

Abstract
Cancer recurrence after surgical treatment is a major concern for patients and doctors. Understanding what makes tumors prone to recurrence would be an important step toward its prevention. Accumulating evidence indicates that the level of membrane‐associated protease regulator reversion‐inducing cysteine‐rich protein with Kazal motifs (RECK) expressed in tumor tissue is a good prognostic indicator in several common cancers. Certain members of the matrix metalloproteinase family are often upregulated in advanced cancers and are known to play important roles in tumor angiogenesis, invasion and metastasis. RECK negatively regulates several matrix metalloproteinases. Therefore, RECK itself may well be considered a promising tool or target molecule to be activated in cancer therapy. Here we review the recent advances in RECK research and discuss some of the important issues to be addressed in future studies. (Cancer Sci 2007; 98: 1659–1665)
What is reversion‐inducing cysteine‐rich protein with Kazal motifs? Early findings
Reversion‐inducing cysteine‐rich protein with Kazal motifs (RECK) was first identified as a cDNA clone inducing morphological reversion (‘flat reversion’) in NIH3T3 cells transformed by the v‐K‐ras oncogene.( 1 ) RECK encodes a glycosylphosphatidylinositol (GPI)‐anchored glycoprotein of approximately 110 kDa, containing multiple serine protease inhibitor‐like motifs. Despite this structural feature, RECK negatively regulates multiple matrix metalloproteinase (MMP) family members (e.g. MMP‐2, MM‐9 and MT1‐MMP) (Fig. 1). RECK is downregulated upon cell transformation by a variety of oncogenes.( 1 , 2 , 3 , 4 ) RECK mRNA is expressed ubiquitously in normal human tissues, but it is undetectable in a number of tumor‐derived cell lines. These cell lines showed reduced tumor angiogenesis, invasion and metastasis when stably transfected with a RECK expression vector.( 5 ) RECK‐deficient mice die at approximately embryonic day 10.5 (E10.5) with reduced extracellular matrix (ECM) integrity (e.g. reduced type I collagen, disrupted basement membranes, cellular disarray, increased tissue fragility) and halted vascular development; this phenotype is partially rescued in the Mmp‐2‐null background,( 6 ) demonstrating that RECK is essential for development and that MMP‐2 is a critical target of RECK in vivo. In contrast, mice lacking the expression of individual MMP family members tend to show little or no obvious developmental abnormalities during embryogenesis, probably due to functional redundancy.( 7 ) Mice lacking RECK, the negative regulator of multiple MMP, may therefore provide an important opportunity for finding clues to understanding the roles of MMP, ECM and ECM remodeling during mammalian development. The implications of these early studies have been discussed in several previous reviews.( 8 , 9 , 10 , 11 , 12 ) Here we focus on more recent work on RECK.
Figure 1.
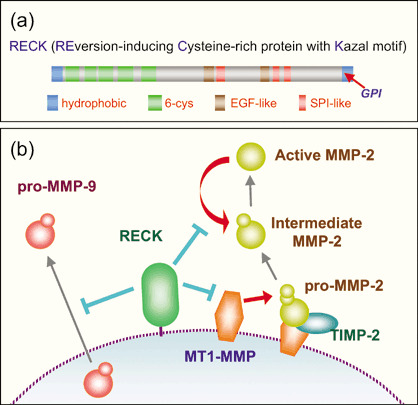
Primary structure and proposed actions of reversion‐inducing cysteine‐rich protein with Kazal motifs (RECK). (a) RECK contains several motifs found commonly in extracellular proteins, such as an N‐terminal signal peptide, C‐terminal glycosylphosphatidylinositol (GPI)‐anchoring signal, 6‐cysteine repeats and epidermal growth factor (EGF)‐like repeats. In addition, RECK contains three Kazal‐type serine protease inhibitor (SPI)‐like motifs. (b) Biochemical studies indicate that RECK regulates matrix metalloproteinases (MMP). Recent evidence suggests that RECK regulates some membrane‐bound proteases, such as MT1‐MMP and CD13, by recruiting them to a specific endocytic pathway (GEEC) and accelerating their degradation.
The more RECK, the better the prognosis
Hepatoma and pancreatic cancer. In their pioneering study, Furumoto et al. compared the levels of RECK mRNA in resected hepatocellular carcinomas and surrounding non‐tumorous tissues (n = 64) by RNA blot hybridization. They noticed that tumors expressing RECK at higher levels showed better prognosis (P = 0.02).( 13 ) They also detected a weak correlation between RECK expression and MMP‐9 immunoreactivity. Subsequently, Masui et al. detected a positive correlation between RECK immunoreactivity in pancreatic carcinomas and survival of the patients (n = 50; P = 0.0463).( 14 ) Consistent with the earlier findings in experimental systems using cultured cells and nude mice,( 13 ) inverse correlations between RECK expression and the extent of MMP‐2 activation as well as the invasive properties of the tumors were also noted.( 14 )
Breast cancer. Span et al. determined the level of RECK mRNA in mammary tumor tissues and surrounding normal tissues (n = 10) by quantitative reverse transcription–polymerase chain reaction (qRT‐PCR), and found a significant reduction of RECK mRNA in tumors (tumor : normal = 1.7 : 3.9; P = 0.028). Their larger study focusing on tumor specimens (n = 278) indicated a significant correlation between the level of RECK expression and survival of the patients (P = 0.037). They also found that the level of RECK expression differed depending on whether samples were taken from patients before menopause or after menopause (7.2 : 5.6; P = 0.045). No correlation was detected between the level of RECK expression and age.( 15 ) These findings are interesting in terms of how RECK is regulated physiologically.
