

Abstract
We analyzed the subchromosomal numerical aberrations of 44 surgically resected pancreatic adenocarcinomas by array‐based comparative genomic hybridization. The aberration profile ranged widely between cases, suggesting the presence of multiple or complementary mechanisms of evolution in pancreatic cancer, and was associated with lymph node metastasis and venous or serosal invasion. A large number of small loci, previously uncharacterized in pancreatic cancer, showed non‐random loss or gain. Frequent losses at 1p36, 4p16, 7q36, 9q34, 11p15, 11q13, 14q32‐33, 16p13, 17p11‐13, 17q11‐25, 18q21‐tel, 19p13, 21q22 and 22q11‐12, and gains at 1q25, 2p16, 2q21‐37, 3q25, 5p14, 5q11‐13, 7q21, 7p22, 8p22, 8q21‐23, 10q21, 12p13, 13q22, 15q13‐22 and 18q11 were identified. Sixteen loci were amplified recurrently. We identified novel chromosomal alterations that were significantly associated with a range of malignant phenotypes. Gain of LUNX, HCK, E2F1 and DNMT3b at 20q11, loss of p73 at 1p36 and gain of PPM1D at 17q23 independently predicted patient outcome. Expression profiling of amplified genes identified Smurf1 and TRRAP at 7q22.1, BCAS1 at 20q13.2‐3, and VCL at 10q22.1 as potential novel oncogenes. Our results contribute to a complete description of genomic structural aberrations and the identification of potential therapeutic targets and genetic indicators that predict patient outcome in pancreatic adenocarcinoma. (Cancer Sci 2007; 98: 392–400)
Abbreviations
- aCGH
array‐based comparative genomic hybridization
- BAC
bacterial artificial chromosome
- CGH
comparative genomic hybridization
- HD
homozygous deletion
- LCM
laser‐capture microdissection
- PCR
polymerase chain reaction
- SNAP
subchromosomal numerical aberration profile
- TSG
tumor suppressor gene.
Pancreatic adenocarcinoma is a leading cause of cancer‐related death worldwide; the 5‐year survival rate for patients that underwent surgery remains below 5%.( 1 ) Pancreatic adenocarcinoma appears to successively acquire genetic aberrations in genes involved in the regulation of cell proliferation, the central ones being early activating mutations of the K‐ras oncogene, followed by inactivation of the p53, p16 and DPC4 TSG.( 2 ) The application of chromosome CGH,( 3 ) karyotype and allelotype studies in pancreatic cancer has also revealed a large number of complex structural and numerical aberrations at the subchromosomal level.( 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 ) Recurrent aberrations reported concern copy number gain on 3q, 5p, 7p, 8q, 11q, 12p, 17q, 19q and 20q and loss on 1p, 3p, 4q, 6q, 8p, 9p, 10q, 12q, 13q, 15q, 17p, 18q, 19p, 21q and 22q.( 8 , 12 ) aCGH methods have recently been developed and used in studies of various malignancies, including pancreatic cancer. The latter used cell lines,( 13 , 14 , 15 , 16 , 17 , 18 ) and a small number of primary cases( 14 , 15 ) or xenografts,( 19 ) to confirm previously described regional alterations and identify novel ones. Although some of these loci are known to contain oncogenes or TSG,( 2 ) the role that copy number alterations of most of the above loci play in pancreatic cancer genesis or progression, if any, is far from being fully evaluated. From these and previous studies, it is also evident that there exists substantial variation in the reported aberrations between studies as well as between individual cases.
The aim of the present study was to examine the SNAP of pancreatic cancer to identify novel loci that contain genes for which copy number status is likely to be relevant to pancreatic carcinogenesis or associated with clinically relevant parameters. For this, we used aCGH to examine a comparatively large number of well‐characterized primary cases and LCM to allow more accurate analysis. In addition, mRNA expression analysis of loci exhibiting amplifications was carried out to identify genes that are amplified recurrently and overexpressed in pancreatic cancer.
Materials and Methods
Tumor samples. Forty‐four methanol‐fixed pancreatic ductal adenocarcinomas from 43 patients were examined (Suppl. Table 1). These included 33 specimens from patients who had undergone surgery at the National Cancer Center Hospital between 1994 and 2003, and 11 xenografts that were produced following the orthotopic implantation of tumors in severe combined immunodeficient mice, as described previously.( 20 ) Forty‐two samples were of primary tumors, one of a liver metastasis and one of a pancreatic xenograft of a liver metastasis, the corresponding primary of which was also examined. Tumor classification was carried out according to the Japan Pancreas Society guidelines.( 21 ) The study was approved by the institutional review board of the National Cancer Center.
