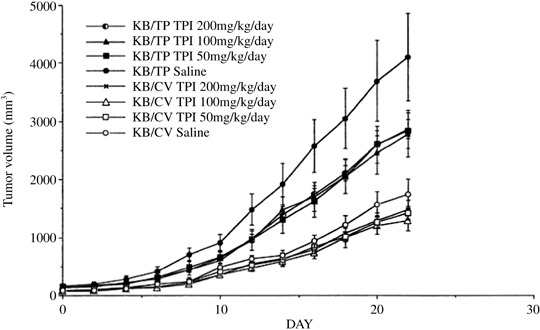
The effects of thymidine phosphorylase inhibitor (TPI) on tumor weight. After the tumor size reached about 50–150 mm3, TPI was administered i.p. for 22 days at the indicated amounts. Statistical significance was determined using multiple regression analysis, adjusting for tumor type and observation period (mean ± standard error of the mean; n = 6). Data were adapted from Matsushita et al. (1999; with permission).( 56 )
The statistically significant correlation between the presence of PD‐ECGF/TP expression and microvessel density in early stage human colon carcinomas was previously reported.( 55 )The depth of tumor invasion is highly related to microvessel density, and therefore PD‐ECGF/TP may have an important role in the early stages in the development of colorectal cancer through angiogenesis. This indicates the existence of an angiogenic switch that may occur in the early development of colorectal cancer, simultaneously to initiation of invasion,( 49 ) which is directly related to liver metastasis, which in turn is associated with poor prognosis and patient survival. TAS‐102 was able to reduce the number of liver metastases in a nude mouse model.( 21 ) TPI alone decreased the chemotactic motility and basement membrane invasion of KB/TP cells,( 57 ) and suppressed the number of liver metastases macroscopically, but also suppressed the level of human β‐globin, which is a marker for liver micrometastases.( 57 ) Sato et al.( 58 ) also determined that, using a matrigel invasion assay, high PD‐ECGF/TP expressing transfected human A549 lung adenocarcinoma cells have higher invasive potential than their mock transfectant cells, and that TPI markedly diminished the invasive activity. In nude mice TPI also suppressed the formation of lung metastases, which together with the previous findings all demonstrate that PD‐ECGF/TP is a key role player in the invasiveness and metastasis of high PD‐ECGF/TP expressing solid tumors. Because of the potent anti‐invasive and antimetastatic activity of TPI, TAS‐102 is a promising candidate for clinical use (see also Table 1).
Table 1.
Summary of studies in which an anti‐invasive effect of TPI on tumor cells expressing TP was demonstrated
| Study | Models | Drug Treatment (tumor cell type) | Antitumor effects | Reference |
|---|---|---|---|---|
| Matsushita et al. (1999) | Tumor implants nude mice; mouse dorsal air sac assay (human epidermoid carcinoma) | TPI alone | Decreased tumor growth rates; increased apoptosis induction; reduced microvessel density | ( 56 ) |
| Fukushima et al. (2000) | Tumor implants nude mice (human gastric carcinoma) | TFT ± TPI | Reduction of tumor volume | ( 34 ) |
| Takao et al. (2000) | Tumor implants nude mice; Matrigel invasion/chemotaxis chamber assays (human epidermoid carcinoma) | TPI alone | Reduced metastasis; inhibition of basement membrane invasion and chemotactic motility | ( 57 ) |
| Sato et al. (2003) | Tumor implants nude mice; Matrigel invasion chamber assay (human lung adenocarcinoma) | TPI alone | Reduced metastasis; inhibition of basement membrane invasion | ( 58 ) |
| Emura et al. (2004) | Tumor implants nude mice (human gastric/pancreatic carcinomas) | TAS‐102 | Decreased tumor growth rates | ( 17 ) |
| Emura et al. (2004) | Tumor implants nude mice (several different human cancers) | TAS‐102 | Decreased tumor growth rates | ( 74 ) |
| Emura et al. (2004) | Tumor implants nude mice (several different human cancers) | TAS‐102 | Decreased tumor growth rates; reduced metastasis | ( 21 ) |
| Emura et al. (2004) | Tumor implants nude rats/mice (human colorectal carcinoma) | TAS‐102 | Decreased tumor growth rates | ( 18 ) |
| Emura et al. (2005) | Tumor implants nude mice (human gastric/colorectal carcinomas) | TFT ± TPI | Decreased tumor growth rates | ( 35 ) |
| Temmink et al. (2007) | Hollow Fiber Model (normal mice) (human colorectal carcinoma) | TFT ± TPI | Decreased tumor growth rates; increased apoptosis induction | ( 61 ) |
TFT, thymidine analog trifluorothymidine; TPI, thymidine phosphorylase inhibitor.
It has been shown that chemotherapeutic drugs themselves often up‐regulate PD‐ECGF/TP,( 59 ) which will not affect TAS‐102, because the enzyme is inhibited by TPI. Additionally, low PD‐ECGF/TP expression might confer resistance to several fluoropyrimidine anticancer agents.( 41 ) The oral 5FU pro‐drug capecitabine (Xeloda; Roche Pharmaceuticals),( 8 , 60 ) is activated by PD‐ECGF/TP, and therefore is extremely dependent on sufficient PD‐ECGF/TP levels at the tumor site. Therefore, low PD‐ECGF/TP levels may negatively influence therapeutic outcome of Xeloda‐based chemotherapy.( 41 ) The in vivo role of PD‐ECGF/TP in the cytotoxicity and short‐term pharmacodynamics of TAS‐102 compared to capecitabine in colon cancer cells was previously studied using the Hollow Fiber Model (HFM).( 61 ) Results showed that TAS‐102 is only effective in inducing cytotoxicity when systemic TPI is present, but acts against both undetectable and high‐TP‐expressing colon cancer cells (Fig. 9), while the capecitabine‐derived metabolite 5‐fluoro‐5′‐deoxyuridine (5′DFUR) needs cellular TP to exert significant activity. However, evidence is accumulating that UP can also play an important role in 5′DFUR conversion into 5FU.( 62 , 63 , 64 ) Although TPI effectively inhibits TP activity, it is a poor inhibitor of UP.( 34 ) However, TFT is a poor substrate for UP, and a high activity of UP is unlikely to affect the efficacy of TFT. This study also showed that the effect of TPI is independent of the level of TP, since a tumor with undetectable TP activity and its TP transfected variant with a very high TP activity were used. ( 61 )
Figure 9.