Lung cancer. Takenaka et al. analyzed RECK expression in resected non‐small cell lung cancers (NSCLC) of various cell types and various stages (n = 171) by immunohistochemistry. They detected a significant correlation (P = 0.016) between high RECK expression in tumors and survival of the patients, especially in the advanced‐stage adenocarcinoma cases. They also found an inverse correlation between RECK expression and intratumoral microvessel density, particularly in tumors positive for vascular endothelial growth factor (VEGF).( 5 ) In a subsequent study focusing on stage IIIA N2 cases with lymph node metastasis (n = 118), they found that a significant correlation between RECK expression and survival was seen only in patients with single N2 node metastasis and not with multiple N2 node metastases. Interestingly, RECK expression was significantly higher in metastatic lesions than in the original tumors.( 16 )
Colorectal cancer. Takeuchi et al. studied another common cancer, colorectal carcinoma (n = 53), and found downregulation of RECK mRNA in tumors. They also detected correlations between higher RECK immunoreactivity in tumors and several other properties including higher differentiation, lower lymph node metastasis, lower Duke's stage, and higher 5‐year survival (P = 0.011). A weak correlation between lower MMP‐9 expression and better prognosis could be detected among these patients (P = 0.053). Importantly, a good correlation was detected when both parameters were considered in combination in the data analysis: patients with tumors expressing low levels of MMP‐9 and high levels of RECK showed a remarkably higher 5‐year survival rate than the high‐MMP‐9/low‐RECK group (89 vs 41%; P = 0.0003). Thus, in this tumor type, RECK and MMP‐9 seem to be regulated independently. In addition, they found correlations between high RECK expression and low VEGF expression as well as low vascular density.( 17 ) van der Jagt et al. also studied RECK expression in resected colon cancers (n = 63) by qRT‐PCR and confirmed: (1) downregulation of RECK in tumors; (2) a correlation between the level of RECK mRNA and survival of the patients; and (3) an inverse correlation between RECK expression and MMP‐2 activation. In addition, they found differential expression of RECK among tumors originating from different areas of the colon (high in sigmoid and rectal, low in descending and ascending colon) and suggested that the ratio of RECK expression between tumors and their surrounding normal tissues, rather than the absolute level of RECK, may serve as an accurate prognostic indicator (i.e. prognosis was better when RECK was high in tumors and low in normal tissues; prognosis was poor when RECK was low in tumors and high in normal tissues).( 18 )
Prostate cancer. Ohl et al. compared RECK mRNA expression between prostate carcinomas, benign prostatic hyperplasia from adenomectomy specimens and normal adjacent tissue of prostate carcinomas by qRT‐PCR. Interestingly, RECK expression was increased in hyperplasia and decreased in carcinoma; the extent of RECK downregulation in carcinoma was correlated with recurrence.( 19 , 20 ) Riddick et al. systematically analyzed the expression profiles of matrix proteases and their inhibitors in prostate tumors, both benign (n = 26) and malignant (n = 44), by qRT‐PCR and detected a correlation between downregulation of RECK and malignancy.( 21 )
Other cancers. A correlation between RECK downregulation and poor prognosis has also been found in other tumor types including hilar cholangiocarcinomas,( 22 ) gastric cancer,( 23 ) ameloblastic tumors( 24 ) and osteosarcomas.( 25 ) However, Nabeshima et al. examined the expression of six MMP, three inhibitors and emmprin, an MMP inducer, in tumors of the peripheral nervous system (14 schwannomas, 14 neurofibromas and 12 malignant peripheral nerve sheath tumors) by immunostaining and found no alteration of RECK expression in these tumors.( 26 )
RECK expression in tumors has also been studied in animal models. Okamura et al. compared gene expression profiles between cancinogen‐induced tumors and non‐treated tissues from mice carrying the wild‐type HRAS transgene under the control of its own promoter. They found consistent downregulation (~1/3) of RECK in tumors induced in three different experiments (7,12‐dimethylbenz[a]anthracene [DMBA]‐induced squamous cell carcinoma, N‐ethyl‐N‐nitrosourea [ENU]‐induced squamous cell carcinoma and urethane‐induced adenoma).( 27 ) Takagi et al. found downregulation of RECK in metastatic osteosarcomas in dogs.( 28 )
Summary. In summary, RECK is downregulated in many types of solid tumors (e.g. those in the pancreas, breast, lung non‐small cell, colorectum, prostate, stomach, bone), and the extent of downregulation often correlates with poor prognosis (Fig. 2a). Correlations between RECK expression and several other parameters have also been reported within certain types of tumors. The generality of such findings, both inside the reported tumor type and among several different tumor types, needs to be confirmed in future studies. These findings may also provide important clues in understanding how RECK regulates tumor formation and progression.
Figure 2.
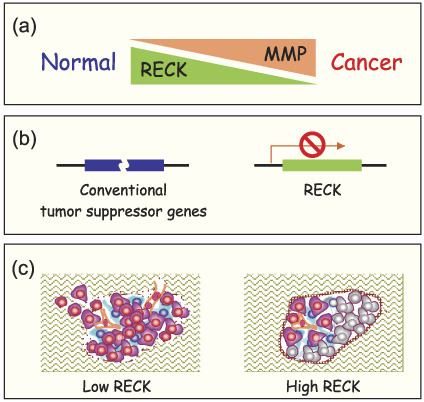
Reversion‐inducing cysteine‐rich protein with Kazal motifs (RECK) in carcinogenesis. (a) Certain matrix metalloproteinases (MMP) are often upregulated in advanced or aggressive tumors. In contrast, RECK is downregulated in tumors of poor prognosis. (b) Conventional tumor suppressor genes are often inactivated or altered by mutations or chromosomal rearrangements. The RECK gene is probably intact in tumor cells but its expression is frequently downregulated. (c) Forced RECK expression in tumor cells improves basement membrane integrity (thick brown capsule) and inhibits vascular invasion and branching (orange), resulting in the death of tumor cells far from blood vessels (gray). Prevention of basement breakdown and angiogenesis may contribute to the observed suppression of tumor invasion and metastasis.
Regulation of RECK expression and activity
Transcription. Chang et al. studied the mechanism of RECK promoter repression by ras or HER‐2/neu oncogene signaling.( 29 , 30 ) They confirmed the earlier finding by Sasahara et al.( 2 ) that the Sp1‐binding sites (GC boxes) located downstream of the transcription start site, termed Sp1(B), is a cis‐element involved in this repression. Furthermore, they demonstrated that phosphorylated Sp1 (Thr453/Thr739 catalyzed by external signal‐regulated kinase [ERK]) and histone deacetylase 1 were recruited to the Sp1(B) site in this process.