LCM and whole‐genome amplification. LCM was carried out with a PixCell II (Arcturus Engineering, Mountain View, CA, USA). At least 5000 tumor cells per sample were recovered. Genomic (test) DNA was extracted by standard procedures. Sex‐matched high molecular weight human genomic DNA (Promega, Madison, WI, USA) was sheared randomly (HydroShear; Gene Machines, San Carlos, CA, USA) and used as reference DNA. Both test and reference DNA were amplified using an adaptor ligation‐mediated whole‐genome PCR, as described previously.( 22 )
Array‐based CGH. A custom‐made CGH array (‘MCG Cancer Array‐800 ver. 2’) was used, consisting of 800 duplicated target BAC clones that correspond to chromosomal loci of potential importance in various cancers (listed at http://www.cghtmd.jp/CGHDatabase/microarray/mcg800_array_e.htm). Labeling of the DNA probes, hybridization, data acquisition and data normalization were carried out as described previously.( 23 , 24 , 25 ) Based on control experiments,( 26 ) we considered a signal ratio <0.75 or >1.25 to indicate loss or gain, respectively, and a ratio of <0.25 or >2.00 to indicate HD or amplification, respectively.
The validity of our aCGH data was confirmed by fluorescence in situ hybridization, PCR (Suppl. Fig. 1), loss of heterozygosity analysis and immunohistochemistry for selected genes.( 26 )
Expression profiling of primary xenografts. We used xenografts for gene expression analysis due to their abundance in tumors cells compared with primary tumors. We focused on the relationship between amplification and overexpression; additional gene expression profiling results will be submitted in a subsequent publication.
Total RNA was extracted from frozen xenograft samples, biotin‐labeled cRNA synthesized and hybridized to a probe array (HG‐U95Av2, Affymetrix) and data acquired as described.( 27 ) A probe set signal log ratio (SLR) of the gene expression level in the tumor relative to the control (normal pancreas) >1.5 was defined as indicating overexpression.
Statistical analysis. The χ2 test was used to assess the statistical significance, set at 0.05, of intergroup differences in the frequency of aberrations of individual loci. The relationship between clinicopathological parameters and the number of aberrations per case was evaluated using Student's unpaired t‐test. Survival curves were calculated using the Kaplan–Meier method, and differences in survival periods were analyzed with the log‐rank test.
Results
Range of numerical aberrations. We constructed and analyzed the genomic profile of 44 pancreatic adenocarcinomas using aCGH. Subchromosomal numerical aberrations were revealed in all but two (42/44) of the tumors examined (Fig. 1). The number of aberrations differed widely between cases (Suppl. Table 2; Suppl. Fig. 2). Apart from the two cases in which no copy number changes were observed, a third case showed changes in only 11 loci (all gains), whereas nine cases (20%) had alterations in more than 50% of loci. In most cases (34/44), the number of gains was higher than the number of losses (P < 10−7). Overall, however, the loss rate was similar to the gain rate (19% of loci altered on average per case for both). Similarly, amplifications were observed more frequently, in terms of number of cases and number of aberrations per case, than HD. Most loci showed aberrations in at least one case, the majority showing loss or gain in 2–25% and 0–20% of cases, respectively.
Figure 1.
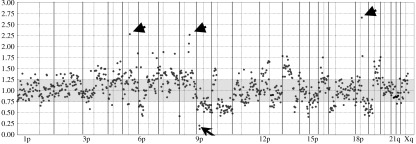
Chromosomal copy number changes revealed by array‐based comparative genomic hybridization. Representative array‐based comparative genomic hybridization profile of a pancreatic adenocarcinoma. Copy number losses (ratio < 0.75) and gains (ratio > 1.25) were detected in both large fractions of the chromosome arms and small chromosomal regions. Amplifications (ratio > 2.00, arrowheads) and homozygous deletions (ratio < 0.25, arrow) were also identified in this tumor. The average signal ratios (test:reference) of two normalized signals from duplicated spots are given from chromosome 1p telomere (left) to Xq telomere (right). The vertical dotted and continuous lines indicate the position of the centromere and telomere of each chromosome, respectively.
Loss. The most frequently lost loci were 17p13.3 (ABR, in 75% of cases), 18qtel (CTDP1, SHGC‐145820, 68%) and 18q21 (SMAD7, 66%). The loci containing the p16 (9p21), p53 (17p13.1), SMAD4 and DCC (both at 18q21) genes were lost in 41, 55, 61 and 30% of cases, respectively. In total, 33 loci with frequent (>50%) losses were identified at 1p36, 4p16.3, 7q36, 9q34.3, 11p15, 11q13, 14q32‐33, 16p13.3, 17p11.2, 17p13.1‐3, 17q11‐qter, 17q21.2, 17q25, 18q21, 18qtel, 19p13.2‐3, 21q22.3, 22q11.23 and 22q12.1‐2 (Fig. 2). The chromosome arms with the highest number of loci lost, taking into account only loci that were lost in >25% of cases, included, in descending order of frequency, 1p, 11q, 17p, 10q, 8p, 18q, 22q, 6q, 9p, 14q and 17q (Suppl. Table 3).
Figure 2.