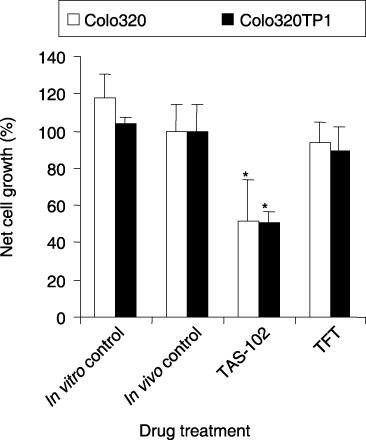
Growth inhibition of Colo320 and Colo320TP1 cells in the hollow fibers implanted s.c. in mice. The mice were treated with the formulation TAS‐102 or with thymidine analog trifluorothymidine (TFT) a; one at the same dose. Four to five fibers per cell line, all implanted in different mice, were assayed. Values are Means ± standard error. In vitro control, fibers were incubated in DMEM culture medium. Compared to in vivo fiber control, *P < 0.05; to Colo320, **P < 0.05. Data were adapted from Temmink et al. (2007; with permission).( 61 )
Combination studies. Novel treatment options often consist of combinations of drugs in which current chemotherapeutic regimens are combined with immunotherapy or radiation therapy. The combination of cytotoxic and antiangiogenic agents would have great potential in improving anticancer therapies. For that reason TAS‐102 is an excellent candidate to be combined with other cytotoxic agents. To investigate this potential, TAS‐102 was combined with other anticancer agents in in vitro combination studies. TFT was combined with other TS inhibitors, oxaliplatin (OHP) or irinotecan (CPT‐11) in different schedules in order to determine their interactions. Combining TFT with FdUrd is no option, since the effects were purely additive and no synergistic interaction was found.( 3 ) At low folate conditions TFT and folate‐based TS inhibitors showed pronounced synergism in growth inhibition, two‐sided TS inhibition and DNA damage induction, whereas at high folate conditions only additive effects were seen.( 65 ) The combination of TFT and raltitrexed (ZD1694) was mainly additive when added simultaneously at both low and high folate conditions.
The sensitivity of colon cancer cells to OHP could be increased by simultaneous exposure to TFT (Fig. 10),( 66 ) resulting in increased Pt‐DNA adduct formation and subsequent increased DNA damage induction and apoptosis induction. The TFT‐OHP combination was dose‐schedule dependent, since TFT preincubation decreased OHP‐induced cytotoxicity to colorectal cancer cells. For the combination of TFT and SN38, the active metabolite of CPT‐11, the most pronounced synergistic interactions were found when colorectal cancer cells were preincubated with TFT before SN38 exposure, which resulted in increased DNA strand break formation and cell death.( 67 )
Figure 10.
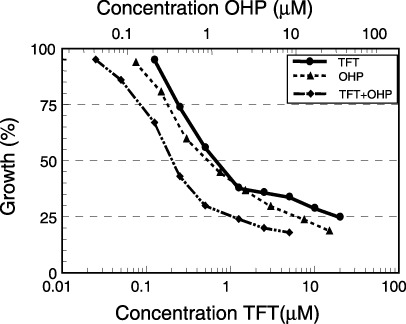
Interaction between thymidine analog trifluorothymidine (TFT) and oxaliplatin (OHP) as tested in the colon carcinoma cell lines H630. Drugs were added in a fixed ratio and cells were exposed for 72 h. This is an example of five cell lines in which a clear synergism was found (reproduced with permission).( 66 )
Pharmacokinetics
TFT catabolism in vivo. The main pharmacokinetics (PK) of TFT resemble those of most antimetabolites with a short half‐life (Table 2).( 68 ) The mean plasma half‐life of single TFT injected to cancer patients was less than 15 min,( 23 ) therefore it was previously assumed that repeated rapid injections of TFT would be more effective with respect to exposing cancer cells to the drug.( 4 ) TFT is phosphorylyzed to TF‐Thy, which was found to have growth inhibitory activity in bacterial but not mammalian biological systems.( 3 ) TF‐Thy can be hydrolyzed to 5‐carboxyuracil with loss of the inorganic fluoride, and early clinical pharmacology studies with TFT showed that the only metabolites detected in urine and serum samples of treated cancer patients were unmodified TFT, TF‐Thy and 5‐carboxyuracil.( 2 , 23 )
Table 2.
Summary of studies in which pharmacokinetic parameters of TFT (± TPI) were determined
| Study (model) | TFT treatment | Pharmacokinetic parameters | Reference | |||
|---|---|---|---|---|---|---|
| Cmax (µg/mL) | Tmax (min) | T1/2 (min) | AUC (µg h/mL) | |||
| Dexter et al. (1972) (human) | 27 mg/kg i.v. | 18 | ( 23 ) | |||
| Takeda et al. (1992) (mouse) | 50 mg/kg oral | 8 | 30 | 30 | 7 | ( 24 ) |
| Fukushima et al. (2000)(monkey) (mouse) | 10 mg/kg oral (+ equi‐M TPI) | 0.23 (15) | 60 (60) | ? | 0.28 (28) | ( 34 ) |
| 50 mg/kg oral (+ equi‐M TPI) | 16 (32) | 10 (30) | ? | 13 (24) | ||
| Yano et al. (2004) (mouse) | 50 mg/kg oral TFT alone | 13 | 30 | ? | 8 | ( 32, 33 ) |
| Hong et al. (2006) (human) (human) | 50 mg/m2/d TAS‐102 (MTD) | 9.1 | 30 | 15.9 | ( 75 ) | |
| 100 mg/m2/d TAS‐102 (DLT) | 13.1 | 45 | 27.8 | |||
AUC, area under the plasma concentration time curve; DLT, dose‐limiting toxicity; MTD, maximum‐tolerated dose; TFT, thymidine analog trifluorothymidine; TPI, thymidine phosphorylase inhibitor.
