

Abstract
Cyclin A1 and cyclin B1 are overexpressed in various tumors but are present at low levels in normal tissues. Cyclin A1 is restricted to germ cells undergoing meiosis. In order to explore the possibility of using cyclin A1 and cyclin B1 as anticancer targets, we knocked them down in two lung cancer cell lines, H157 and H596, using siRNA. As with cyclin A1 siRNA in lung cancer cell lines, cyclin B1, Cdc2 and CDK2 were all significantly downregulated. The S phase fraction increased significantly, and they eventually underwent apoptosis by way of downregulated intrinsic apoptotic pathways and modulators with upregulated extrinsic apoptotic pathways. Our study suggests that cyclin A1 might be a promising anticancer target specific to lung cancer. (Cancer Sci 2006; 97: 1082–1092)
Abbreviations:
- CDK1
cyclin‐dependent kinase 1 (equal to Cdc2)
- CDK2
cyclin‐dependent kinase 2
- DMEM
Dulbecco's Modified Eagle's Medium
- FBS
fetal bovine serum
- FITC
fluorescein‐isothiocyanate
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- HDAC
histone deacetylase
- p‐CDK2
threonine 160 phosphorylation of cyclin‐dependent kinase 2
- PBS‐T
0.1% Tween‐20 in 1 × phosphate‐buffered saline
- PCR
polymerase chain reaction
- PI
propidium iodide
- RNAi
RNA interference
- RT‐PCR
reverse transcription–polymerase chain reaction
- SDS
sodium dodecylsulfate
- siRNA
small interfering RNA
- SSC
sodium chloride/sodium citrate buffer
- TNF
tumor necrosis factor.
Non‐small cell lung cancer is the most common cause of cancer death in developed countries. There are various novel therapeutic strategies currently under evaluation, as the clinical use of cytotoxic drugs is limited due to intrinsic or acquired resistance and toxicity.( 1 ) A better understanding of the molecular mechanisms of cytotoxic drug action and the identification of markers for individual patient chemosensitivity and prognosis has shed light on individualization of therapy, and novel agents that target specific intracellular pathways related to the distinctive properties of cancer cells continue to be developed. Among these agents, angiogenesis inhibitors, epidermal growth factor receptor targeting agents, COX2 inhibitors and protein kinase C inhibitors have received particular attention.( 2 , 3 ) Gene therapy, such as replacement of defective tumor suppressor genes, has also demonstrated a transient effect, and limitation of systemic delivery.( 4 ) However, few of these methods have been successful, thus it is urgent to explore novel therapies to maximize specificity and minimize toxicity.( 5 ) RNAi, a novel therapy that aims to silence a specific target molecule at the post‐transcriptional level, has been recently designed to leave the normal cell physiology undisturbed. RNAi is a potent novel tool for gene‐specific therapeutics based on the fact that it targets specific molecular alterations unique to tumor cells,( 6 , 7 , 8 , 9 , 10 , 11 ) and the apoptotic pathways, including the extrinsic and the intrinsic pathways.( 12 )
Human cyclin A binds to CDK2 and CDK1 and the resultant cyclin A–CDK1 and cyclin A–CDK2 complexes are required for entry into the S and M phases of the cell cycle, respectively.( 13 ) A second human cyclin A, cyclin A1, was found to be highly expressed in the testis and in several leukemic cell lines.( 14 ) An association of human cyclin A1 with CDK2 has been proved in vitro and in vivo. The recently cloned murine cyclin A1 is also expressed specifically in the normal testis, and binds to both CDK2 and CDK1. In situ hybridization has shown that murine cyclin A1 is expressed only in germ cells undergoing meiosis in the testis, suggesting that cyclin A1 plays a role in meiotic cell division.( 15 ) Moreover, Xenopus cyclin A1 is expressed in eggs and early embryonic cells, but not in late embryonic or cultured cells.( 16 ) It was demonstrated that cyclin A1 expression is regulated in the mitotic cell cycle in MG63 cells, and binds to the transcription factor, E2F‐1, the Rb family of proteins and the p21 family of CDK inhibitors.( 15 , 16 ) Its associated H1 kinase activity is regulated during the cell cycle, and the cyclin A1–CDK2 complex of insect cells has been shown to have kinase activity on the Rb family of proteins and E2F‐1.( 17 )
Cyclin B1 is the regulatory subunit of CDK1 Ser/Thr kinase. In mammalian cells, the accumulation of cyclin B1 is a prerequisite for mitotic initiation in the late G2 phase.( 18 , 19 , 20 ) As cyclin B1 has a direct effect on mitosis, the overexpression of cyclin B1 might lead to uncontrolled cell proliferation, neoplastic transformation and tumorigenesis.( 21 , 22 , 23 )
We aimed to explore the potency and specificity of cyclin A1 or cyclin B1 as a novel target by using siRNA in lung cancer.
Materials and Methods
Cell lines and culture conditions. Two lung cancer cell lines, H157, which contained a p53 deletion, and H596, which contained a p53 mutation, were kindly donated by the Dr YS Jang at the Department of Pulmonary Medicine (Yonsei University College of Medicine, Seoul, Korea). They were cultured in RPMI‐1640 medium (Gibco‐BRL, Grand Island, NY) and DMEM (Gibco‐BRL), respectively, supplemented with 10% heat‐inactivated FBS and maintained at 37°C in 5% CO2.
mRNA and protein expression of cyclin A1 in lung cancer cell lines.
Quantitative RT‐PCR Total RNA was extracted from cell lines using an UltraspecTM‐II RNA isolation kit (Biotecx, Houston, TX) according to the manufacturer's protocol. The following primers were used: forward primer for cyclin A1, 5′‐TAATACGACTCACTATAGGGAGA TGAAGTAGACACGG AC‐3′ reverse primer for cyclin A1, 5′‐TAATACGACTCACTATAGGGAG ATTCGAAGCCAAAAGCATGC‐3′ forward primer for cyclin A, 5′‐CCATTCATGTG AAGCA‐3′ and reverse primer for cyclin A, 5′‐AGTGCCCACAAGCTGAAGTT‐3′.
The amplification conditions were as follows: denaturation at 95°C for 15 min; 40 cycles of 95°C for 40 s, 53.2°C (for cyclin A1) and 61.3°C (cyclin A) for 1 min, 72°C for 1 min; and a final 10 min extension at 72°C.
Cell cycle synchronization. For G0/G1 arrest, cells were incubated in DMEM or RPMI‐1640 supplemented with 5% FBS for 24 h. Subsequently, they were incubated in DMEM or RPMI‐1640 supplemented with 1% FBS for 24 h before finally moving cells into serum‐free conditions. For S phase and G2/M arrest, cells were incubated in cell culture medium containing 2 mM thymidine (Sigma, St Louis, MO) and 0.4 µg/µL nocodazole (Sigma) for 16 h, respectively.