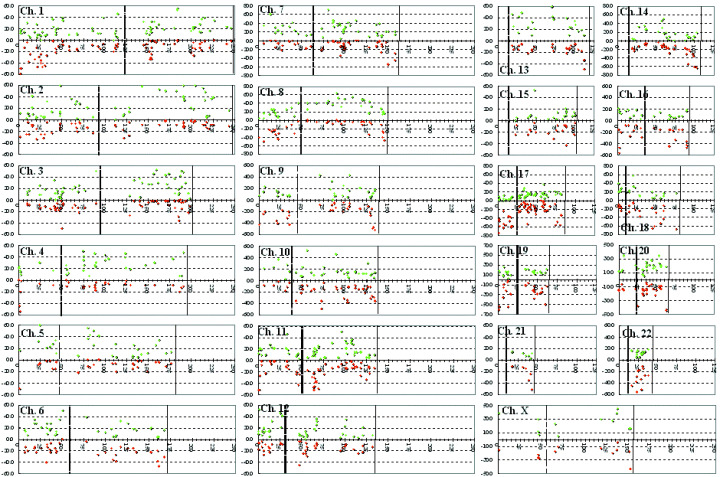
Distribution of chromosomal copy number aberrations in pancreatic cancer. The horizontal axis indicates the physical distance (Mb) of the chromosomal loci from the telomere of the short arm. The vertical axis indicates the frequency (%) of tumors with chromosomal alterations (green, gain; red, loss). The vertical dotted and continuous lines indicate the positions of the centromere and telomere of each chromosome, respectively.
Homozygous deletions. Twenty‐six loci with HD were detected, nine of which were in more than one case (Table 1). HD were detected in 11 cases (25%), seven of which in only one locus. The 1p35‐36.33 region contained the highest number of loci deleted (six). The most frequently deleted locus was 9p21 spanning the p16 gene, whereas the locus containing SMAD4 (18q21) was deleted in one case.
Table 1.
Loci deleted homozygously in more than one case
| Locus† | No. cases | Percentage of cases |
|---|---|---|
| 9p21 (p16) | 5 | 11 |
| 16p13.3 (ABCA3) | 3 | 7 |
| 1p36.1 (p73) | 2 | 5 |
| 5p15 (TERT) | 2 | 5 |
| 11p15 (HRAS) | 2 | 5 |
| 17q25 (MAFG) | 2 | 5 |
| 18q21 (SMAD7) | 2 | 5 |
| 18qtel (CTDP1,SHGC‐145820) | 2 | 5 |
| 19p13.3 (ABCA7) | 2 | 5 |
Known cancer‐related genes contained in the respective clones are shown in parentheses.
Gains. Loci with frequent (>50% cases) gains were identified at 1q25.2‐q25.3, 2p16, 2q21.2, 2q23‐q37, 2q31, 2q33, 2q34, 3q25.1, 5p14.2, 5q11.2‐q13.2, 7q21.1, 7p22, 8p22, 8q21, 8q22‐q23, 10q21.1, 12p13.33, 13q22, 15q13‐q22 and 18q11.2 (Fig. 2; Suppl. Table 4). The most frequently gained locus was 7q21.1 (71%) containing the HGF gene. The loci spanning the KRAS2 (12p12.1) and KRAG (12p11.2) genes were gained in 45 and 20% of cases, respectively. The NRAS (1p13), MYC (8q24), MDM2 (12q14.3) and AKT1 (14q32.2)( 28 ) loci were gained in 45, 43, 36 and 18% of cases, respectively.
Amplifications. Amplifications were observed in 37 tumors. The seven cases in which no amplification was observed included six with few aberrations, and, interestingly, one case with 419 aberrations. Nineteen cases had amplifications in more than 1%, and three cases in more than 5% of loci examined.
Sixteen loci were amplified in five cases or more (>10%) (Table 2). The most frequently amplified locus was 18q11.2 containing RBBP8. 7q34 (BRAF) was amplified in four cases, whereas 12p12.1 (KRAS2), 1p13 (NRAS) and 8q24 (MYC) were amplified in two cases each.
Table 2.
Loci amplified in more than 10% of cases
| Locus† | No. cases | Percentage of cases |
|---|---|---|
| 18q11.2 (RBBP8) | 10 | 23 |
| 7q21.1 (HGF) | 9 | 20 |
| 2q31 (PMS1) | 7 | 16 |
| 11q13.3 (BCL1,FGF4) | 7 | 16 |
| 2q34 (ERBB4) | 6 | 14 |
| 11q13 (CCND1) | 6 | 14 |
| 7q22.1 (Smurf1) | 6 | 14 |
| 8q21 (NBS1) | 5 | 11 |
| 2p16 (GTBP) | 5 | 11 |
| 7p22 (ETV1) | 5 | 11 |
| 2q21.2 (LRP1B) | 5 | 11 |
| 2q35 (HUP2) | 5 | 11 |
| 6q22 (ROS1) | 5 | 11 |
| 8p11.2‐p11.1 (FGFR1) | 5 | 11 |
| 7q22.1 (CYP3A4) | 5 | 11 |
| 7q22.1 (TRRAP) | 5 | 11 |
Known cancer‐related genes contained in the respective clones are shown in parentheses.