When studying TFT metabolism in rat brain using 19F‐nuclear magnetic resonance spectroscopy,( 69 ) it was demonstrated that TFT can cross the blood–brain barrier in low micromolar concentrations and is predominantly metabolized to TF‐Thy. Reduction of the nitrogen base moiety and generation of 5–6‐dihydro‐species followed by ring degradation is the major pathway for hepatic metabolism. Besides TF‐Thy, the dihydro‐species of TFT and TF‐Thy, the reductive catabolites α‐trifluoromethyl‐β‐ureidopropionic acid (F 3MUPA) and α‐trifluoromethyl‐β‐alanine (F 3MBA) were detected in liver extracts. Low levels of fluoride ions were detected in serum and urine, but also in brain and liver extracts. So far no validation experiments in humans have been reported.
In contrast to TF‐Thy, TFT itself has considerable biological activity as described in the previous sections. TPI was able to increase the AUC of TFT when TAS‐102 was orally administered to rodents or monkeys.( 34 , 35 ) This increase was more dramatic in monkeys due to differences in expression of pyrimidine phosphorylases in these animals.( 41 , 64 , 70 ) PD‐ECGF/TP is predominantly expressed in monkeys, while UP is higher than PD‐ECGF/TP in rodents. Tsuchiya et al.( 71 ) developed a PK model in order to evaluate TFT bioavailability in monkeys after co‐administration with TFT. Their setup for hepatic intrinsic clearance was time‐ or concentration dependent, since TFT catabolism is inhibited by TPI competitively, thereby taking into account the time‐courses of both TFT and TPI in plasma. Using this model Tsuchiya et al. were able to provide a quantitative explanation for the dose‐dependent enhancement of TPI for the AUC of TFT in monkeys and they observed a much smaller AUC increase in rats.
It was also important to establish an optimal molar drug ratio of TFT:TPI to reach maximal TFT plasma levels. With a TFT:TPI molar ratio of 2:1 the highest TFT concentrations in the plasma were obtained.( 35 ) At a co‐administration of 1 M TFT (10 mg/kg) and >0.5 M TPI a maximum and constant level of 15 µg TFT/mL was found (4, 6). Using this molar drug ratio, a maximal augmented antitumor acivity of TFT was found for gastrointestinal cancer cells xenografted into mice, with ED50 values between 10 and 25 mg/kg. In addition to optimal TFT plasma concentrations, the incorporation of TFT into DNA is an important pharmacological discriminating feature in contrast to 5FU metabolites.( 17 ) Steady levels of TFT in plasma obtained with divided dosing of TFT within an adequate time‐period results in optimal DNA incorporation and subsequent antitumor activity.
The enzyme activities of PD‐ECGF/TP, TK and TS are the most important in TFT metabolism and cytotoxic activity in cancer cells. In vivo PD‐ECGF/TP mediates TFT catabolism, and TK levels are often higher in human tumor tissues than normal tissues,( 27 , 72 , 73 ) which would lead to more rapid TFT activation. The TS expression levels in xenografts of human tumors were not directly related to the antitumor activity, neither were TK and PD‐ECGF/TP.( 74 ) However, the ratio of TK and PD‐ECGF/TP was correlated with the antitumor activity, demonstrating that TK and PD‐ECGF/TP were important factors in the in vivo efficacy of TAS‐102, and a high TK:PD‐ECGF/TP ratio may predict the response to TAS‐102 therapy. Besides TFT breakdown, TdR levels may theoretically reduce the growth inhibiting effect of TFT, because inhibition of both TFT and TdR degradation by TPI can lead to an increase of intracellular TdR.( 74 ) TAS‐102 or TPI administration can increase plasma TdR levels, but not that of Urd, dUrd or deoxycytidine (dCyt), however, no evidence for a protective effect of TdR was found.
Clinical trials. The initial antitumor effects of TFT against colon cancer were reported in 1971, showing that repeated administration of TFT can produce a reduction in tumor size of patients with breast and colon cancer.( 4 ) However, systemic administration of TFT alone (2.5 mg/kg per day) in divided doses every 3 h for 8–13 days, resulted in severe bone marrow depression.( 4 ) TFT‐mediated cytotoxicity was primarily confined to the hematopoietic system with less damage of the gastrointestinal tract compared to 5FU and FdUrd.( 3 , 23 ) The TFT early clinical trials were discontinued because of the short half‐life due to its rapid clearance and extensive degradation by PD‐ECGF/TP in vivo. This likely resulted in the moderate antitumor efficacy that was observed.( 4 )
Thymidine analog trifluorothymidine re‐entered clinical development as TAS‐102, and three phase I clinical trials have been completed (summarized in Table 3). The first phase I trial of TAS‐102 orally administered to patients with solid tumors once daily for 14 days every 3 weeks with a determined maximum‐tolerated dose (MTD) of 50 mg/m2 per day and with granulocytopenia as the dose‐limiting toxicity (DLT) at 100 mg/m2 per day.( 75 ) In this schedule drug accumulation was observed, with the AUC increasing twofold to 2.5‐fold after 2 weeks, possibly explaining the toxicity. In the second trial once‐daily TAS‐102 was given orally (50–80 mg/m2 per day) to patients with gastrointestinal malignancies for 5 days a week for 2 weeks repeated every 4 weeks.( 76 ) This schedule with the 2‐day treatment rest allowed to administer higher doses of TAS‐102 compared to the continuous daily schedule. In a third phase I study, patients with solid tumors received TAS‐102 doses ranging from 100 to 140 mg/m2 per day for 5 days every 3 weeks in order to determine the MTD.( 77 ) At 120 mg/m2 per day, patients experienced severe granulocytopenia which was considered to be the DLT. Other toxicities observed included neutropenia, mild to moderate nausea, vomiting, diarrhea, fatigue and rash.