Cell cycle distribution observation Cell cycle distribution was monitored by flow cytometry (FACSCalibur; Becton Dickinson, San Jose, CA) using PI staining (Sigma), according to the manufacturer's instructions.
Western blot analysis Sample proteins and controls were resolved by polyacrylamide gel electrophoresis. Proteins were then transferred to nitrocellulose membranes using standard methods (Invitrogen, Carlsbad, CA). Blots were blocked with 5% non‐fat dried milk freshly made in 1 × PBS‐T, rocked on a rotating shaker for 1 h at room temperature, and rinsed three times in 1 × PBS‐T. The blots were incubated for 1 h at room temperature with the following primary antibodies diluted to 1 : 500 in PBS‐T: cyclin A1 (rabbit, polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA); cyclin A (rabbit, polyclonal; Santa Cruz Biotechnology); and cyclin B1 (rabbit, polyclonal; Santa Cruz Biotechnology), and were subsequently rinsed three times in PBS‐T. The blots were then probed with an enzyme‐linked secondary antibody (horseradish peroxidase) in 1 × PBS‐T (1 : 25 000–100 000) for 1 h at room temperature. Excess secondary antibody was rinsed off with three 5 min rinses in 100 mL PBS‐T. Finally, chemiluminescent detection reagents were used to reveal the results. Immunoblotting for GAPDH (goat, polyclonal; Santa Cruz Biotechnology) was carried out to verify equivalent protein loading. Densitometric analyses of Western blots were carried out using TINA 2.0 software (Raytest Isotopenmessgerate, Straubenhardt, Germany).
siRNA generation and transfection. Cyclin A1 and cyclin B1 siRNAs were prepared based on the manufacturer's instructions. Briefly, siRNA was obtained from the transcription and digestion of PCR‐derived templates carrying a T7 promoter (TAATACGACTCACTATAGGGAGA) using a Silencer siRNA Cocktail Kit (Ambion, Austin, TX). The following primers for were used: cyclin A1 forward primer, 5′‐TAATACGACTCACTATAGGGAGAT GAAGTAGACA CCGGCACAC‐3′ cyclin A1 reverse primer, 5′‐TAATACGACTCACTATAGGGAGATTCGAAGCCAAAAGCATGC‐3′ cyclin B1 forward primer, 5′‐TAATACGACTCACTATA GGGAGAGGCCAAAATGCC TATGAAGA‐3′; and cyclin B1 reverse primer, 5′‐TAATACGACTCACTATAGGGAGATGGCTCT CATGTTTCC AGTG‐3′. The amplification conditions were as follows: denaturation at 95°C for 15 min; 40 cycles of 95°C for 40 s, 53.2°C (for cyclin A1) and 61°C (cyclin B1) for 1 min, 72°C for 1 min; and a final 10 min extension at 72°C. Both sequences used for generating the siRNA were subjected to a BLAST (http://www.ncbi.nlm.nih.gov/BLAST/) search to ensure that they did not have significant sequence homology with other genes. Cells were transfected with siRNA using siPORT Amine (Ambion) according to the manufacturer's instructions.
siRNA effect on cell morphology: cell proliferation and viability analysis. Cells were seeded in a flat 96‐well plate and incubated at 37°C in 5% CO2. Cell proliferation was measured using a WelCount Cell Viability Assay Kit (WelGENE, Seoul, Korea) at 0, 12, 24, 48 and 72 h after transfection with cyclin A1 siRNA or negative control siRNA. Briefly, we defrosted XTT reagent and phenozine methosulfate at 37°C, prepared a reaction mixture (40 µL phenozine methosulfate and 2 mL XTT reagent per plate), added 20 µL of the reaction mixture to each well (each containing 100 µL media), and incubated at 37°C for 4 h. After observing a change in color of the solution, we measured these samples at a wavelength of 490 nm using a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA). All experiments were performed in triplicate.
SiRNA effect on cyclin A1 and cyclin B1‐related genes.
Western blot analysis All procedures were carried out as described above. The primary antibodies used are as follows: cyclin A1 (rabbit, polyclonal; Santa Cruz Biotechnology); cyclin A (rabbit, polyclonal; Santa Cruz Biotechnology); cyclin B1 (rabbit, polyclonal; Santa Cruz Biotechnology); Cdc2 (mouse, monoclonal; Santa Cruz Biotechnology); CDK2 (rabbit, polyclonal; Santa Cruz Biotechnology) and p‐CDK2 (Thr160, rabbit, polyclonal; Cell Signaling, Beverly, MA).
Cell cycle distribution observation All procedures were carried out as described above.
cDNA microarray The cDNA microarray was carried out using a 17K human cDNA microarray (GT‐CMRC, Seoul, Korea) with an amplified RNA in an indirect design. The test sample RNA and the lung cancer cells treated with siRNA for 12, 24, and 48 h, were labeled with Cy5 and individually cohybridized with the Cy3‐labeled the H596 without siRNA treatment. The microarray experiments were carried out in triplicate.
RNA extraction . All procedures were carried out as described above.
RNA amplification . Four micrograms of the total RNA was used in one round of amplification. The RNA template was mixed with 2 µg of the oligo‐dT/T7 primer (5′‐GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGG‐3′ Genotech, Daejun, Korea) and denatured at 65°C for 10 min. The first‐strand cDNA was synthesized by reverse transcription with 4 µL of the 5 × first‐strand buffer (Invitrogen), 2 µL of 100 mM dithiothreitol, 2 µL of 10 mM dNTP mix (Invitrogen), 2 µL of RNAsin (Promega, Madison, WI) and 2 µL of SuperScript II (Invitrogen) at 42°C for 1 h. For the second‐strand cDNA synthesis, 30 µL of a 5 × second‐strand buffer, 3 µL of 10 mM dNTP mix, 10 U of DNA ligase, 4 U of DNA polymerase I and 2 U of RNase H were added to the first‐strand cDNA product then incubated at 16°C for 2 h. The double‐stranded cDNA was extracted with an equal volume of phenol : chloroform : isoamyl alcohol (25 : 24 : 1), and precipitated with ethanol in the presence of 1 µL linear acrylamide (0.1 µg/µL, Ambion, Austin, TX, USA). The dried pellet was re‐suspended in 8 µL of RNase‐free water. The mRNA was transcribed from the double‐stranded cDNA using a T7 MEGAscript kit (Ambion). Briefly, 2 µL each of 75 mM NTP, 2 µL of an enzyme mix and 2 µL of a 10 × reaction buffer were added to 8 µL of the double‐stranded cDNA. The reaction mixture was then incubated at 37°C for 5 h. The amplified mRNA was cleaned using an RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. The quantity of the amplified RNA was measured by a NanoDrop (NanoDrop Technologies, Rockland, DE), and agarose gel electrophoresis was used to assess the integrity.