Association of SNAP with clinicopathological parameters. A number of clinicopathological parameters were associated with the degree and type of aberration (Suppl. Table 5). Overall, cases with a phenotype indicating increased malignant potential had a higher degree of aberrations. Smaller tumors and tumors with higher venous or perineural invasion histological scores had a higher total number of aberrations than tumors that were larger or with lower invasion scores. No other clinicopathological parameters examined, such as the sex, primary tumor location, macroscopic type (infiltrative or nodular), degree of differentiation (Suppl. Table 6), infiltration or otherwise of certain neighboring tissues, pattern of such infiltration (INF α, β, or γ), or spread within the main pancreatic duct had significant correlation with SNAP (data not shown).
Association with venous invasion. Venous invasion‐negative tumors had markedly different SNAP than venous invasion‐positive tumors, although it should be noted that only a small number of negative tumors was examined. The loci lost or gained more frequently in the venous invasion‐positive tumors are shown in Table 3. HD were not observed in the venous invasion‐negative tumors (0/5 vs 19/37, P = 0.03). In the venous invasion‐negative tumors, 178 and 84 loci, respectively, were lost or gained more frequently than in the positive tumors; these included frequently amplified loci (3/5) such as the ones containing FGF7 (15q13‐q22), BRAF (7q34) (P ≤ 4.1 × 10−5), ROS1 (6q22), GTBP (2p16) (P = 0.0004) and HGF (7q21.1) (P = 0.02).
Table 3.
Loci altered frequently in the venous invasion‐positive pancreatic adenocarcinomas
| Chromosomal locus | Contained cancer‐related gene | Sub‐chromosomal loss detected | P value† | |||
|---|---|---|---|---|---|---|
| Venous invasion‐positive cases | Venous invasion‐negative cases | |||||
| n | % | n | % | |||
| 19p13.3 | ABCA7 | 26 | 70 | 0 | 0 | 0.002 |
| 9q34.3 | ABCA2 | 25 | 68 | 1 | 20 | 0.040 |
| 1p36.33 | TP73 | 22 | 59 | 0 | 0 | 0.012 |
| 11q13 | FGF3 | 22 | 59 | 0 | 0 | 0.012 |
| 4p16 | GAK | 20 | 54 | 0 | 0 | 0.023 |
| 11q12 | LTBP3 | 20 | 54 | 0 | 0 | 0.023 |
| 20q13 | Livin | 20 | 54 | 0 | 0 | 0.023 |
| 18q22 | BCL2 | 19 | 51 | 0 | 0 | 0.030 |
| 5p14.2 | CDH10 | 25 | 68 | 0 | 0 | 0.004 |
| 8q24 | OPG | 21 | 57 | 0 | 0 | 0.017 |
| 3q27‐q29 | TP63 | 20 | 54 | 0 | 0 | 0.023 |
| 8q24.1 | NOV | 20 | 54 | 0 | 0 | 0.023 |
| 14q22.3 | RBBP1 | 19 | 51 | 0 | 0 | 0.030 |
χ2 test.
Association with lymph node metastasis. Unlike venous invasion, few differences were observed when the genomic profiles of lymph node metastasis‐positive and lymph node metastasis‐negative tumors were compared, although only six negative tumors were examined. Only three loci showed significant differences in their signal ratios, 9p13 (SCYA21), 11q22 (ATM) and 17q12 (RAD51L3). Xq28 (MAGEA2) was lost more frequently in the lymph node metastasis‐negative group (3/6 vs 5/36, P = 0.037). Four loci, all on 7q21‐22 (containing the HGF, DMTF1, MLL5 and CDK6 genes), were gained more frequently in the lymph node metastasis‐positive group (all P < 0.05).
Association with survival. Thirty‐two cases had survival data amenable to analysis. The genomic profiles of cases with a survival period shorter (n = 13) or longer than (n = 19) 1 year were compared. Four and 13 loci, respectively, were lost or gained more frequently in the short‐compared with the long‐survival group (Table 4). In contrast, only two loci were lost (6q25/ESR1 and 22q11.23/ADRBK2) and none gained more frequently in the long‐compared with the short‐survival group. Loss of 1p36 (p73) and 11q121‐3 was associated with both short‐term survival and evidence of venous invasion, whereas gain of 7q21‐22 was associated with both short‐term (<3 years) survival and the presence of lymph node metastases.
Table 4.