Certain histone deacetylase [HDAC] inhibitors are know to induce flat reversion and suppress tumor invasion.( 31 , 32 ) Liu et al. reported that trichostatin A, the first discovered HDAC inhibitor, activated the RECK promoter three‐fold and reduced the level of secreted MMP‐2 in a RECK‐dependent manner.( 33 ) Chang et al. also proposed another mechanism of RECK silencing in response to Ras/ERK signaling. Based on the experiments using a DNA methylation inhibitor (5′‐azacytidine) and small interfering RNA (siRNA), they proposed the involvement of DNA methyltransferase 3b in this process.( 34 )
To extend this finding, Cho et al. determined the levels of RECK mRNA, RECK protein, and methylation of the RECK promoter in resected colon cancers (n = 25) and found a significant correlation between RECK downregulation and RECK promoter methylation (P = 0.028). In addition, treatment of colon cancer‐derived cell lines (SW480 and SW620) with 5′‐azacytidine resulted in the suppression of invasive activity, and upregulation of RECK was required for this suppression.( 35 ) In their study on non‐small cell lung cancers (n = 55), they detected significant correlations between RECK downregulation and RECK promoter methylation (P = 5 × 10−6), between RECK downregulation and lymph node metastasis (P = 0.038), and between KRAS mutation (condon 12) and promoter methylation (P = 0.047) as well as RECK downregulation (P = 0.023). In cultured cells (H520, H358, A549), they found RECK upregulation and invasion suppression caused by 5′‐azacytidine treatment.( 36 )
Upstream pathways. Liu et al. identified a couple of other RECK expression modulators. Epstein–Bar virus latent membrane protein 1, known as a tumor metastasis inducer, was found to repress the RECK promoter via the ERK–Sp1 pathway.( 37 ) Non‐steroidal anti‐inflammatory drugs, NS398 and aspirin, were found to upregulate RECK mRNA and downregulate MMP‐2 in a lung cancer cell line; this effect seems to be independent of cycloxygenase‐2 (COX‐2) inhibition, as prostaglandin E2 and COX‐2 overexpression failed to reverse the effect.( 38 )
Oh et al. investigated the up‐stream signaling that regulates RECK expression and found an interesting link between a classical MMP regulator, tissue inhibitor of metalloproteinases‐2 (TIMP‐2), and RECK.( 39 ) TIMP‐2 inhibits migration of human microvascular endothelial cells, and previously identified mechanisms include direct inhibition of MMP and of VEGF signaling.( 40 ) Their new study indicates the involvement of RECK induction via the Crk–C3G–Rap1 pathway in this process.( 39 ) They also reported some evidence supporting the model that TIMP‐2 stimulates the phosphorylation of Src at Tyr‐527 by C‐terminal SRC kinase (CSK), leading to underphosphorylation of paxillin at Tyr‐31/118; this reduces the level of active Rac1 due to dissociation of the paxillin–Crk–DOCK180 complexes, and the concomitant increase in paxillin–Crk–C3G complexes leads to an increase in the level of active Rap1.( 41 ) Subsequent studies using adenoviral vectors indicated that TIMP‐2 promotes Rap1 expression and its association with actin.( 42 )
Correa et al. reported on the upregulation of RECK and gelatinases by type I collagen in glioma cells.( 43 ) In contrast, Takagi et al. observed downregulation of RECK by Matrigel, a reconstituted basement membrane preparation from the murine Englebreth–Holm–Swarm sarcoma, probably due to the upregulation of gelatinases.( 44 ) ECM components seem to have activities to induce both RECK and MMP, and the final outcome may depend on the balance between the two. Data obtained using cultured cells should therefore be interpreted with caution, taking into account the possibility that a slight shift in balance between RECK and MMP (and other targets; see below) may lead to robust changes in cell behavior.
Post‐translational regulation. Shimizu et al. reported another mode of RECK regulation: glycosylation. They introduced point mutations at five putative N‐glycosylation sites in RECK and found that glycosylation at three of the sites (Asn86, Asn297, Asn352) was required for its biological activities. They also found functional segregations among these mutants: the glycosylation at Asn297 is required for suppression of MMP‐9‐secretion, whereas glycosylation at Asn352 is required for inhibition of pro‐MMP‐2‐processing.( 45 )
Mori et al. isolated a cDNA exhibiting transforming activity in NIH3T3 cells from an adult T‐cell leukemia cDNA library. Its product, named Tgat, contains a Rho guanine nucleotide exchange factor (RhoGEF) domain and a unique C‐terminal 15 amino acids. They used this C‐terminal fragment as bait in a yeast two‐hybrid screening and cloned a fragment of RECK cDNA. They showed that RECK partially suppresses Tgat‐mediated NIH3T3 transformation and that full‐length Tgat, but not its mutant lacking the C‐terminal portion, reversed the RECK‐mediated inhibition of Matrigel invasion of HT1080 (fibrosarcoma) cells. Based on these findings, they proposed that Tgat may inhibit the activity of RECK through its C terminus.( 46 ) Although the evidence suggests an interesting link between cytoskeletal dynamics and RECK, the important questions as to when and where this cytoplasmic protein with a RhoGEF domain can interact with the GPI‐anchored external protein RECK need to be clarified.
Summary. As RECK is probably inactivated in tumors through epigenetic mechanisms in most cases (Fig. 2b), artificial activation of the dormant RECK gene is likely to have beneficial effects (Fig. 2c). It is therefore important to continue our efforts to elucidate how RECK is regulated in vivo and to find reagents that upregulate RECK but not other unwanted molecules.