Probe labeling and hybridization . cDNA microarray experiment was carried out based on the protocol of Cancer Metastasis Reseach Center (CMRC), Yonsei University, Seoul, Korea.( 24 ) Two micrograms of the amplified mRNA was labeled with Cy3‐ or Cy5‐dUTP during reverse transcription. The RNA was mixed with 6 §¶ of the random primer (Invitrogen) and incubated at 65°C for 10 min. Eight microliters of the 5 × first strand buffer, 4 §¡ of 100 mM dithiothreitol, 2 §¡ of SuperScript II RT, 2 §¡ of 20 × low‐dT/dNTP mix, and 1 §¡ of RNasin (Promega) were added to the RNA/random primer mixture and incubated at 42°C for 2 h. The residual RNA was hydrolyzed by incubation at 65°C for 30 min in 15 §¡ of a 1.0 M NaOH solution. The reaction was neutralized with 15 §¡ of 1.0 M HCl. The Cy3‐ and Cy5‐labeled probes were purified using a QIAquick PCR Purification Kit (Qiagen). The purified probes were combined and mixed with 20 §¶ of Human Cot‐1 DNA (Invitrogen), 20 §¶ of yeast tRNA (Invitrogen), and 20 §¶ of poly(A) RNA (Sigma). The final probe was concentrated to 50 §¡ using a Microcon YM‐30 column (Millipore, Bedford, MA) then denatured at 100°C for 2 min. The cDNA microarrays were prehybridized in 3.5 × SSC, 0.1% SDS, and 10 §·/§¢ bovine serum albumin at 42°C for 1 h prior to probe application. The probe from the amplified mRNA was hybridized in 25% formamide, 5 × SSC and 0.1% SDS at 42°C for 16 h. Following hybridization, the arrays were washed in 2 × SSC with 0.1% SDS, 1 × SSC with 0.1% SDS, 0.2 × SSC, and 0.05 × SSC, sequentially washed for 2 min each, then spun dried at 500 g .
Image scanning and data processing . The fluorescence signals on the microarrays were acquired using a GenePix 4000B scanner (Axon Instruments, Foster City, CA). The scanned images were processed using GenePix Pro 4.0 software (Axon Instruments). Systemic errors were corrected by normalization of the log2‐transformed data using an intensity‐dependent, within‐print tip normalization based on the Lowess function.( 25 ) After normalization, the genes without any missing values were obtained for further analysis.
Data analysis The Pearson correlation coefficient among the triplicate microarrays using the cell lines was measured using S‐Plus 2000 software (Insightful, Seattle, WA). Hierarchical clustering analysis was carried out with cluster software and the resulting dendrogram was visualized using TreeView software (http://rana.lbl.gov/EisenSoftware.htm). To search the influence of siRNA on apoptosis, 301 apoptosis‐related genes out of our 17K cDNA microarray were selected. The genes with significant changes in expression after siRNA treatment were selected using significant microarray analysis. Then, an annotation of the selected genes was done using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://apps1.niaid.nih.gov/david) and Stanford Online Universal Resource for Clones and Expressed (SOURCE) sequence tags (http://genome‐www5.stanford.edu/egi‐bin/source/sourcesearch).
Validation using Western blot analysis Western blot analysis was done for validation of candidates screened from cDNA microarray. The primary antibodies that we used are as follows: Survivin (rabbit, polyclonal; Santa Cruz Biotechnology); E2F1 (rabbit, polyclonal; Santa Cruz Biotechnology); HDAC1 (goat, polyclonal; Santa Cruz Biotechnology); and HDAC3 (goat, polyclonal; Santa Cruz Biotechnology).
siRNA effect on cell apoptosis.
Annexin V–FITC staining . Annexin V is a Ca2+‐dependent phospholipid‐binding protein that has a high affinity for phosphatidylserine. In apoptotic cells, phosphatidylserine is translocated from the inner leaflet of the plasma membrane to the outer leaflet. PI, a standard flow cytometric viability probe, was used to distinguish viable from non‐viable cells. Cells that were stained were detected by a FACScan flow cytometer (Becton Dickinson). At 0, 12, 24, 48, and 72 h after the transfection of cyclin A1 siRNA, 1–105 cells were harvested, washed with ice‐cold PBS containing 100 mg/L CaCl2, resuspended in 100 µL of binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM CaCl2), and incubated with 5 µL of Annexin V conjugated with FITC in the absence of light for 10 min at room temperature. An additional 400 µL of binding buffer was added to the samples, and the samples were analyzed by flow cytometry. Cells that showed up as Annexin V–/PI+ were recognized as necrotic, those that showed up as Annexin V+/PI+ were taken to be late apoptotic or secondarily necrotic, and Annexin V+/PI– cells were recognized as apoptotic. All procedures stated above were carried out according to the manufacturer's instructions (Annexin V‐FITC Apoptosis Kit I; Becton Dickinson).
Apoptotic DNA ladder assays After transfecting cyclin A1 siRNA, cyclin B1 siRNA, or negative control siRNA for 48 h, we isolated and analyzed cellular DNA on a 1% agarose gel using an apoptotic DNA ladder kit (Roche Molecular Biochemicals, Mannheim, Germany). Adherent cells were pelleted and lyzed with lysis solution. After the lysis of cultured cells in binding buffer, the lysate was applied to a filter tube with glass fiber fleece and passaged through by centrifugation. Residual impurities were removed by a wash step, then DNA was eluted from the column with elution buffer according to the manufacturer's instructions. Samples were separated by electrophoresis on a 1% agarose/Tris–acetate–EDTA gel. The size of the DNA standards used to evaluate DNA fragmentation ranged from 100 to 2072 bp (Roche, Basel, Switzerland). Electrophoresis was carried out for 1 h at 100 V in TBE buffer (80 mmol/L Tris–borate (pH 8.0), 2 mmol/L EDTA containing 0.1 mg/mL ethidium bromide). After electrophoresis, visualization of the DNA band was done by staining with ethidium bromide and viewing with an ultraviolet transilluminator (Bio‐Rad, Hercules, CA). The gel was photographed under ultraviolet light with Polaroid film.
Caspase assay The activation of caspase‐3/7 was measured with Apo‐ONE assay (Promega) and all procedures were carried out according to the manufacturer's instructions. Briefly, the harvested cells were washed twice with PBS then incubated for 2–10 h in a buffer–substrate mix. The fluorescence of the cleaved rhodamine group was measured at 535 nm emission with a fluorescence reader.
Statistics. A t‐test was performed using SAS statistical software; a P value of < 0.05 was considered statistically significant.