Table 3.
Phase I studies of TAS‐102 orally given in patients with solid tumors to determine toxicity and efficacy profiles
| Study (reference) | Treatment | Evaluation | |||
|---|---|---|---|---|---|
| Dose (mg/m2 per day) | Schedule | No. patients | Toxicity | Efficacy (best response) | |
| Hong et al.( 75 ) | 100 | q1d×14 | 2 | °3/4 bone marrow suppression (2/2) # | ND |
| 50 | q1d×14 | 6 | °3/4 toxicities (0/6) | SD (2/6), PD (4/6) ## | |
| 60 | q1d×14 | 6 | °3/4 granulocytopenia (3/6) | SD (2/6), PD (4/6) | |
| Dwivedy et al.( 76 ) | 50 | q1d×5 | 3 | °3/4 toxicities (0/3) | ND |
| 70 | q1d×5 | 6 | °4 granulocytopenia (1/6), °3 toxicities (0/6) | SD (1/6), NE (5/6) | |
| 80 | q1d×5 | NK | NK | NK | |
| Thomas et al.( 77 ) | 120 | q1d×5 | NK | °4 granulocytopenia (NK) | NK |
| Wolff et al. (unpublished) | 60 | TID×5 | 3 | °3 neutropenia (3/3) | SD (9/15)* |
| 70 | TID×5 | 6 | °3 neutropenia (5/6) | ||
| 80 | TID×5 | 6 | °3/4 neutropenia (6/6), °3 thrombocytopenia (1/6) | ||
| Green et al.( 78 ) | 80 | BID×5 | 3 | °3/4 granulocytopenia (2/3) | SD (7/19)* |
| 90 | BID×5 | 16 | °3/4 granulocytopenia (9/16), °4 thrombocytopenia (1/16) | ||
Dose not specified, median duration of SD was >4 months;
# number of patients with toxicity/number of patients enrolled.
number of patients/number of patients enrolled; BID×5: twice per day for 5 days followed by 2 days rest for 2 weeks, repeated every 4 weeks; ND, not determined; NE, not evaluable; NK, not known; PD, progressive disease. q1d×14, once per day for 14 days, repeated every 3 weeks; q1d×5, once per day for 5 days followed by 2 days rest for 2 weeks, repeated every 4 weeks (Dwivedy et al. 76 ) or for 1 week, repeated every 3 weeks (Thomas et al. 77 ); SD, stable disease; TID×5, three times per day for 5 days followed by 2 days rest for 2 weeks, repeated every 4 weeks.
In more recent TAS‐102 phase I trials two or three times a day administration schedules were used, because divided daily dosing of TFT resulted in higher antitumor activity in preclinical studies due to increased incorporation of TFT into DNA. Furthermore, the previous once‐daily TAS‐102 phase I trials showed a short TFT half‐life. In an unpublished phase I trial conducted at the MD Anderson Cancer Center (Houston, TX, US) using a three times a day (60–80 mg/m2 per day) schedule (5 days per week for 2 weeks, repeated every 4 weeks) with patients with solid tumors receiving TAS‐102 orally, it was concluded that TAS‐102 was well‐tolerated with manageable hematological (most common grade 3/4 neutropenia) and non‐hematological (e.g. nausea, vomiting, fatigue, colitis) toxicities. The suggested phase II TAS‐102 dose is 70 mg/m2 per day. Another phase I trial was conducted to determine the phase II dose and the DLT in patients with metastatic breast cancer.( 78 ) Patients received TAS‐102 orally at an initial dose level of 80 mg/m2 per day in a twice daily schedule (5 days per week for 2 weeks, repeated every 4 weeks). Clinical activity was observed including prolonged disease control (>12 weeks). It was concluded that TAS‐102 is an active agent against heavily pretreated metastatic breast cancer patients, with primarily hematologic toxicities (mainly grade 3/4 granulocytopenia and grade 4 thrombocytopenia), and the recommended dose was 50 mg/m2/d using this schedule.
Single and/or combination regimens of TAS‐102 are currently planned against gastrointestinal cancer in phase II studies (in US and Japan).
Conclusions and future directions
The novel formulation TAS‐102 is promising because of its dual‐targeted properties: both antiangiogenic and cytotoxic effects, which may be beneficial against gastrointestinal malignancies. TAS‐102 has also great potential in future clinical drug combination studies, both with cytotoxic agents and targeted therapy. A combination of these treatments may help to prevent recurrence of tumors due to either drug resistance, or to increased vascularization of the tumor between chemotherapeutic treatments. Current strategies favor attacking multiple targets, such as angiogenesis inhibitors in combination with radio‐ or chemotherapeutic treatment.