Functions and mechanisms of action
Bioactivity. Suppression of tumor invasion and MMP activation in cultured cells have been used widely to assess RECK bioactivity. Kang et al. observed an inverse correlation between the level of RECK mRNA (determined by qRT‐PCR) and pro‐MMP‐2 activation (gelatin zymography) among osteosarcoma samples (n = 23; P = 0.037). They also observed reduced MMP‐2 activity, morphological changes and suppression of invasion in vitro in an osteosarcoma‐derived cell line (HOS) after RECK transfection.( 25 ) This study not only supports earlier findings with the HT1080 fibrosarcoma cell line but also demonstrates their clinical relevance.( 25 ) Takagi et al. cloned dog RECK cDNA, measured the expression of RECK mRNA by qRT‐PCR, and found its high expression in normal lung and testis. They also found low expression of RECK in various dog tumors (n = 36), including mast cell tumors, and relatively high expression in osteosarcoma. They could also detect the ability of RECK to suppress invasion when transfected into transitional cell carcinoma.( 47 ) Such studies in animals may be important in both scientific and practical terms; comparative studies among different species may yield important scientific insights, whereas dogs may be useful in testing the effectiveness and safety of RECK‐targeted therapies in future.
Tissue distribution. Precise knowledge of the temporal and spatial patterns of RECK expression in our body will provide an important foundation for future studies aiming to understand RECK's physiological functions and its roles in various disorders. Nuttall et al. carried out a systematic survey on the expression patterns of a whole series of molecules (i.e. 23 MMP family members, seven a disintegrin and metalloproteinase (ADAM) family members, five endogenous metalloproteinase inhibitors) in various organs from mice at several time points (E11.5–P28) during development by qRT‐PCR.( 48 ) The data indicate that at the organ level, RECK is widely (most abundant in lung) expressed throughout the developmental stages.
Guo and Zou collected placental tissue from 52 normal pregnant women (27 in early pregnancy, 25 in term pregnancy) and determined RECK protein level, its localization and MMP‐2 activation. They detected a significant increase in RECK expression and a decrease in MMP‐2 activation in the term pregnancy group compared with the early pregnancy group. Immunohistochemistry showed that RECK expression was found in villous tissues of both the early pregnancy and term pregnancy, localized mainly in the cell membrane and cytoplasm of cytotrophoblasts and syncytiotrophoblasts, increasing with gestational time, and significantly lower in cellular column with invasion ability.( 49 ) Thus, interplay between MMP and RECK may play a role in the regulation of trophoblast invasion. This study also demonstrates the dynamic nature of RECK expression inside an organ, which is likely to be undetectable by RNA blot hybridization or qRT‐PCR using RNA extracted from whole organs.
Physiological functions. Srivastava et al. used Drosophila genetics to explore the molecular pathways regulating MMP‐mediated basement membrane degradation during developmental invasion (i.e. imaginal disc evasion) and tumor invasion. They found that c‐Jun N‐terminal kinase (JNK) mediates MMP upregulation in both processes. Interestingly, although ectopic expression of TIMP was sufficient to inhibit developmental invasion, both TIMP and RECK were required to stop tumor invasion.( 50 ) Insects are evolutionally the most remote organisms known to carry RECK orthologues; no RECK is found in Caenorhabditis elegans. RECK is a single gene both in Drosophila and mammals. Drosophila would therefore serve as a good model system to explore conserved functions of RECK, including invasion suppression.
As mentioned above, RECK‐deficient mice die at mid‐gestation around E10.5,( 5 ) making it difficult to elucidate the functions of RECK in later stages of development using these mice. To address this issue, we analyzed the expression patterns of RECK protein and mRNA in mouse embryos at later stages. When immunohistochemistry was used, abundant RECK expression was found in skeletal muscles in E13.5–E14.5 embryos, and the staining pattern was reminiscent of that of a myogenic transcription factor, MRF4, but differed from that of MyoD. Experiments using cultured myogenic cells indicated that ectopic expression of RECK inhibits myotube formation and that the RECK gene promoter was inhibited by MyoD and activated by MRF4 in luciferase reporter assays. These data are consistent with the model (Fig. 3a) that RECK is downregulated by MyoD at the early stage of skeletal muscle development where myoblasts actively migrate and fuse to form myotubes, whereas RECK is upregulated by MRF4 at the stage when myotubes are sheathed by basement membranes to form myofibers and around the sites where mechanical strength is required (e.g. myo‐tendinous junctions).( 51 )
Figure 3.
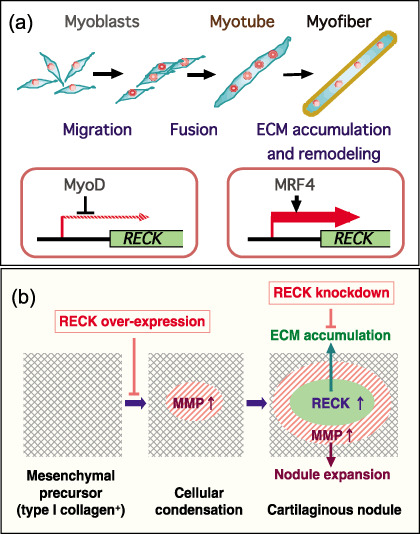
Reversion‐inducing cysteine‐rich protein with Kazal motifs (RECK) in tissue morphogenesis. (a) RECK in skeletal muscle development. The RECK gene promoter is inhibited by MyoD and stimulated by MRF4. Indeed, MRF4, but not MyoD, shares similar expression patterns with RECK in mouse embryos. RECK overexpression suppresses myotube formation, and RECK‐deficient embryonic cells show enhanced myogenic activity. These observations are consistent with the model that RECK is downregulated by MyoD during the early phase of muscle development in areas where cells actively migrate and fuse to form myotubes, whereas RECK is activated in the later phase in areas where myotubes become sheathed by basement membrane and acquire mechanical strength. (b) RECK as a key player in cartilage differentiation. Matrix metalloproteinase (MMP) expression is high and RECK expression is low in the initial stage of chondrogenic differentiation of ATDC5 cells. At this stage, cells migrate and form aggregates (cellular condensation). In the later stage, RECK expression is activated inside the large cell aggregates (now called cartilaginous nodules) where type IV collagen also accumulates, whereas MMP stay active in the periphery of the expanding nodules. RECK overexpression suppresses initial cellular condensation, whereas RECK knockdown inhibits extracellular matrix (ECM) accumulation and consolidation of the nodules. In both systems, RECK stays low in the initial phase and is upregulated in the later phase, suggesting that RECK plays a general role in helping to accumulate ECM accumulation and stabilizing tissue architecture.