Results
Lung cancer cell lines express both cyclin A1 mRNA and proteins. To determine the effectiveness of cyclin A1 as a target for siRNA in lung cancer, mRNA and protein expression of cyclin A1 were analyzed together with cell cycle synchronization. Both lung cancer cell lines, H157 and H596, showed high expression of cyclin A1 mRNA (Fig. 1a) and protein (Fig. 1b), particularly in the S and G2 phases following cell cycle synchronization (Fig. 1c). With serum starvation, approximately 90% of cells were arrested in the G0/G1 phase. We found that treatment with 2 mM thymidine for 16 h arrested approximately 54.5% and 52.9% of H157 and H596 cell lines with S phase DNA contents, respectively. We also found that treatment with 0.4 µg/mL nocodazole for 16 h arrested approximately 46.8% and 78.3% of H157 and H596 cell lines with G2 phase DNA contents, respectively (Fig. 1c).
Figure 1.
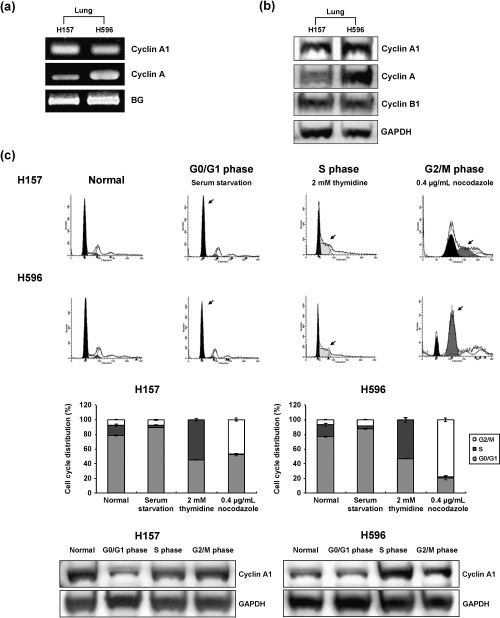
The mRNA and protein expression of cyclin A1 in lung cancer cell lines. (a) Quantitative RT‐PCR for cyclin A1 and cyclin A demonstrated higher mRNA expression for cyclin A1 in H157 lung cancer cell lines. BG, beta globulin. (b) Western blot analysis for cyclin A1, cyclin A and cyclin B1. Lung cancer cell lines H157 and H596 showed increased expression of cyclin A1, cyclin A and cyclin B. (c) Cyclin A1 expression in H157 and H596 cells after cell cycle synchronization. Both cell lines showed the highest expression of cyclin A1 in the S phase. Arrows indicate each fraction.
Downregulation of cyclin A1 reduced the cell growth in lung cancer cell lines. As cyclin A1 is one of the key components in the regulation of cell division, we attempted to analyze the antiproliferative impact of cyclin A1 RNAi. Treatment with cyclin A1 siRNA in lung cancer cell lines resulted in a decreased absorbance at 490 nm after 12 h, whereas no change of absorbance was noted in the NIH3T3 cell line ( Fig. 2a). H157 and H596 cells treated with cyclin A1 were floated markedly after 24–48 h and showed the alteration of cell morphology leading to cell apoptosis (Fig. 2b).
Figure 2.

The effect of cyclin A1 siRNA on the proliferation, viability and growth ability of H157 and NIH3T3 cells. (a) The results shown are the mean ± standard deviation of triplicate microcultures. Cyclin A1 siRNA significantly inhibited the proliferation and viability of H157 and H596 cells, in contrast to NIH3T3 cells. Note the siRNA effect of cyclin A1 on lung cancer cell lines. Cyclin A1 targeted for siRNA is specific to the lung cancer cell lines. (b) Both lung cancer cell lines showed alterations in cell morphology, which was apparent after 48 h. Some anchoring cells are remarkably shrunken with irregular contours, indicating they are undergoing cellular apoptosis (inset: magnification, × 400). Note markedly irregular cell membranes especially after 24–48 h (insets with red lines).
Cyclin A1 siRNA effectively reduced the expression of cyclin A1 and induced S phase arrest. Treatment with each siRNA in H157 and H596 cells showed a marked decrease in the protein levels of cyclin A1 and B1 after 12–24 h, however, the effect of siRNA on cyclin A1 and B1 levels lessened after 48–72 h (Fig. 3). CDK2 levels in both lung cancer cell lines were reduced after siRNA treatment, however, the patterns of the knockdown effects in H157 and H596 cell lines differed. In H157, the knockdown effect of cyclin A1 siRNA on CDK2 showed a similar pattern to that on cyclin A1, and downregulation of active phosphorylated CDK2 was seen 24–48 h after treatment. In H596, siRNA treatment resulted in a marked reduction of CDK2 levels between 12–48 h. p‐CDK2 (Thr160) was markedly decreased after 24–48 h. Cyclin A was unaffected. Cyclin B1/Cdc2 was downregulated in parallel with cyclin A1 in H157. Cdc2 in H596 was markedly downregulated 24–48 h after cyclin A1 siRNA treatment (Fig. 3).
Figure 3.
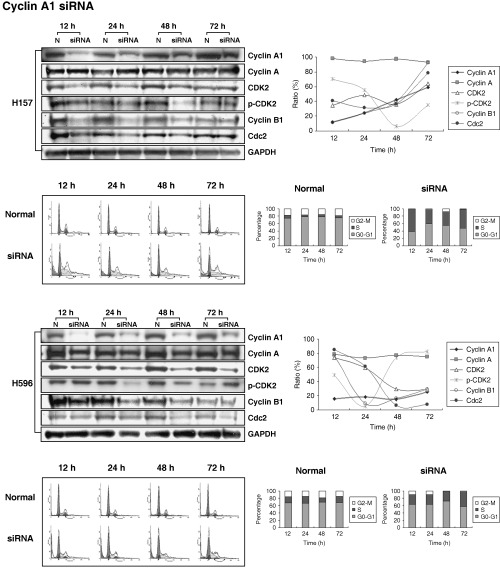
Reduction rate of target genes in lung cancer cell lines with cyclin A1 RNAi. All cell lines demonstrated a marked decrease in cyclin A1 protein levels after 12–24 h, however, the effect of siRNA on cyclin A1 levels lessened after 48–72 h. The figure represents relative protein levels using quantification of Western blot analysis, standardized to GAPDH. Each value was obtained from the ratio of the corresponding normal value (P < 0.05). H157: lanes 1, 3, 5 and 7 represent samples at 12, 24, 48 and 72 h after negative control siRNA treatment, respectively; lanes 2, 4, 6 and 8 represent samples at 12, 24, 48 and 72 h after cyclin A1 siRNA treatment. H596: lanes 1, 3, 5 and 7 represent samples at 12, 24, 48 and 72 h after negative control siRNA treatment, respectively; lanes 2, 4, 6 and 8 represent samples at 12, 24, 48 and 72 h after cyclin A1 siRNA treatment. DNA histogram demonstrates the influence of cyclin A1 and cyclin B1 siRNA on cell cycle fractions. This figure compares the S and G2/M phase fraction of cyclin A1 RNAi treated group and the negative control group. Notice the decreased S phase fraction in both lung cancer cell lines. H157: the S phase percentages were 61.27%, 35.97%, 35.46% and 51.91% at 12, 24, 48 and 72 h after siRNA for cyclin A1, respectively; H596: 24.51%, 25.59%, 29.95% and 39.52% at 12, 24, 48 and 72 h after cyclin A1 siRNA treatment, respectively.