Loci altered frequently in pancreatic adenocarcinoma cases with short‐survival (<1 year) compared with long‐survival periods
| Chromosomal locus | Contained cancer‐ related gene | Sub‐chromosomal loss detected | P value† | |||
|---|---|---|---|---|---|---|
| Venous invasion‐positive cases | Venous invasion‐negative cases | |||||
| n | % | n | % | |||
| 1p36.33 | TP73 | 9 | 69 | 4 | 21 | 0.006 |
| 8q24.3 | GLI4 | 5 | 38 | 1 | 5 | 0.018 |
| Xq12 | AR | 5 | 38 | 1 | 5 | 0.018 |
| 11q13 | STIP1, FOLR1 | 8 | 62 | 5 | 26 | 0.046 |
| 20q11.2 | LUNX, TOP1 | 5 | 38 | 0 | 0 | 0.003 |
| 18p11.3 | TGIF | 6 | 46 | 1 | 5 | 0.006 |
| 4q13‐q21 | AREG | 4 | 31 | 0 | 0 | 0.010 |
| 6q21 | CCNC | 4 | 31 | 0 | 0 | 0.010 |
| 10q21.1 | PCDH15 | 10 | 77 | 6 | 32 | 0.012 |
| 1p32 | RLF | 5 | 38 | 1 | 5 | 0.018 |
| 2q36 | Cul3 | 8 | 62 | 4 | 21 | 0.020 |
| 17q23 | PPM1D | 8 | 62 | 4 | 21 | 0.020 |
| 4q21 | GRO1 | 6 | 46 | 2 | 11 | 0.022 |
| 1p36.2 | KIAA0591(KIF1B) | 7 | 54 | 3 | 16 | 0.023 |
| 4q21 | GRO2 | 7 | 54 | 3 | 16 | 0.023 |
| 13q32 | GPC5 | 7 | 54 | 3 | 16 | 0.023 |
| 8q22‐q23 | EIF3S6 | 9 | 69 | 6 | 32 | 0.036 |
χ2 test.
Kaplan–Meier analysis showed that loss of 1p36 (p73) (P = 0.02; Fig. 3a), gain of 17q23 (PPM1D) (P < 0.05; Fig. 3b) and particularly gain of the LUNX locus at 20q111‐12 (P < 0.0001; Fig. 3c) were significantly associated with prognosis, whereas loss of the STIP1 or FOLR1 locus (11q13), gain of the TOP1 (20q11‐12) and gain of MUC3 or Smurf1 loci (7q21‐22) were not. Loci adjacent to LUNX on 20q11 were further analyzed; gain of the HCK (P < 0.001; Fig. 3d), E2F1 (P < 0.005; data not shown) and DNMT3b loci (P < 0.05; data not shown), but not TGIF2, were also associated with prognosis, albeit not as closely as LUNX.
Figure 3.
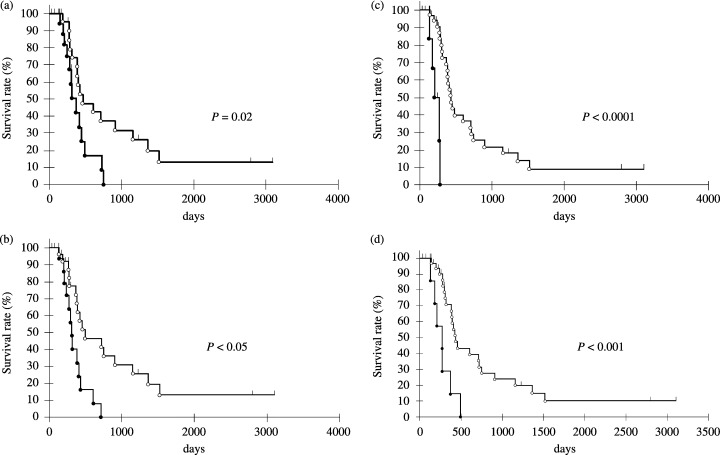
Overall survival rate of pancreatic cancer patients according to the absence or presence of chromosomal abnormalities. (a) Overall survival rates of cases with chromosomal loss of the p73 locus on 1p36 (indicated as black dots) and cases without such loss (indicated as white dots). (b) Overall survival rates of cases with chromosomal gain of the PPM1D locus on 17q23 (indicated as black dots) and cases without such gain (indicated as white dots). Overall survival rates of cases with chromosomal gain of the (c) LUNX and (d) HCK loci on 20q11 (indicated as black dots) and cases without such gain (indicated as white dots). Survival curves were calculated by the Kaplan–Meier method.
Potential oncogenes revealed by expression profiling analysis. Eighty‐one loci were amplified in at least one case in the group examined; these loci contained 15 genes that were overexpressed in at least one case (Table 5). Of the individual amplifications observed, 14.7% (20/136) resulted in overexpression. Only four genes were amplified and overexpressed in more than one case: Smurf1 (7q22.1), BCAS1 (20q13.2‐3), which was the most frequently overexpressed, VCL (10q22.1) and TRRAP (7q22.1) (Table 5). Genes that were contained in loci frequently amplified but not overexpressed included RBBP8 (18q11.2), LRP1B (2q21.2) and HGF (7q21.1). It should be noted that Smurf1 protein overexpression was also detected in pancreatic cancer clinical samples, as part of a separate study (F. Suzuki, T. Shibata, S. Hirohashi, J. Inazawa, I. Imoto, unpublished data).
Table 5.