Although inhibition of angiogenesis can reduce microvessel density, it does not necessarily have to result in reduced delivery of chemotherapeutic drugs to the tumor cells.( 79 ) However, solid tumors often have increased interstitial fluid pressure, which will result in decreased delivery of chemotherapeutic drugs.( 80 ) Anti‐angiogenic treatment has considerable antitumor efficacy, but several studies showed that a combination of angiostatic and cytotoxic agents can increase this efficacy. TAS‐102 has both properties, and has great potential to improve the treatment of gastrointestinal malignancies when combined with other angiostatic or cytotoxic agents, such as with the VEGF inhibitor bevacizumab (Avastin; Genentech) or the DNA synthesis inhibitors OHP (Eloxatin; Sanofi‐Synthelabo) or CPT‐11 (Camptosar; Pfizer Pharmaceuticals), respectively. Other possibilities are combining TAS‐102 with cetuximab (IMC‐C225; Erbitux; ImClone Systems), which targets the human epidermal growth factor receptor (EGFR), or with the small‐molecule inhibitors of the EGFR tyrosine kinase domain gefitinib (ZD1839; Iressa; AstraZeneca Pharmaceuticals) or erlotinib (OSI‐774; Tarceva; OSI Pharmaceuticals).
Acknowledgment
We thank Dr Shin‐ichi Akiyama (Institute for Cancer Research, Kagoshima University, Kagoshima, Japan) for providing data to illustrate the antiangiogenic properties of TPI (7, 8).
References
- 1. Heidelberger C, Parsons DG, Remy DC. Syntheses of 5‐trifluoromethyluracil and 5‐trifluoromethyl‐2′‐deoxyuridine. J Med Chem 1964; 55: 1–5. [DOI] [PubMed] [Google Scholar]
- 2. Heidelberger C, Boohar J, Kampschroer B. Fluorinated pyrimidines. XXIV. In vivo metabolism of 5‐trifluoromethyluracil‐2‐C‐14 and 5‐trifluoromethyl‐2′‐deoxyuridine‐2‐C‐14. Cancer Res 1965; 25: 377–81. [PubMed] [Google Scholar]
- 3. Heidelberger C, Anderson SW. Fluorinated pyrimidines. XXI. The tumor inhibitory activity of 5‐trifluoromethyl‐2′‐deoxyuridine. Cancer Res 1964; 24: 1979–85. [PubMed] [Google Scholar]
- 4. Ansfield FJ, Ramirez G. Phase I and II studies of 2′‐deoxy‐5‐(trifluoromethyl)‐uridine (NSC‐75520). Cancer Chemother Rep 1971; 55: 205–8. [PubMed] [Google Scholar]
- 5. De Clercq E. Antiviral drugs in current clinical use. J Clin Virol 2004; 30: 115–33. [DOI] [PubMed] [Google Scholar]
- 6. Carmine AA, Brogden RN, Heel RC, Speight TM, Avery GS. Trifluridine: a review of its antiviral activity and therapeutic use in the topical treatment of viral eye infections. Drugs 1982; 23: 329–53. [DOI] [PubMed] [Google Scholar]
- 7. De Bruin M, Temmink OH, Hoekman K, Pinedo HM, Peters GJ. Role of platelet derived endothelial cell growth factor/thymidine phosphorylase in health and disease. Cancer Ther 2006; 4: 99–124. [Google Scholar]
- 8. Hoff PM, Ansari R, Batist G et al. Comparison of oral capecitabine versus intravenous fluorouracil plus leucovorin as first‐line treatment in 605 patients with metastatic colorectal cancer: results of a randomized phase III study. J Clin Oncol 2001; 19: 2282–92. [DOI] [PubMed] [Google Scholar]
- 9. Reyes P, Heidelberger C. Fluorinated pyrimidines. XXVI. Mammalian thymidylate synthetase: its mechanism of action and inhibition by fluorinated nucleotides. Mol Pharmacol 1965; 1: 14–30. [PubMed] [Google Scholar]
- 10. Van Triest B, Peters GJ. Thymidylate synthase: a target for combination therapy and determinant of chemotherapeutic response in colorectal cancer. Oncology 1999; 57: 179–94. [DOI] [PubMed] [Google Scholar]
- 11. Eckstein JW, Foster PG, Finer‐Moore J, Wataya Y, Santi DV. Mechanism‐based inhibition of thymidylate synthase by 5‐(trifluoromethyl)‐2′‐deoxyuridine 5′‐monophosphate. Biochemistry 1994; 33: 15 086–94. [DOI] [PubMed] [Google Scholar]
- 12. Santi DV, Sakai TT. Thymidylate synthetase. Model studies of inhibition by 5‐trifluoromethyl‐2′‐deoxyuridylic acid. Biochemistry 1971; 10: 3598–607. [DOI] [PubMed] [Google Scholar]
- 13. Temmink OH, Comijn EM, Fukushima M, Peters GJ. Intracellular thymidylate synthase inhibition by trifluorothymidine in FM3A cells. Nucleosides Nucleotides Nucleic Acids 2004; 23: 1491–4. [DOI] [PubMed] [Google Scholar]
- 14. Peters GJ, Backus HH, Freemantle S et al. Induction of thymidylate synthase as a 5‐fluorouracil resistance mechanism. Biochim Biophys Acta 2002; 1587: 194–205. [DOI] [PubMed] [Google Scholar]
- 15. Fujiwara Y, Heidelberger C. Fluorinated pyrimidines. 38. The incorporation of 5‐trifluoromethyl‐2′‐deoxyuridine into the deoxyribonucleic acid of vaccinia virus. Mol Pharmacol 1970; 6: 281–91. [PubMed] [Google Scholar]
- 16. Temmink OH, De Bruin M, Comijn EM, Fukushima M, Peters GJ. Determinants of trifluorothymidine sensitivity and metabolism in colon and lung cancer cells. Anticancer Drugs 2005; 16: 285–92. [DOI] [PubMed] [Google Scholar]