Flow cytometry was carried out to observe the influence of cyclin A1 siRNA on the cell cycle. An increase in S phase fraction and a marked decrease in G2/M fraction were seen in both H157 and H596 cell lines in comparison to the negative controls after treatment with cyclin A1 RNAi (Fig. 3).
Treatment with siRNA triggered apoptosis in lung cancer cell lines. Cell death was observed after treatment with siRNA, mainly through apoptosis and a minor proportion of necrosis. Lung cancer cell lines H157 and H596 underwent apoptosis 48 h after siRNA for cyclin A1. Treatment of siRNA for cyclin A1 in H157 induced less apoptosis than in H596. The fractional rates of early (LR quadrant) and late apoptosis/necrosis (UR quadrant) in H157 were 4.32% and 8.45%, respectively. In H596 cells, the rates of early (LR quadrant) and late apoptosis/necrosis (UR quadrant) were 8.32% and 16.45%, respectively (Fig. 4a). A typical apoptosis DNA ladder was obtained for the cell lines treated with cyclin A1 siRNA in lung cancer cell lines, but not for the negative control group (Fig. 4b). The DNA ladder in H157 was less remarkable than in H596. On caspase assay, the activity of the caspase‐3 and ‐7 in each cell line at 12, 24, 48 and 72 h after siRNA treatment was intensively increased in lung cancer cell lines (Fig. 4c).
Figure 4.
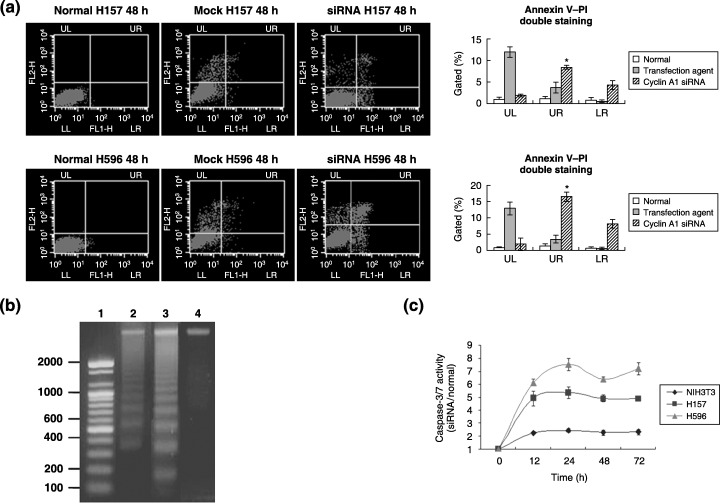
The influence of cyclin A1 siRNA on apoptosis. (a) Annexin V–FITC staining pattern. This diagram shows that siRNA‐ treated H157 cells underwent cell death through apoptosis (*P < 0.05). Cells that showed up as annexin V+/PI– cells were recognized as early apoptotic and annexin V+/PI+ were taken to be late apoptotic or secondarily necrotic (*P < 0.05). LL, lower left (quadrant); LR, lower right; UL, upper left; UR, upper right. (b) Apoptotic DNA ladder. H157 and H596 cells treated with cyclin A1 siRNA showed the typical DNA ladder pattern, which represents cells undergoing apoptosis. Lane 1, size marker (bp); lane 2, DNA from H157 cells treated with cyclin A1 siRNA; lane 3, DNA from H596 cells treated with cyclin A1 siRNA; lane 4, negative control. (c) Caspase assay. The activity of caspase‐3 and ‐7 in each cell line at 12, 24, 48 and 72 h after siRNA treatment was intensively increased in lung cancer cell lines.
Differential expression profiles of cell cycle‐ or apoptosis‐related genes were matched with siRNA effect in lung cancer cell lines. Compared with the cDNA array, several genes related to apoptosis and the cell cycle were strongly associated with the siRNA cyclin A1 effect in lung cancer cell lines. Functional annotation of genes of more than twofold at 48 h after siRNA cyclin A1 in lung cancer was 155 genes on clustering of gene expression profiles (Fig. 5a). The genes with the most apparent decrease in expression at 48 h after siRNA were apoptosis inhibitor 5 of the intrinsic apoptosis pathway, baculoviral IAP repeat‐containing 5 (survivin) related to the common apoptosis pathway, v‐myb avian myeloblastosis viral oncogene homolog‐like 2 in the category of oncogene, protein phosphatase 2, and regulatory subunit (PR65) alpha‐isoform in terms of the modulator. The most upregulated gene was TNF member 10 with regards to the extrinsic pathway (Table 1). On validation for candidates using Western blot analysis, we found that siRNA‐treated lung cancer cell lines showed lower expression of E2F1, HDAC1 and HDAC3 than in mock cell lines. Survivin expression in H157 and H596 was diminished 12 h after siRNA treatment, and remained low for 72 h.
Figure 5.
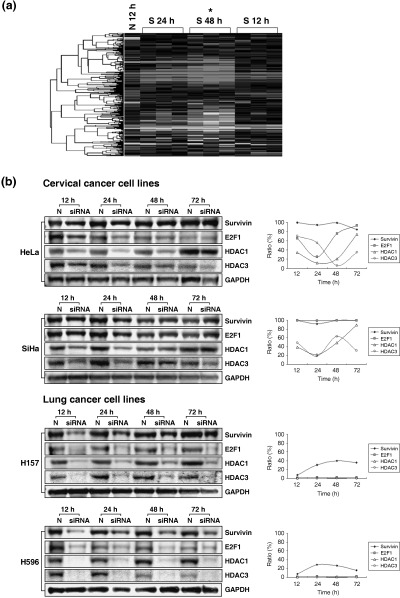
The influence of cyclin A1 siRNA on gene expression profiles in lung cancer. (a) cDNA microarray analysis. Functional annotation of 155 genes that were changed more than twofold 48 h after cyclin A1 siRNA out of the 17K cDNA microarray containing 301 apoptosis‐related genes. A hierarchical cluster analysis was carried out by complete linkage clustering using EisenSoftware. Downregulation (red) of tested genes in comparison with the normal control (N 12 h) is apparent. Note: The signified values mean log2R/G ratio (R: Cy‐5, red intensity; G: Cy‐3, green intensity). *statistically significant in comparison with the control. (b) Validation analysis. siRNA‐treated lung cancer cell lines showed lower expression of E2F1, HDAC1 and HDAC3 than in mock cell lines. Survivin expression in H157 and H596 cells was diminished 12 h after siRNA treatment, and kept in low expression for 72 h.