When in situ hybridization was used, however, abundant RECK mRNA was detected in cartilage in E13.5–E16.5 embryos. The mouse embryonal carcinoma‐derived cell line ATDC5 is known to be able to recapitulate cartilage development in culture. At the initial stage, ATDC5 cells actively migrate and form small aggregates or foci, which we call ‘cellular condensation’. At the later stage, these foci continue to grow and form larger ridges called cartilaginous nodules. We found that during cellular condensation, RECK expression is repressed and MMP expression is elevated especially at the center of the foci. At the later stage, RECK becomes upregulated in cartilaginous nodules where chondrocyte‐specific type II collagen is accumulated; MMP remain active in the periphery of the expanding nodules. Ectopic expression of RECK suppresses cellular condensation, whereas RECK siRNA inhibits ECM accumulation and nodule consolidation( 52 ) (Fig. 3b). Thus, in both skeletal muscle and cartilage, the level of RECK is low in the early phase of tissue morphogenesis in areas where cell migration and dynamic cell–cell interaction take place, whereas the level of RECK becomes elevated in the later phase in areas where ECM accumulation is required.
We recently identified a developmental abnormality, including precocious neuronal differentiation and reduction in the number of nestin‐positive neural precursor cells, in the central nervous system of RECK‐deficient mice. This and the vascular phenotype( 6 ) of RECK‐deficient mice is reminiscent of the phenotype seen in mice deficient in Notch signaling. Indeed, remarkable downregulation of Notch signaling was observed, and the mechanism probably involves excessive ectodomain shedding of Notch ligands mediated by ADAM10.( 53 ) Thus, RECK regulates an ADAM family member and supports cell proliferation in this system. The Notch pathway is conserved across species and is involved in various biological events. It would be interesting to address whether RECK plays any roles in other species or in other biological events.
We also characterized the effects of RECK on other molecules on the cell surface. RECK was found to form a complex with MT1‐MMP and inhibit its proteolytic activity. When expressed in HT1080 cells, RECK increased the amount of MT1‐MMP that associated with the membrane microdomain corresponding to the ‘lipid raft’ during sucrose density gradient ultracentrifugation, and this effect was abolished when membrane cholesterol was perturbed. Thus, RECK may regulate MT1‐MMP function by modulating its behavior on the cell surface as well as inhibiting its enzymatic activity. A subsequent study indicated that RECK interacts with CD13/aminopeptidase N and inhibits the proteolytic activity of CD13 in a cholesterol perturbation‐sensitive manner. Moreover, RECK was found to recruit these membrane proteases to the specific endocytic pathway known as GPI‐anchored protein‐enriched early endosomal compartment (GEEC), resulting in accelerated degradation.( 54 )
These new findings have widened our view on the biological functions of RECK and expanded the repertoire of RECK targets as well as substrates protected by RECK.
Future directions
Overview. As summarized above, compelling evidence indicates a good correlation between the extent of RECK downregulation and poor prognosis in a wide variety of solid tumors. Knowledge on the transcriptional regulation of RECK is also accumulating. However, the number of reports on the mechanisms of action of RECK has been limited. One of the important issues at the molecular level is to clarify the spectrum of RECK targets. We also need to find molecules that interact physically with RECK, to determine the 3‐dimensional structure of RECK, and to understand the nature of interactions between RECK and its targets at the atomic level. At the single‐cell level, the role of RECK in cell differentiation and migration needs to be clarified. Signaling pathways and drugs regulating RECK expression should also be explored. At the system level, involvement of RECK in animal development and various diseases has to be studied. Various model systems including simple organisms (e.g. fruit fly) and genetically engineered mice will be valuable in such studies.
Human genetics. Studies on the possible involvement of RECK in various diseases may yield information of both clinical and biological importance. Single nucleotide polymorphism (SNP) markers may serve as powerful tools for such studies. For instance, Lei et al. analyzed SNP in the RECK promoter region in healthy control (n = 952) and mammary tumor patients (n = 959) and found a correlation between C/T heterozygosity at the –420 position and better prognosis.( 55 ) Eisenberg et al. established the entire genomic structure of the human RECK gene (mapped at 9p13→p12) which spans more than 87 kb and consists of 21 exons and 20 introns. They identified 13 SNP: four in the coding region (exons 1, 9, 13 and 15) and nine in introns (introns 5, 8, 10, 12, 15 and 17).( 56 ) These and other SNP markers will be valuable in future studies.
Other disorders. Involvement of MMP in the pathology of rheumatoid arthritis has been well documented. van Lent et al. analyzed RECK expression in the synovial membrane by both qRT‐PCR and immunohistochemistry; they found that RECK expression is reduced in patients with rheumatoid arthritis whereas MMP‐14 shows no significant difference.( 57 ) Milner et al. used small pieces of bovine nasal septum cartilage in culture to analyze the effects of inflammatory cytokines, interleukin‐1 and oncostatin M, on the expression of several MMP and MMP inhibitors to understand their roles in cartilage resorption. In this system, no significant alteration in the level of RECK expression was detected.( 58 ) In light of the suggested roles of RECK in chondrogenesis( 52 ) it should be important to clarify the roles of RECK in rheumatoid arthritis and other disorders affecting cartilaginous tissues.