- 17. Emura T, Nakagawa F, Fujioka A et al. An optimal dosing schedule for a novel combination antimetabolite, TAS‐102, based on its intracellular metabolism and its incorporation into DNA. Int J Mol Med 2004; 13: 249–55. [PubMed] [Google Scholar]
- 18. Emura T, Suzuki N, Yamaguchi M, Ohshimo H, Fukushima M. A novel combination antimetabolite, TAS‐102, exhibits antitumor activity in FU‐resistant human cancer cells through a mechanism involving FTD incorporation in DNA. Int J Oncol 2004; 25: 571–8. [PubMed] [Google Scholar]
- 19. Inaba M, Mitsuhashi J, Sawada H et al. Reduced activity of anabolizing enzymes in 5‐fluorouracil‐resistant human stomach cancer cells. Jpn J Cancer Res 1996; 87: 212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murakami Y, Kazuno H, Emura T, Tsujimoto H, Suzuki N, Fukushima M. Different mechanisms of acquired resistance to fluorinated pyrimidines in human colorectal cancer cells. Int J Oncol 2000; 17: 277–83. [DOI] [PubMed] [Google Scholar]
- 21. Emura T, Murakami Y, Nakagawa F, Fukushima M, Kitazato K. A novel antimetabolite, TAS‐102 retains its effect on FU‐related resistant cancer cells. Int J Mol Med 2004; 13: 545–9. [PubMed] [Google Scholar]
- 22. De Bruin M, Van Capel T, Van Der Born K et al. Role of platelet‐derived endothelial cell growth factor/thymidine phosphorylase in fluoropyrimidine sensitivity. Br J Cancer 2003; 88: 957–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dexter DL, Wolberg WH, Ansfield FJ, Helson L, Heidelberger C. The clinical pharmacology of 5‐trifluoromethyl‐2′‐deoxyuridine. Cancer Res 1972; 32: 247–53. [PubMed] [Google Scholar]
- 24. Takeda S, Wierzba K, Yamashita J et al. Potentiation of the antitumor activity of 5‐trifluoromethyl‐2′‐deoxyuridine by the use of depot forms of the parent compound. Cancer Chemother Pharmacol 1992; 30: 360–4. [DOI] [PubMed] [Google Scholar]
- 25. Yamashita J, Takeda S, Matsumoto H, Unemi N, Yasumoto M. Studies on antitumor agents. 8. Antitumor activities of O‐alkyl derivatives of 2′‐deoxy‐5‐(trifluoromethyl) uridine and 2′‐deoxy‐5‐fluorouridine. J Med Chem 1989; 32: 136–9. [DOI] [PubMed] [Google Scholar]
- 26. Yamashita J, Takeda S, Matsumoto H, Terada T, Unemi N, Yasumoto M. Studies on antitumor agents. VI. Syntheses and antitumor activities of acyl derivatives of 2′‐deoxy‐5‐trifluoromethyluridine. Chem Pharm Bull (Tokyo) 1987; 35: 2090–4. [DOI] [PubMed] [Google Scholar]
- 27. Peters GJ, Van Groeningen CJ, Laurensse EJ, Pinedo HM. A comparison of 5‐fluorouracil metabolism in human colorectal cancer and colon mucosa. Cancer 1991; 68: 1903–9. [DOI] [PubMed] [Google Scholar]
- 28. Langen P, Etzold G, Barwolff D, Preussel B. Inhibition of thymidine phosphorylase by 6‐aminothymine and derivatives of 6‐aminouracil. Biochem Pharmacol 1967; 16: 1833–7. [DOI] [PubMed] [Google Scholar]
- 29. Reigan P, Gbaj A, Chinje E, Stratford IJ, Douglas KT, Freeman S. Synthesis and enzymatic evaluation of xanthine oxidase‐activated prodrugs based on inhibitors of thymidine phosphorylase. Bioorg Med Chem Lett 2004; 14: 5247–50. [DOI] [PubMed] [Google Scholar]
- 30. Cole C, Reigan P, Gbaj A et al. Potential tumor‐selective nitroimidazolylmethyluracil prodrug derivatives: inhibitors of the angiogenic enzyme thymidine phosphorylase. J Med Chem 2003; 46: 207–9. [DOI] [PubMed] [Google Scholar]
- 31. Balzarini J, Gamboa AE, Esnouf R et al. 7‐Deazaxanthine, a novel prototype inhibitor of thymidine phosphorylase. FEBS Lett 1998; 438: 91–5. [DOI] [PubMed] [Google Scholar]
- 32. Yano S, Kazuno H, Suzuki N et al. Synthesis and evaluation of 6‐methylene‐bridged uracil derivatives. Part 1: discovery of novel orally active inhibitors of human thymidine phosphorylase. Bioorg Med Chem 2004; 12: 3431–41. [DOI] [PubMed] [Google Scholar]
- 33. Yano S, Kazuno H, Sato T et al. Synthesis and evaluation of 6‐methylene‐bridged uracil derivatives. Part 2: optimization of inhibitors of human thymidine phosphorylase and their selectivity with uridine phosphorylase. Bioorg Med Chem 2004; 12: 3443–50. [DOI] [PubMed] [Google Scholar]
- 34. Fukushima M, Suzuki N, Emura T et al. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2′‐deoxyribonucleosides. Biochem Pharmacol 2000; 59: 1227–36. [DOI] [PubMed] [Google Scholar]
- 35. Emura T, Suzuki N, Fujioka A, Ohshimo H, Fukushima M. Potentiation of the antitumor activity of alpha, alpha, alpha‐trifluorothymidine by the co‐administration of an inhibitor of thymidine phosphorylase at a suitable molar ratio in vivo. Int J Oncol 2005; 27: 449–55. [PubMed] [Google Scholar]
- 36. Ellis LM. Angiogenesis and its role in colorectal tumor and metastasis formation. Semin Oncol 2004; 31: 3–9. [DOI] [PubMed] [Google Scholar]