Table 1.
List of genes with specific regards to the apoptosis pathway influenced by cyclin A1 siRNA
| Genes | GenBank accession no. | Ratio (12 h) | Ratio (24 h) | Ratio (48 h) |
|---|---|---|---|---|
| Upregulated genes | ||||
| Apoptosis: The Extrinsic Pathway | ||||
| TNF receptor superfamily, member 5 | AA886208 | 14 | 7.67 | 5 |
| TNF (ligand) superfamily, member 10 | H54629 | −4.44 | 121.41 | 169.71 |
| TNF receptor‐associated factor 1 | R71691 | −3.08 | 36.13 | 46.25 |
| Downregulated genes | ||||
| Apoptosis: The Intrinsic Pathway | ||||
| BCL2‐antagonist of cell death | AA460291 | −0.73 | −1.16 | −2.00 |
| BCL2‐associated athanogene 5 | AI765232 | −0.74 | −0.86 | −1.63 |
| Apoptosis inhibitor 5 | AI972925 | 4.83 | −14.50 | −16.67 |
| Death‐associated transcription factor 1 | AA004823 | 1.19 | −0.48 | −3.86 |
| Defender against cell death 1 | AA455281 | −0.27 | −3.13 | −4.33 |
| E2F transcription factor 1* | AA424950 | −0.25 | −0.89 | −2.58 |
| Catenin (cadherin‐associated protein), alpha‐like 1 | AA621315 | 0.20 | −0.37 | −0.53 |
| HDAC1 * | AA465353 | 0.58 | −0.96 | −3.42 |
| HDAC3 * | H79779 | 2.00 | 1.44 | −0.44 |
| Apoptosis: The Common Pathway | ||||
| Baculoviral IAP repeat‐containing 5 (survivin)* | AA460685 | 1.00 | −4.13 | −12.07 |
| Baculoviral IAP repeat‐containing 2 | AA702174 | −0.09 | −0.78 | −0.79 |
| Oncogenes v‐myb avian myeloblastosis viral oncogene homolog‐like 2 | AA457034 | −2.78 | −5.56 | −11.06 |
| V‐rel avian reticuloendotheliosis viral oncogene homolog A | AA443547 | 0.67 | 0.03 | −2.41 |
| Cullin 2 | AA454094 | −0.43 | −0.73 | −4.73 |
| Forkhead box O1A (rhabdomyosarcoma) | AA448277 | 0.57 | −1.10 | −2.16 |
| Modulators | ||||
| Dual specificity phosphatase 6 | AA455254 | −2.57 | −3.29 | −5.81 |
| Protein phosphatase 2 (formerly 2 A), catalytic subunit, alpha isoform | AA599092 | 2.10 | −1.29 | −3.57 |
| Protein phosphatase 2 (formerly 2 A), regulatory subunit A (PR 65), alpha isoform | AA427433 | −0.33 | −8.17 | −25.67 |
| Protein phosphatase 2 (formerly 2 A), regulatory subunit A (PR 65), beta isoform | H57850 | 0.93 | −4.47 | −7.47 |
| Protein phosphatase 2 (formerly 2 A), regulatory subunit A (PR 65), beta isoform | H99771 | 1.15 | 0.05 | −1.52 |
| Protein phosphatase 2 (formerly 2 A), catalytic subunit, beta isoform | AA490473 | 0.20 | −0.80 | −0.82 |
Log2R/G ratio (R: Cy‐5, red intensity; G: Cy‐3, green intensity; P < 0.05).
Discussion
The selective silencing of endogenous genes, especially oncogenes, by RNAi is particularly important in functional genomics and cancer therapy.( 6 , 7 , 8 , 9 , 10 , 11 ) Some of the greatest advantages of RNAi are that synthetic siRNA are relatively easily manufactured and that they are designed to meet mutation changes within target sequences. Moreover, RNAi is very efficient; a single dose of siRNA can sustain RNAi for long enough to allow recovery of the cellular regulatory system. However, there are limitations to gene silencing, such as the transient nature of the response and the technical difficulties in transduction procedures. As siRNAs seem to be relatively resistant to degradation, the transient nature of the knockdown response is determined by the rate of cell growth and by the dilution of the siRNAs below a crucial threshold level necessary to inhibit gene expression.( 11 )
Cyclins deserve to be powerful candidates for human cancer therapy, as almost all human tumors involve cell cycle aberrations.( 26 , 27 ) Cyclin and CDK complexes play important roles in cell cycle control.( 28 ) Cyclin A or A1 was originally indistinguishable from true mitotic B‐type cyclins in its ability to induce mitosis in various assays, and was thought to be one of the cyclins required for initiation of mitosis.( 13 , 14 , 15 , 16 , 17 ) However, several differences between cyclin A and cyclin B1 have recently been documented. Cyclin A1 expression precedes that of cyclin B1, cyclin A1 is localized in the nucleus, and cyclin A1 binds to Cdc2 and the Cdc2‐related kinase, p33, a gene product of CDK2.( 21 , 22 , 23 , 26 , 27 , 28 ) The physiologic function of cyclin A1 remains obscure. Moreover, microinjection of an antisense cyclin A1‐encoding plasmid leads to inhibition of DNA synthesis, suggesting that cyclin A might be important in the G1–S transition. The resultant CDK2–cyclin A–p107–E2F complex begins to appear around the G1–S transition and disappears after S phase, suggesting its role in S phase progression.( 21 , 22 ) Cyclin B1 can bind to and activate CDK2 in addition to Cdc2.( 19 , 20 ) The phase of cyclin B1 accumulation overlaps with very late DNA replication,( 29 ) whereas inhibition of CDK2 in the S phase triggers the destruction of cyclin B1.( 30 ) Thus, it is logical to infer that cyclin B1 has the ability to orchestrate with CDK2 as well as with cyclin A1, the partner of CDK2. The downregulation of cyclin B1 following cyclin A1 siRNA treatment in the present study can be explained by the cobinding of CDK2 with cyclin B1 and cyclin A1.