Correlation of amplification with overexpression in pancreatic cancer genes both amplified and overexpressed in at least one xenograft
| Gene | Locus | Amplified cases (%) | Overexpressed cases (%) | Amplified and overexpressed in the same case (%) | Amplified or gained and overexpressed in the same case (%) |
|---|---|---|---|---|---|
| Smurf1 | 7q22.1 | 36 | 25 | 25 | 25 |
| BCAS1 | 20q13.2‐q13.3 | 18 | 83 | 17 | 33 |
| VCL | 10q22.1 | 18 | 33 | 17 | 17 |
| TRRAP | 7q22.1 | 36 | 17 | 17 | 17 |
| SRI | 7q21.1 | 9 | 83 | 8 | 33 |
| Cul3 | 2q36 | 27 | 42 | 8 | 33 |
| TPD52 | 8q21 | 9 | 42 | 8 | 33 |
| EFNB2 | 13q33 | 9 | 83 | 8 | 17 |
| PDAP1 | 7q22 | 27 | 17 | 8 | 17 |
| ZNF217 | 20q13 | 9 | 25 | 8 | 17 |
| PLAU | 10q24 | 18 | 17 | 8 | 8 |
| WHSC1 | 4p16.3 | 9 | 17 | 8 | 8 |
| CDK4 | 12q14 | 9 | 17 | 8 | 8 |
| CYP3A4 | 7q22.1 | 36 | 8 | 8 | 8 |
| CCNE1 | 19q11 | 18 | 8 | 8 | 8 |
| OPG | 8q24 | 9 | 33 | 0 | 33 |
| BARD1 | 2q34 | 9 | 25 | 0 | 25 |
| ELE1,MSMB | 10q11.2 | 9 | 42 | 0 | 17 |
| RAP1B | 12q14 | 9 | 25 | 0 | 17 |
| KRAS2 | 12p12.1 | 18 | 17 | 0 | 17 |
| DHFR,MSH3 | 5q11.2‐q13.2 | 9 | 17 | 0 | 17 |
| TPR | 1q25 | 9 | 17 | 0 | 8 |
| MLL5 | 7q22.3 | 27 | 8 | 0 | 8 |
| SSXT | 18q11.2 | 27 | 8 | 0 | 8 |
| PEG10 | 7q21.3 | 9 | 8 | 0 | 8 |
| NBS1 | 8q21 | 9 | 8 | 0 | 8 |
The expression levels of genes on 20q11 were examined in more detail, because of the close association of four loci on 20q11 with survival. Eleven genes on 20q11 (BLCAP, RALY, GSS, ID1, NCOA6, TPX2, COX4I2, EPB41L1, BCL2L1, DNCL2A, CTNNBL1) were overexpressed. BCL2L1 (or BCL‐x1) expression was also associated with lymph node metastasis of the xenografted tumors in mice (data not shown). It should be noted that two adjacent loci, TNFRSF6B and ZNF217 (20q13), were amplified in three cases each.
Discussion
This study represents the first genome‐wide analysis of the subchromosomal numerical aberration profile (here designated SNAP) of a substantial number of pancreatic cancer cases by aCGH and is the first to establish its relationship with particular clinicopathological parameters of known prognostic value. It examined a number of primary tumors large enough to exclude randomly observed alterations from being considered as likely candidates, as would be the case in smaller‐scale studies. In all previous studies except one,( 29 ) case selection was based on the exclusion of samples that did not possess a high degree of neoplastic cellularity, which translates to a high copy number ratio error probability. In the present study, tumors were subjected to LCM so as to exclude non‐tumor DNA from the analysis and thus increase both the number of available cases and the accuracy of the derived copy number ratio.
One of the striking findings of the study was the wide range in the number and pattern of aberrations observed between cases. Whereas many cases showed few aberrations and two had none whatsoever, 20% of cases showed alterations in more than 50% of loci examined. Importantly, the loss rate and range reported here (17%, 0–46%) is in very close agreement with the one reported in a comprehensive genome‐wide allelic loss study of pancreatic cancer (15%, 1.5–32%).( 30 ) It should be noted that it was not possible to know whether alterations of adjacent loci represented single amplicons or losses or whether they were independent events. Our results indicate that in the majority of pancreatic adenocarcinomas genomic instability occurs at the subchromosomal level, affecting a varying but large number of genes, and suggests the presence of multiple or complementary patterns of tumor evolution. Based on the association of SNAP with clinicopathological parameters revealed here, it is fair to assume that some of these aberrations contribute to tumor progression whereas others are the result of it. For the remaining cases showing a low SNAP or absence of aberrations, alternative mechanisms leading to tumor progression may be in place, such as DNA methylation or mismatch repair system aberrations, mutations or small deletions, or chromosomal translocations and rearrangements not accompanied by numerical aberrations. These mechanisms may act in a way complementary to that of numerical aberrations in pancreatic carcinogenesis, so that in cases with high or low SNAP the above‐mentioned mechanisms would be expected to play a minor role whereas other alternative mechanisms would be expected to play a minor or major role respectively.