Some respiratory disorders are accompanied by tissue destruction and might involve an imbalance between RECK and MMP. Indeed, Paulissen et al. reported that the level of RECK mRNA was significantly decreased in the cells in the sputum from patients with asthma (n = 21) compared with healthy controls (n = 17), and that RECK expression was positively correlated with forced expiratory volume in the first second (FEV1), an indicator of maximal exercise capacity (r = 0.45, P = 0.01).( 59 )
Recent findings indicate that MMP act on pro‐inflammatory cytokines, chemokines and other proteins to regulate various aspects of inflammation and immunity.( 60 ) RECK may well be involved in the regulation of these events, but we have no evidence for or against this possibility at the moment. Expression of RECK in macrophages has been reported.( 57 ) A reduction in the phagocytotic activity of macrophages has been found in endometriosis. Wu et al. reported that this phenomenon involves prostaglandin E2 (PGE2)‐mediated downregulation of MMP‐9 but not changes in the expression of TIMP or RECK.( 61 ) Thus, the functions of RECK in macrophages and its role in endometriosis remain unclear.
Conclusions. Here we have cited most, if not all, of the papers on RECK published since the announcement of its discovery in 1998.( 1 ) These limited number of studies already illuminate the importance of RECK (or its absence) in some physiological processes and diseases, especially those involving ECM remodeling. Further studies on various aspects of RECK will lead to a better understanding of how this molecule works to prevent tumor angiogenesis, invasion and metastasis and how we can selectively manipulate these processes.
Acknowledgments
We thank Regina M. Sasahara, Sachiko Nishimura, Junseo Oh, Shunya Kondo, Michiko Echizenya, Teruyuki Muraguchi, Takao Miki and Hitoshi Kitayama for their contributions to the studies from our laboratory. We also thank Rei Takahashi, Motoharu Seiki and Shigeyoshi Itohara for continued collaborations. Our studies are supported by a Grant‐in‐Aid for Creative Scientific Research from JSPS and a Grant‐in‐Aid on Priority Areas from MEXT.
References
- 1. Takahashi C, Sheng Z, Horan TP et al . Regulation of matrix metalloproteinase‐9 and inhibition of tumor invasion by the membrane‐anchored glycoprotein RECK. Proc Natl Acad Sci USA 1998; 95: 13 221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sasahara RM, Takahashi C, Noda M. Involvement of the sp1 site in ras‐mediated downregulation of the RECK metastasis suppressor gene. Biochem Biophys Res Commun 1999; 264: 668–75. [DOI] [PubMed] [Google Scholar]
- 3. Sasahara RM, Takahashi C, Sogayar MC, Noda M. Oncogene‐mediated downregulation of RECK, a novel transformation suppressor gene. Braz J Med Biol Res 1999; 32: 891–5. [DOI] [PubMed] [Google Scholar]
- 4. Sasahara RM, Brochado SM, Takahashi C et al . Transcriptional control of the RECK metastasis/angiogenesis suppressor gene. Cancer Detect Prev 2002; 26: 435–43. [DOI] [PubMed] [Google Scholar]
- 5. Takenaka K, Ishikawa S, Kawano Y et al . Expression of a novel matrix metalloproteinase regulator, RECK, and its clinical significance in resected non‐small cell lung cancer. Eur J Cancer 2004; 40: 1617–23. [DOI] [PubMed] [Google Scholar]
- 6. Oh J, Takahashi R, Kondo S et al . The membrane‐anchored MMP inhibitor reck is a key regulator of extracellular matrix integrity and angiogenesis. Cell 2001; 107: 789–800. [DOI] [PubMed] [Google Scholar]
- 7. Oh J, Takahashi R, Adachi E et al . Mutations in two matrix metalloproteinase genes, MMP‐2 and mt1‐MMP, are synthetic lethal in mice. Oncogene 2004; 23: 5041–8. [DOI] [PubMed] [Google Scholar]
- 8. Rhee JS, Coussens LM. Recking MMP function: implications for cancer development. Trends Cell Biol 2002; 12: 209–11. [DOI] [PubMed] [Google Scholar]
- 9. Welm B, Mott J, Werb Z. Developmental biology: vasculogenesis is a wreck without RECK. Curr Biol 2002; 12: R209–11. [DOI] [PubMed] [Google Scholar]
- 10. Weaver VM. Membrane‐associated MMP regulators: novel cell adhesion tumor suppressor proteins? Dev Cell 2002; 2: 6–7. [DOI] [PubMed] [Google Scholar]
- 11. Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci 2002; 115: 3719–27. [DOI] [PubMed] [Google Scholar]
- 12. Noda M, Oh J, Takahashi R, Kondo S, Kitayama H, Takahashi C. RECK: a novel suppressor of malignancy linking oncogenic signaling to extracellular matrix remodeling. Cancer Metastasis Rev 2003; 22: 167–75. [DOI] [PubMed] [Google Scholar]
- 13. Furumoto K, Arii S, Mori A et al . RECK gene expression in hepatocellular carcinoma: correlation with invasion‐related clinicopathological factors and its clinical significance. Reverse‐inducing cysteine‐rich protein with kazal motifs. Hepatology 2001; 33: 189–95. [DOI] [PubMed] [Google Scholar]
- 14. Masui T, Doi R, Koshiba T et al . RECK expression in pancreatic cancer: Its correlation with lower invasiveness and better prognosis. Clin Cancer Res 2003; 9: 1779–84. [PubMed] [Google Scholar]
- 15. Span PN, Sweep CG, Manders P, Beex LV, Leppert D, Lindberg RL. Matrix metalloproteinase inhibitor reversion‐inducing cysteine‐rich protein with kazal motifs: a prognostic marker for good clinical outcome in human breast carcinoma. Cancer 2003; 97: 2710–15. [DOI] [PubMed] [Google Scholar]