The S phase fraction gradually increased in the cyclin A1 RNAi group in comparison to the negative control group. This was probably associated with the repression of cyclin A1 and CDK2 after cyclin A1 RNAi treatment. Cyclin A1 mRNA levels were low before the S phase but increased significantly during the S and G2/M phases.( 17 ) Cyclin A1 might possess S phase promoting potential, and might even activate CDK2 kinase, contributing to S phase progression, as with cyclin B1.( 19 , 20 ) In lung cancer cell lines, it was found that cyclin A1 protein levels were low before the S phase but increased significantly during the S and G2/M phases.
The present study shows that cyclin A1 RNAi inhibited tumor cell proliferation and impaired tumor cell viability, whereas cyclin B1 RNAi had no effect on lung cancer cell lines. Furthermore, cyclin B1 RNAi inhibited proliferation and impaired viability of HeLa cell lines, in contrast to cyclin A1 RNAi which showed no effect, emphasizing that RNAi has cell‐line specificity according to the target molecule. Moreover, the results were independent of the functional status and mutational aberration of p53.
The central role of p53 in the regulation of cell apoptosis by caspase‐3, caspase‐9, Bcl‐2, and Bax is well established.( 31 , 32 , 33 ) However, apoptosis, with specific regards to Bcl‐2 or caspase, is also triggered in a p53‐independent manner, through the Par‐4 or p21 WAF1/CIP1 pathway.( 34 , 35 , 36 ) There is no convincing proof that cyclin A1 overexpression is usually caused by the inactivation of p53, although it is known that the C‐terminal regulatory domain of p53 contains a functional docking site for cyclin A.( 29 ) Moreover, cyclin A1 binds to the p21 family and pRb family, p107. The depletion of cyclin A1, which can lead to the downregulation of CDK2, is especially applicable for tumors with functional p53 inactivation, as the inhibition of CDK activity can induce apoptotic cell death in a p53‐independent manner.( 37 ) We also found that cyclin A1 siRNA in lung cancer cell lines induced apoptosis in a caspase‐3/7‐dependent manner.
On analysis of cDNA array mainly regarding apoptosis, the most significant changes included an upregulation of TNF member 10 of the extrinsic pathway and downregulation of apoptosis inhibitor 5 of the intrinsic apoptotic pathway, baculoviral IAP repeat‐containing 5 (survivin) of the common apoptosis pathway, v‐myb avian myeloblastosis viral oncogene homolog‐like 2 of the oncogenic category, protein phosphatase 2, and regulatory subunit (PR65) alpha‐isoform in terms of the modulator. There are many known pathways and proteins controlling the apoptosis machinery, including the intrinsic mitochondrial pathways, extrinsic cytoplasmic pathways, final common pathways, various modulators and oncogenes,( 11 ) and from our results, we suggest that cyclin A1 siRNA might induce apoptosis in lung cancer by affecting all three apoptotic pathways, as depicted in Fig. 6. Apoptosis might have been induced by an upregulation of the extrinsic pathway by way of the Fas death receptor, a member of the TNF receptor family,( 38 ) downregulation of the intrinsic pathway and decreased survivin, a baculovirus inhibitor of apoptosis repeat protein domain of the final common pathway.( 39 ) Inhibition of Cdc2 activity through the downregulation of cyclin A1 has been reported to trigger apoptosis through subsequent survivin downregulation,( 40 ) which is compatible with the present result.
Figure 6.
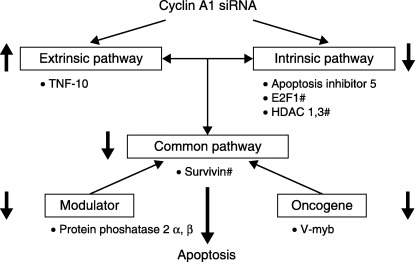
A suggested scheme for the influence of cyclin A1 siRNA on specific apoptotic pathways in lung cancer. siRNA for cyclin A1 in lung cancer leads to apoptosis through the upregulation of a member of the TNF receptor family in the extrinsic pathway, downregulation of the intrinsic pathway, decreased survivin in the final common pathway and v‐myb avian myeloblastosis viral oncogene homolog‐like 2 in the oncogene group, protein phosphatase 2 in the modulator category. # Log2R/G ratio, P < 0.05.
The use of RNAi in therapeutic practice is currently being investigated and it is mandatory to search for the most specific target molecule.
In conclusion, this study shows that loss of cyclin A1 resulted in the downregulation of CDK2, S phase delay, cell apoptosis, reduced cell viability, and p53‐independent proliferation in lung cancer cell lines. Therefore cyclin A1 could be a promising anticancer target molecule in lung cancers.
Acknowledgments
This study was supported by the CMB‐YUHAN research fund (06‐2004‐0005; NHC) and the Brain Korea 21 Project for Medical Science, Yonsei University College of Medicine (NHC), Seoul, Korea.
References
- 1. Petty RD, Nicolson MC, Kerr KM, Collie‐Duguid E, Murray GI. Gene expression profiling in non‐small cell lung cancer: from molecular mechanisms to clinical application. Clin Cancer Res 2004; 10: 3237–48. [DOI] [PubMed] [Google Scholar]
- 2. Ferreira CG, Huisman C, Giaccone G. Novel approaches to the treatment of non‐small cell lung cancer. Crit Rev Oncol Hematol 2002; 41: 57–77. [DOI] [PubMed] [Google Scholar]
- 3. Bunn PA Jr, Shepherd FA, Sandler A et al. Ongoing and future trials of biologic therapies in lung cancer. Lung Cancer 2003; 41 (Suppl. 1): S175–86. [DOI] [PubMed] [Google Scholar]
- 4. Nemunaitis J, Swisher SG, Timmons T et al. Adenovirus‐mediated p53 gene transfer in sequence with cisplatin to tumors of patients with non‐small cell lung cancer. J Clin Oncol 2000; 18: 609–22. [DOI] [PubMed] [Google Scholar]
- 5. Wadler S. Perspectives for cancer therapies with cdk2 inhibitors. Drug Resist Updat 2001; 4: 347–67. [DOI] [PubMed] [Google Scholar]
- 6. Tuschl T. RNA interference and small interfering RNAs. Chembiochem 2001; 2: 239–45. [DOI] [PubMed] [Google Scholar]
- 7. Castanotto D, Li H, Rossi JJ. Functional siRNA expression from transfected PCR products. RNA 2002; 8: 1454–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Novina CD, Sharp PA. The RNAi revolution. Nature 2004; 430: 161–4. [DOI] [PubMed] [Google Scholar]