A number of clinicopathological parameters was associated with SNAP, including, importantly, survival probability. Overall, cases with a phenotype indicating increased malignant potential had a higher SNAP. Specific loci, the loss or gain of which is associated with particular clinicopathological characteristics, were identified and are delineated in detail in the results section. Although only a small number of negative tumors was examined, it is noteworthy that loci associated with venous invasion were different from those associated with lymph node metastasis. Our results therefore appear to indicate that invasiveness and metastatic ability result from diverse and distinct molecular mechanisms in pancreatic cancer.
Despite the aforementioned genomic complexity, we identified genes the copy number status of which is associated with survival and may therefore be of prognostic value. Gains of the LUNX (20q11.2), AREG (4q13‐q21) and CCNC (6q21) loci were detected exclusively in the short‐survival group. Loss of 1p36 (p73) and 11q12‐13 was associated with both short‐term survival and evidence of venous invasion, whereas gain of 7q21‐22 was associated with both short‐term survival and the presence of lymph node metastases. Combining the above observations, we identified candidates most likely to yield clinically relevant results. A strong association was revealed between the copy number status of a number of loci at 20q11 and prognosis, mainly concerning the LUNX (PLUNC) locus (P < 0.0001) but also including adjacent loci containing HCK, E2F1 and DNMT3b. LUNX is upregulated and has been proposed as a marker for detection of micrometastases in non‐small‐cell lung cancer.( 31 ) E2F1 activates the transcription of genes that encode proteins necessary for DNA replication, and is deregulated in most tumors.( 32 ) DNMT3b may contribute to tumorigenesis by improper de novo methylation and silencing of the promoters of growth‐regulatory genes, and its expression may be of clinical significance in breast cancer.( 33 ) Although our data refer to loci rather than individual genes, the significance of the copy aberrations of the above loci has not been described previously in pancreatic or other cancers. Two loci on 20q13 were amplified in three cases each, whereas a further 12 genes on 20q11, including BCL2L1, were overexpressed. BCL2L1 is a BCL2‐independent apoptosis regulator located in close proximity to LUNX. Its overexpression has already been linked to short survival times in pancreatic cancer( 34 , 35 ) and other malignancies, and was also found to be associated with lymph node metastasis in the present study. Amplification and overexpression of BCL10 and BCL6 were also recently described in pancreatic carcinoma.( 15 ) The 11q13.3 locus, containing another BCL family member, BCL1, was found to be amplified frequently in our study, which together with our findings on BCL2L1 described above may indicate a role for the BCL family in pancreatic carcinogenesis. The BCL2L1 overexpression and association with the metastatic phenotype may partially explain the effect the 20q11 region copy number status has on survival. However, we tend to think, in agreement with a similar proposal,( 36 ) that our findings are more indicative of the fact that many (but not all) genes collectively confer selective advantage, in varying degrees of involvement, within the 20q11 region.
Loss of 1p36 (p73) and gain of 17q23 (PPM1D) were also significantly associated with prognosis. As mentioned earlier, 1p36/p73 loss was also associated with evidence of venous invasion in our study. p73, like its homolog p53, is able to induce apoptosis and has been reported to predict clinical outcome in bladder cancer.( 37 ) PPM1D amplification abrogates p53 tumor‐suppressor activity. PPM1D is located within one of the most commonly amplified regions in breast cancer.( 38 ) Gain of 17q21‐q24 has also been associated with poor prognosis in ovarian clear cell adenocarcinomas, in which both PPM1D and APPBP2 were identified as likely amplification targets,( 39 ) but, like p73, the PPM1D locus has not been previously reported to be of prognostic significance in pancreatic cancer.
Examination of the association between SNAP and expression provided a satisfactory filter for candidate genes. Only 15 of the 81 loci amplified and 14.7% (20/136) of individual amplifications observed contained genes that were overexpressed concurrently. This concordance level lies between those observed in breast cancer( 40 , 41 ) and colon cancer,( 42 ) in which 44–62% and 4%, respectively, of genes showing amplifications were overexpressed. It is, however, significantly lower than the one recently reported for pancreatic cancer cell lines, in which 60% of the genes within highly amplified genomic regions displayed associated overexpression,( 14 ) a discrepancy that may partially be explained by the different source used (primary tumors vs cell lines) and the fact that we examined loci rather than genes. More than one target gene was overexpressed in some amplicons in our study, a finding not in disagreement with the above study.( 14 ) We identified four genes contained in loci that were amplified and that were overexpressed recurrently: Smurf1 and TRRAP, both at 7q22.1, BCAS1 (20q13.2‐3), and VCL (10q22.1). Smurf1 acts as a negative regulator of transfroming growth factor β signaling.( 43 ) It was amplified in six cases overall and overexpressed concurrently in four. Although, as mentioned, gain of the Smurf1 locus was not associated with poor prognosis, 7q21‐22 gain was associated with the presence of lymph node metastasis and was detected significantly more frequently in the short‐term (<3 years) survival group. TRRAP is an essential cofactor for both the c‐Myc and E1A/E2F oncogenic transcription factor pathways and interacts specifically with the E2F‐1 transactivation domain. Its inclusion among the four genes both amplified and overexpressed lends further support to the association between E2F1 gain and poor survival revealed here. The fact that Smurf1 and TRRAP are amplified in pancreatic cancer was reported recently, albeit only in cell lines.( 16 ) We show that amplifications of these genes also occurs in primary tumors and that they are recurrently accompanied by overexpression, therefore presenting as very likely novel oncogenes in pancreatic cancer. BCAS1 (20q13), reported to be amplified and overexpressed in breast cancer,( 44 ) was the most frequently overexpressed gene among the ones contained in loci recurrently amplified, and may therefore have a similar role in pancreatic cancer; 20q13 was also one of the most frequently amplified loci in a recent aCGH study on pancreatic cancer.( 14 , 15 ) Finally, 10q22‐24 contained another novel candidate, vinculin, an intracellular protein with a crucial role in the maintenance and regulation of cell adhesion and migration.( 45 ) KRAS2 and 20 other genes have recently been identified as potential target genes on 12p.( 36 ) This finding is in partial agreement with our study, in which five loci on 12p were amplified and KRAS2 was amplified in two cases.