- 16. Takenaka K, Ishikawa S, Yanagihara K et al . Prognostic significance of reversion‐inducing cysteine‐rich protein with kazal motifs expression in resected pathologic stage IIIa n2 non‐small‐cell lung cancer. Ann Surg Oncol 2005; 12: 817–24. [DOI] [PubMed] [Google Scholar]
- 17. Takeuchi T, Hisanaga M, Nagao M et al . The membrane‐anchored matrix metalloproteinase (MMP) regulator RECK in combination with MMP‐9 serves as an informative prognostic indicator for colorectal cancer. Clin Cancer Res 2004; 10: 5572–9. [DOI] [PubMed] [Google Scholar]
- 18. Van Der Jagt MF, Sweep FC, Waas ET et al . Correlation of reversion‐inducing cysteine‐rich protein with kazal motifs (RECK) and extracellular matrix metalloproteinase inducer (emmprin), with MMP‐2, MMP‐9, and survival in colorectal cancer. Cancer Lett 2006; 237: 289–97. [DOI] [PubMed] [Google Scholar]
- 19. Ohl F, Jung M, Xu C et al . Gene expression studies in prostate cancer tissue: Which reference gene should be selected for normalization? J Mol Med 2005; 83: 1014–24. [DOI] [PubMed] [Google Scholar]
- 20. Rabien A, Burkhardt M, Jung M et al . Decreased RECK expression indicating proteolytic imbalance in prostate cancer is associated with higher tumor aggressiveness and risk of prostate‐specific antigen relapse after radical prostatectomy. Eur Urol 2007; 51: 1259–66. [DOI] [PubMed] [Google Scholar]
- 21. Riddick AC, Shukla CJ, Pennington CJ et al . Identification of degradome components associated with prostate cancer progression by expression analysis of human prostatic tissues. Br J Cancer 2005; 92: 2171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Y, Zhang Y, Zheng Q. Expression of RECK gene and MMP‐9 in hilar cholangiocarcinoma and its clinical significance. J Huazhong Univ Sci Technolog Med Sci 2005; 25: 552–4. [DOI] [PubMed] [Google Scholar]
- 23. Song SY, Son HJ, Nam E, Rhee JC, Park C. Expression of reversion‐inducing‐cysteine‐rich protein with kazal motifs (RECK) as a prognostic indicator in gastric cancer. Eur J Cancer 2006; 42: 101–8. [DOI] [PubMed] [Google Scholar]
- 24. Kumamoto H, Ooya K. Immunohistochemical detection of mt1‐MMP, RECK, and emmprin in ameloblastic tumors. J Oral Pathol Med 2006; 35: 345–51. [DOI] [PubMed] [Google Scholar]
- 25. Kang HG, Kim HS, Kim KJ et al . RECK expression in osteosarcoma: correlation with matrix metalloproteinases activation and tumor invasiveness. J Orthop Res 2007; 25: 696–702. [DOI] [PubMed] [Google Scholar]
- 26. Nabeshima K, Iwasaki H, Nishio J, Koga K, Shishime M, Kikuchi M. Expression of emmprin and matrix metalloproteinases (MMPs) in peripheral nerve sheath tumors. Emmprin and membrane‐type (mt) 1‐MMP expressions are associated with malignant potential. Anticancer Res 2006; 26: 1359–67. [PubMed] [Google Scholar]
- 27. Okamura M, Unami A, Moto M et al . The possible mechanism of enhanced carcinogenesis induced by genotoxic carcinogens in rash2 mice. Cancer Lett 2007; 245: 321–30. [DOI] [PubMed] [Google Scholar]
- 28. Takagi S, Kato Y, Asano K et al . Matrix metalloproteinase inhibitor RECK expression in canine tumors. J Vet Med Sci 2005; 67: 761–7. [DOI] [PubMed] [Google Scholar]
- 29. Chang HC, Liu LT, Hung WC. Involvement of histone deacetylation in ras‐induced down‐regulation of the metastasis suppressor reck. Cell Signal 2004; 16: 675–9. [DOI] [PubMed] [Google Scholar]
- 30. Hsu MC, Chang HC, Hung WC. Her‐2/neu represses the metastasis suppressor RECK via ERK and SP transcription factors to promote cell invasion. J Biol Chem 2006; 281: 4718–25. [DOI] [PubMed] [Google Scholar]
- 31. Kwon HJ, Owa T, Hassig CA, Shimada J, Schreiber SL. Depudecin induces morphological reversion of transformed fibroblasts via the inhibition of histone deacetylase. Proc Natl Acad Sci USA 1998; 95: 3356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim MS, Son MW, Kim WB, In Park Y, Moon A. Apicidin, an inhibitor of histone deacetylase, prevents h‐ras‐induced invasive phenotype. Cancer Lett 2000; 157: 23–30. [DOI] [PubMed] [Google Scholar]
- 33. Liu LT, Chang HC, Chiang LC, Hung WC. Histone deacetylase inhibitor up‐regulates RECK to inhibit MMP‐2 activation and cancer cell invasion. Cancer Res 2003; 63: 3069–72. [PubMed] [Google Scholar]
- 34. Chang HC, Cho CY, Hung WC. Silencing of the metastasis suppressor RECK by ras oncogene is mediated by DNA methyltransferase 3b‐induced promoter methylation. Cancer Res 2006; 66: 8413–20. [DOI] [PubMed] [Google Scholar]
- 35. Cho CY, Wang JH, Chang HC, Chang CK, Hung WC. Epigenetic inactivation of the metastasis suppressor RECK enhances invasion of human colon cancer cells. J Cell Physiol 2007; 213: 65–9. [DOI] [PubMed] [Google Scholar]
- 36. Chang HC, Cho CY, Hung WC. Downregulation of RECK by promoter methylation correlates with lymph node metastasis in non‐small cell lung cancer. Cancer Sci 2007; 98: 169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu LT, Peng JP, Chang HC, Hung WC. RECK is a target of Epstein–Barr virus latent membrane protein 1. Oncogene 2003; 22: 8263–70. [DOI] [PubMed] [Google Scholar]
- 38. Liu LT, Chang HC, Chiang LC, Hung WC. Induction of RECK by nonsteroidal anti‐inflammatory drugs in lung cancer cells. Oncogene 2002; 21: 8347–50. [DOI] [PubMed] [Google Scholar]