- 37. Ishigami SI, Arii S, Furutani M et al. Predictive value of vascular endothelial growth factor (VEGF) in metastasis and prognosis of human colorectal cancer. Br J Cancer 1998; 78: 1379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manetti F, Botta M. Small‐molecule inhibitors of fibroblast growth factor receptor (FGFR) tyrosine kinases (TK). Curr Pharm Des 2003; 9: 567–81. [DOI] [PubMed] [Google Scholar]
- 39. George D. Platelet‐derived growth factor receptors: a therapeutic target in solid tumors. Semin Oncol 2001; 28: 27–33. [PubMed] [Google Scholar]
- 40. Miyazono K, Okabe T, Urabe A, Takaku F, Heldin CH. Purification and properties of an endothelial cell growth factor from human platelets. J Biol Chem 1987; 262: 4098–103. [PubMed] [Google Scholar]
- 41. Ackland SP, Peters GJ. Thymidine phosphorylase: its role in sensitivity and resistance to anticancer drugs. Drug Resist Updat 1999; 2: 205–14. [DOI] [PubMed] [Google Scholar]
- 42. Moghaddam A, Bicknell R. Expression of platelet‐derived endothelial cell growth factor in Escherichia coli and confirmation of its thymidine phosphorylase activity. Biochemistry 1992; 31: 12 141–6. [DOI] [PubMed] [Google Scholar]
- 43. Moghaddam A, Zhang HT, Fan TP et al. Thymidine phosphorylase is angiogenic and promotes tumor growth. Proc Natl Acad Sci USA 1995; 92: 998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haraguchi M, Miyadera K, Uemura K et al. Angiogenic activity of enzymes. Nature 1994; 368: 198. [DOI] [PubMed] [Google Scholar]
- 45. Takebayashi Y, Yamada K, Miyadera K et al. The activity and expression of thymidine phosphorylase in human solid tumours. Eur J Cancer 1996; 32A: 1227–32. [DOI] [PubMed] [Google Scholar]
- 46. Takebayashi Y, Akiyama S, Akiba S et al. Clinicopathologic and prognostic significance of an angiogenic factor, thymidine phosphorylase, in human colorectal carcinoma. J Natl Cancer Inst 1996; 88: 1110–17. [DOI] [PubMed] [Google Scholar]
- 47. Van Triest B, Pinedo HM, Blaauwgeers JL et al. Prognostic role of thymidylate synthase, thymidine phosphorylase/platelet‐derived endothelial cell growth factor, and proliferation markers in colorectal cancer. Clin Cancer Res 2000; 6: 1063–72. [PubMed] [Google Scholar]
- 48. Takahashi Y, Bucana CD, Liu W et al. Platelet‐derived endothelial cell growth factor in human colon cancer angiogenesis: role of infiltrating cells. J Natl Cancer Inst 1996; 88: 1146–51. [DOI] [PubMed] [Google Scholar]
- 49. Takahashi Y, Ellis LM, Mai M. The angiogenic switch of human colon cancer occurs simultaneous to initiation of invasion. Oncol Rep 2003; 10: 9–13. [PubMed] [Google Scholar]
- 50. Amaya H, Tanigawa N, Lu C et al. Association of vascular endothelial growth factor expression with tumor angiogenesis, survival and thymidine phosphorylase/platelet‐derived endothelial cell growth factor expression in human colorectal cancer. Cancer Lett 1997; 119: 227–35. [DOI] [PubMed] [Google Scholar]
- 51. Kabbinavar FF, Hambleton J, Mass RD, Hurwitz HI, Bergsland E, Sarkar S. Combined analysis of efficacy: the addition of bevacizumab to fluorouracil/leucovorin improves survival for patients with metastatic colorectal cancer. J Clin Oncol 2005; 23: 3706–12. [DOI] [PubMed] [Google Scholar]
- 52. Zondor SD, Medina PJ. Bevacizumab: an angiogenesis inhibitor with efficacy in colorectal and other malignancies. Ann Pharmacother 2004; 38: 1258–64. [DOI] [PubMed] [Google Scholar]
- 53. Uchimiya H, Furukawa T, Okamoto M et al. Suppression of thymidine phosphorylase‐mediated angiogenesis and tumor growth by 2‐deoxy‐L‐ribose. Cancer Res 2002; 62: 2834–9. [PubMed] [Google Scholar]
- 54. Ikeda R, Furukawa T, Kitazono M et al. Molecular basis for the inhibition of hypoxia‐induced apoptosis by 2‐deoxy‐D‐ribose. Biochem Biophys Res Commun 2002; 291: 806–12. [DOI] [PubMed] [Google Scholar]
- 55. Akiyama S, Furukawa T, Sumizawa T et al. The role of thymidine phosphorylase, an angiogenic enzyme, in tumor progression. Cancer Sci 2004; 95: 851–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Matsushita S, Nitanda T, Furukawa T et al. The effect of a thymidine phosphorylase inhibitor on angiogenesis and apoptosis in tumors. Cancer Res 1999; 59: 1911–6. [PubMed] [Google Scholar]
- 57. Takao S, Akiyama SI, Nakajo A et al. Suppression of metastasis by thymidine phosphorylase inhibitor. Cancer Res 2000; 60: 5345–8. [PubMed] [Google Scholar]
- 58. Sato J, Sata M, Nakamura H et al. Role of thymidine phosphorylase on invasiveness and metastasis in lung adenocarcinoma. Int J Cancer 2003; 106: 863–70. [DOI] [PubMed] [Google Scholar]
- 59. Endo M, Shinbori N, Fukase Y et al. Induction of thymidine phosphorylase expression and enhancement of efficacy of capecitabine or 5′‐deoxy‐5‐fluorouridine by cyclophosphamide in mammary tumor models. Int J Cancer 1999; 83: 127–34. [DOI] [PubMed] [Google Scholar]