- 9. Silva J, Chang K, Hannon G, Rivas F. RNA‐interference‐based functional genomics in mammalian cells: reverse genetics coming of age. Oncogene 2004; 23: 8401–9. [DOI] [PubMed] [Google Scholar]
- 10. Semizarov D, Kroeger P, Fesik S. siRNA‐mediated gene silencing: a global genome view. Nucleic Acids Res 2004; 32: 3836–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ichim TE, Li M, Qian H et al. RNA interference: a potent tool for gene specific therapeutics. Am J Transplantation 2004; 4: 1227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin 2005; 55: 178–94. [DOI] [PubMed] [Google Scholar]
- 13. Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J 1991; 11: 961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang R, Morosetti R, Koeffler HP. Characterization of a second human cyclin A that is highly expressed in testis and in several leukemic cell lines. Cancer Res 1997; 57: 913–20. [PubMed] [Google Scholar]
- 15. Sweeney C, Murphy M, Kubelka M et al. A distinct cyclin A is expressed in germ cells in the mouse. Development 1996; 122: 53–64. [DOI] [PubMed] [Google Scholar]
- 16. Howe JA, Howell M, Hunt T, Newport JW. Identification of a developmental timer regulating the stability of embryonic cyclin A and a new somatic A‐type cyclin at gastrulation. Genes Dev 1995; 9: 1164–76. [DOI] [PubMed] [Google Scholar]
- 17. Xu M, Sheppard KA, Peng CY, Yee AS, Piwnica‐Worms H. Cyclin A/CDK2 binds directly to E2F‐1 and inhibits the DNA‐binding activity of E2F‐1/DP‐1 by phosphorylation. Mol Cell Biol 1994; 14: 8420–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Porter LA, Singh G, Lee JM. Abundance of cyclin B1 regulates gamma‐radiation‐induced apoptosis. Blood 2000; 95: 2645–50. [PubMed] [Google Scholar]
- 19. Moore JD, Kirk JA, Hunt T. Unmasking the S‐phase‐promoting potential of cyclin B1. Science 2003; 300: 987–90. [DOI] [PubMed] [Google Scholar]
- 20. Pan ZQ, Amin A, Hurwitze J. Characterization of the in vitro reconstituted cyclin A or B1‐dependent CDK2 and Cdc2 kinase activities. J Biol Chem 1993; 268: 20443–51. [PubMed] [Google Scholar]
- 21. Sarafan‐Vasseur N, Lamy A, Bourguignon J et al. Overexpression of B‐type cyclins alters chromosomal segregation. Oncogene 2002; 2: 2051–7. [DOI] [PubMed] [Google Scholar]
- 22. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004; 23: 2825–37. [DOI] [PubMed] [Google Scholar]
- 23. Yin XY, Grove L, Datta NS, Katula K, Long MW, Prochownik EV. Inverse regulation of cyclin B1 by c‐Myc and p53 and induction of tetraploidy by cyclin B1 overexpression. Cancer Res 2001; 61: 6487–93. [PubMed] [Google Scholar]
- 24. Kim TM, Jeong HJ, Seo MY et al. Determination of genes related to gastrointestinal tract origin cancer cells using a cDNA microarray. Clin Cancer Res 2005; 11: 79–86. [PubMed] [Google Scholar]
- 25. Yang YH, Dudoit S, Luu P et al. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 2002: 30: e15. [DOI] [PMC free article] [PubMed]
- 26. Motokura T, Arnold A. Cyclins and oncogenesis. Biochim Biophys Acta 1993; 1155: 63–78. [DOI] [PubMed] [Google Scholar]
- 27. Ivanchuk SA, Rutka JT. The cell cycle: accelerators, brakes and checkpoints. Neurosurgery 2004; 54: 692–700. [DOI] [PubMed] [Google Scholar]
- 28. Doree M, Galas S. The cyclin‐dependent protein kinases and the control of cell division. FASEB J 1994; 8: 1114–21. [DOI] [PubMed] [Google Scholar]
- 29. Luciani MG, Hutchins JR, Zheleva D, Hupp TR. The C‐terminal regulatory domain of p53 contains a functional docking site for cyclin A. J Mol Biol 2000; 300: 503–18. [DOI] [PubMed] [Google Scholar]
- 30. Lukas C, Sorensen CS, Kramer E et al. Accumulation of cyclin B1 requires E2F and cyclin‐A‐dependent rearrangement of the anaphase‐promoting complex. Nature 1999; 401: 815–8. [DOI] [PubMed] [Google Scholar]
- 31. Fan G, Ma X, Wong PY, Rodrigues CM, Steer CJ. p53 dephosphorylation and p21 (Cip1/Waf1) translocation correlate with caspase‐3 activation in TGF‐β1‐induced apoptosis of HuH‐7 cells. Apoptosis 2004; 9: 211–21. [DOI] [PubMed] [Google Scholar]
- 32. Wang Y, Prives C. Increased and altered DNA binding of human p53 by S and G2/M but not G1 cyclin‐dependent kinases. Nature 1995; 376: 88–91. [DOI] [PubMed] [Google Scholar]
- 33. Zhen W, Lijun W, Linhao L, Shinichi T, Satoshi O, Takashi I. P53‐mediated cell cycle arrest and apoptosis induced by shikonin via a caspase‐9‐dependent mechanism in human malignant melanoma A375–S2 cells. J Pharmacol Sci 2004; 94: 166–76. [DOI] [PubMed] [Google Scholar]
- 34. Rangnekar VM. Apoptosis mediated by a novel leucine zipper protein Par‐4. Apoptosis 1998; 3: 61–6. [DOI] [PubMed] [Google Scholar]
- 35. Park SE, Choi HJ, Yee SB et al. Synthetic bile acid derivatives inhibit cell proliferation and induce apoptosis in HT‐29 human colon cancer cells. Int J Oncol 2004; 25: 231–6. [PubMed] [Google Scholar]
- 36. Shao RG, Shimizu T, Pommier Y. Brefeldin A is a potent inducer of apoptosis in human cancer cells independently of p53. Exp Cell Res 1996; 227: 190–6. [DOI] [PubMed] [Google Scholar]
- 37. Shibata Y, Nishimura S, Okuyama A, Nakamura T. p53‐independent induction of apoptosis by cyclin‐dependent kinase inhibition. Cell Growth Differ 1996; 7: 887–91. [PubMed] [Google Scholar]
- 38. Zapata JM, Pawlowski K, Haas E, Ware CF, Godzik A, Reed JC. A diverse family of proteins containing tumor necrosis factor receptor associated factor domains. J Biol Chem 2001; 276: 24242–52. [DOI] [PubMed] [Google Scholar]
- 39. Li F. Role of survivin and its splice variants in tumorigenesis. Br J Cancer 2005; 92: 212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim EH, Kim SU, Choi KS. Rottlerin sensitizes glioma cells to TRAIL‐induced apoptosis by inhibition of Cdc2 and the subsequent downregulation of survivin and XIAP. Oncogene 2005; 24: 838–49. [DOI] [PubMed] [Google Scholar]