- 39. Oh J, Seo DW, Diaz T et al . Tissue inhibitors of metalloproteinase 2 inhibits endothelial cell migration through increased expression of RECK. Cancer Res 2004; 64: 9062–9. [DOI] [PubMed] [Google Scholar]
- 40. Vincent L, Varet J, Pille JY et al . Efficacy of dendrimer‐mediated angiostatin and TIMP‐2 gene delivery on inhibition of tumor growth and angiogenesis: In vitro and in vivo studies. Int J Cancer 2003; 105: 419–29. [DOI] [PubMed] [Google Scholar]
- 41. Oh J, Diaz T, Wei B, Chang H, Noda M, Stetler‐Stevenson WG. TIMP‐2 upregulates RECK expression via dephosphorylation of paxillin tyrosine residues 31 and 118. Oncogene 2006; 25: 4230–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chang H, Lee J, Poo H et al . TIMP‐2 promotes cell spreading and adhesion via upregulation of Rap1 signaling. Biochem Biophys Res Commun 2006; 345: 1201–6. [DOI] [PubMed] [Google Scholar]
- 43. Correa TC, Brohem CA, Winnischofer SM et al . Downregulation of the reck‐tumor and metastasis suppressor gene in glioma invasiveness. J Cell Biochem 2006; 99: 156–67. [DOI] [PubMed] [Google Scholar]
- 44. Takagi S, Hoshino Y, Osaki T, Okumura M, Fuginaga T. Expression of membrane‐anchored matrix metalloproteinase inhibitor reversion inducing cysteine rich protein with kazal motifs in murine cell lines. Exp Oncol 2007; 29: 30–4. [PubMed] [Google Scholar]
- 45. Shimizu S, Takagi S, Tamura Y, Osada H. RECK‐mediated suppression of tumor cell invasion is regulated by glycosylation in human tumor cell lines. Cancer Res 2005; 65: 7455–61. [DOI] [PubMed] [Google Scholar]
- 46. Mori T, Moriuchi R, Okazaki E, Yamada K, Katamine S. Tgat oncoprotein functions as an inhibitor of RECK by association of the unique c‐terminal region. Biochem Biophys Res Commun 2007; 355: 937–43. [DOI] [PubMed] [Google Scholar]
- 47. Takagi S, Kitamura T, Hosaka Y et al . Molecular cloning of canine membrane‐anchored inhibitor of matrix metalloproteinase, RECK. J Vet Med Sci 2005; 67: 385–91. [DOI] [PubMed] [Google Scholar]
- 48. Nuttall RK, Sampieri CL, Pennington CJ, Gill SE, Schultz GA, Edwards DR. Expression analysis of the entire MMP and TIMP gene families during mouse tissue development. FEBS Lett 2004; 563: 129–34. [DOI] [PubMed] [Google Scholar]
- 49. Guo J, Zou L. Correlation of RECK with matrix metalloproteinase‐2 in regulation of trophoblast invasion of early pregnancy. J Huazhong Univ Sci Technolog Med Sci 2006; 26: 738–40. [DOI] [PubMed] [Google Scholar]
- 50. Srivastava A, Pastor‐Pareja JC, Igaki T, Pagliarini R, Xu T. Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc Natl Acad Sci USA 2007; 104: 2721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Echizenya M, Kondo S, Takahashi R et al . The membrane‐anchored MMP‐regulator RECK is a target of myogenic regulatory factors. Oncogene 2005; 24: 5850–7. [DOI] [PubMed] [Google Scholar]
- 52. Kondo S, Shukunami C, Morioka Y et al . Dual effects of the membrane‐anchored MMP regulator RECK on chondrogenic differentiation of atdc5 cells. J Cell Sci 2007; 120: 849–57. [DOI] [PubMed] [Google Scholar]
- 53. Muraguchi T, Takegami Y, Chandana EPS et al . RECK modulates Notch signaling during cortical neurogenesis by regulating ADAM10 activity. Nat Neurosci 2007; 10: 838–45. [DOI] [PubMed] [Google Scholar]
- 54. Miki T, Takegami Y, Okawa K, Muraguchi T, Noda M, Takahashi C. The reversion‐inducing cysteine‐rich protein with Kazal motifs (RECK) interacts with membrane type 1 matrix metalloproteinase and CD13/aminopeptidase N and modulates their endocytic pathways. J Biol Chem 2007; 282: 12 341–52. [DOI] [PubMed] [Google Scholar]
- 55. Lei H, Hemminki K, Altieri A et al . Promoter polymorphisms in matrix metalloproteinases and their inhibitors: few associations with breast cancer susceptibility and progression. Breast Cancer Res Treat 2007; 103: 61–9. [DOI] [PubMed] [Google Scholar]
- 56. Eisenberg I, Hochner H, Sadeh M, Argov Z, Mitrani‐Rosenbaum S. Establishment of the genomic structure and identification of thirteen single‐nucleotide polymorphisms in the human RECK gene. Cytogenet Genome Res 2002; 97: 58–61. [DOI] [PubMed] [Google Scholar]
- 57. Van Lent PL, Span PN, Sloetjes AW et al . Expression and localisation of the new metalloproteinase inhibitor RECK (reversion inducing cysteine‐rich protein with kazal motifs) in inflamed synovial membranes of patients with rheumatoid arthritis. Ann Rheum Dis 2005; 64: 368–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Milner JM, Rowan AD, Cawston TE, Young DA. Metalloproteinase and inhibitor expression profiling of resorbing cartilage reveals pro‐collagenase activation as a critical step for collagenolysis. Arthritis Res Ther 2006; 8: R142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Paulissen G, Rocks N, Quesada‐Calvo F et al . Expression of ADAMs and their inhibitors in sputum from patients with asthma. Mol Med 2006; 12: 171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Parks WC, Wilson CL, Lopez‐Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 2004; 4: 617–29. [DOI] [PubMed] [Google Scholar]
- 61. Wu MH, Shoji Y, Wu MC et al . Suppression of matrix metalloproteinase‐9 by prostaglandin E2 in peritoneal macrophage is associated with severity of endometriosis. Am J Pathol 2005; 167: 1061–9. [DOI] [PMC free article] [PubMed] [Google Scholar]