- 60. Van Cutsem E, Twelves C, Cassidy J et al. Oral capecitabine compared with intravenous fluorouracil plus leucovorin in patients with metastatic colorectal cancer: results of a large phase III study. J Clin Oncol 2001; 19: 4097–106. [DOI] [PubMed] [Google Scholar]
- 61. Temmink OH, Prins HJ, Van Gelderop E, Peters GJ. The Hollow Fibre Assay as a model for in vivo pharmacodynamics of fluoropyrimidines in colon cancer cells. Br J Cancer 2007; 96: 61–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Temmink OH, De Bruin M, Turksma AW, Cricca S, Laan AC, Peters GJ. Activity and substrate specificity of pyrimidine phosphorylases and their role in fluoropyrimidine sensitivity in colon cancer cell lines. Int J Biochem Cell Biol 2007; 39: 565–75. [DOI] [PubMed] [Google Scholar]
- 63. Temmink OH, De Bruin M, Laan AC et al. The role of thymidine phosphorylase and uridine phosphorylase in (fluoro) pyrimidine metabolism in peripheral blood mononuclear cells. Int J Biochem Cell Biol 2006; 38: 1759–65. [DOI] [PubMed] [Google Scholar]
- 64. Cao D, Pizzorno G. Uridine phosphorylase: an important enzyme in pyrimidine metabolism and fluoropyrimidine activation. Drugs Today (Barc) 2004; 40: 431–43. [DOI] [PubMed] [Google Scholar]
- 65. Temmink OH, Hoogeland MF, Fukushima M, Peters GJ. Low folate conditions may enhance the interaction of trifluorothymidine with antifolates in colon cancer cells. Cancer Chemother Pharmacol 2006; 57: 171–9. [DOI] [PubMed] [Google Scholar]
- 66. Temmink OH, Hoebe EK, Van Der Born K, Ackland SP, Fukushima M, Peters GJ. Mechanism of trifluorothymidine potentiation of oxaliplatin‐induced cytotoxicity to colorectal cancer cells. Br J Cancer 2007; 96: 231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Temmink OH, Hoebe EK, Fukushima M, Peters GJ. Irinotecan‐induced cytotoxicity to colon cancer cells in vitro is stimulated by pre‐incubation with trifluorothymidine. Eur J Cancer 2007; 43: 175–83. [DOI] [PubMed] [Google Scholar]
- 68. Peters GJ, Schornagel JH, Milano GA. Clinical pharmacokinetics of anti‐metabolites. Cancer Surv 1993; 17: 123–56. [PubMed] [Google Scholar]
- 69. Pouremad R, Bahk KD, Shen YJ, Knop RH, Wyrwicz AM. Quantitative 19F NMR study of trifluorothymidine metabolism in rat brain. NMR Biomed 1999; 12: 373–80. [DOI] [PubMed] [Google Scholar]
- 70. Pugmire MJ, Ealick SE. Structural analyses reveal two distinct families of nucleoside phosphorylases. Biochem J 2002; 361: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tsuchiya H, Kuwata K, Nagayama S, Yamashita K, Kamiya H, Harashima H. Pharmacokinetic modeling of species‐dependent enhanced bioavailability of trifluorothymidine by thymidine phosphorylase inhibitor. Drug Metab Pharmacokinet 2004; 19: 206–15. [DOI] [PubMed] [Google Scholar]
- 72. Maehara Y, Nakamura H, Nakane Y et al. Activities of various enzymes of pyrimidine nucleotide and DNA syntheses in normal and neoplastic human tissues. Gann 1982; 73: 289–98. [PubMed] [Google Scholar]
- 73. Gordon HL, Bardos TJ, Chmielewicz ZF, Ambrus JL. Comparative study of the thymidine kinase and thymidylate kinase activities and of the feedbach inhibition of thymidine kinase in normal and neoplastic human tissue. Cancer Res 1968; 28: 2068–77. [PubMed] [Google Scholar]
- 74. Emura T, Nakagawa F, Fujioka A, Ohshimo H, Kitazato K. Thymidine kinase and thymidine phosphorylase level as the main predictive parameter for sensitivity to TAS‐102 in a mouse model. Oncol Rep 2004; 11: 381–7. [PubMed] [Google Scholar]
- 75. Hong DS, Abbruzzese JL, Bogaard K et al. Phase I study to determine the safety and pharmacokinetics of oral administration of TAS‐102 in patients with solid tumors. Cancer 2006; 107: 1383–90. [DOI] [PubMed] [Google Scholar]
- 76. Dwivedy S, Hoff PM, Dumas P et al. Safety and pharmacokinetics (PK) of an antitumor/antiangiogenic agent, TAS‐102: a phase I study for patients (PTS) with solid tumors. Proc Am Soc Clin Oncol 2001; 20: Abstract 386. [Google Scholar]
- 77. Thomas MB, Hoff PM, Carter S et al. A dose‐finding, safety and pharmacokinetics study of TAS‐102, an antitumor/antiangiogenic agent given orally on a once‐daily schedule for five days every three weeks in patients with solid tumors. Proc Am Assoc Cancer Res 2002; 43: Abstract 2754. [Google Scholar]
- 78. Green MC, Pusztai L, Theriault RL et al. Phase I study to determine the safety of oral administration of TAS‐102 on a twice daily (BID) schedule for five days a week (wk) followed by two days rest for two wks, every (Q) four wks in patients (pts) with metastatic breast cancer (MBC). Proc Am Soc Clin Oncol 2006; 25: Abstract 10 576. [Google Scholar]
- 79. Wildiers H, Guetens G, De Boeck G et al. Effect of antivascular endothelial growth factor treatment on the intratumoral uptake of CPT‐11. Br J Cancer 2003; 88: 1979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer 2004; 4: 806–13. [DOI] [PubMed] [Google Scholar]