Numerous recurrent, non‐random, patterns of subchromosomal aberrations have emerged through our analysis. Thirty‐three loci with frequent losses (>50% cases) were identified. The chromosome arms found to contain the highest number of loci lost are in agreement with allelic loss( 30 ) and chromosome CGH studies, with the additional detection of losses at 5p, 8q, 9q, 11p, 16p and 20q. The most frequently lost loci were: 17p13.3 (in 75% of cases), containing ABR, a multifunctional cellular signaling regulator and a putative TSG in medulloblastoma, 18qtel (CTDP1, SHGC‐145820, 68%) and 18q21 (66%), containing SMAD7, a member of the SMAD family, although all three are close to either the p53 or the DPC4 locus. HD were detected in 25% of cases, affecting 26 loci. The 1p353‐6.33 region contained the highest number of loci deleted (six) or lost (16). The most frequently deleted locus was 9p21 spanning the p16 gene, the inactivation of which is known to play an established role in pancreatic carcinogenesis. The above regions (17p, 18q and 1p353‐6) have been reported previously to show frequent loss.( 19 ) The most frequently gained locus was 7q21.1 (71%) containing the HGF gene, which encodes a cytokine involved in initiating cell migration. Some regions in which gains were observed frequently, at 6p21 (2 loci), 11q22 (7 loci) 12p12 (three loci) and 17q12 (Suppl. Table 4), have been previously proposed as novel amplicons.( 16 ) Sixteen loci were amplified frequently (>10%), although, again, the possibility of another gene being amplified within these loci cannot be excluded. Two of the most frequently amplified loci were on 11q13, in agreement with a report by Holzmann et al. on 13 pancreatic cancer cell lines and six primary tumors.( 15 )
Novel loci likely to play important roles in pancreatic carcinogenesis and in the acquisition of certain malignant phenotypes were identified. Genes associated with prognosis or established histopathological indicators of malignancy, or showing both numerical aberrations and overexpression, may represent novel oncogenes. The copy number alterations of the p73 and PPM1D loci, the 20q11 region, including LUNX, and the loci amplified that contained genes concurrently overexpressed, particularly Smurf1, shown here may be of great importance for predicting clinical outcomes and setting new therapeutic targets in pancreatic cancer but will require prospective studies in order to be firmly established.
Supporting information
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We thank T. Sakiyama for helping with the aCGH data analysis, T. Kondo for advice on statistical analysis, and Y. Arai, S. Uryu and Y. Kuwabara for advice on the hybridization technique. This study was supported in part by a Grant‐in‐Aid for the Second Term Comprehensive 10‐Year Strategy for Cancer Control from the Ministry of Health, Labor and Welfare of Japan; the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NiBio), Japan; and by a Grant‐in‐Aid from CREST of JST. P. L. was a recipient of a Research Fellowship from the Program for Invitation of Foreign Researchers from the Foundation for Promotion of Cancer Research in Japan. H. K. was a recipient of a Research Resident Fellowship from the Foundation for Promotion of Cancer Research.
References
- 1. Conlon KC, Klimstra DS, Brennan MF. Long‐term survival after curative resection for pancreatic ductal adenocarcinoma. Clinicopathologic analysis of 5‐year survivors. Ann Surg 1996; 223: 273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Torrisani J, Buscail L. Molecular pathways of pancreatic carcinogenesis. Ann Pathol 2002; 22: 349–55. [PubMed] [Google Scholar]
- 3. Kallioniemi A, Kallioniemi OP, Sudar D et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992; 258: 818–21. [DOI] [PubMed] [Google Scholar]
- 4. Johansson B, Bardi G, Heim S et al. Nonrandom chromosomal rearrangements in pancreatic carcinomas. Cancer 1992; 69: 1674–81. [DOI] [PubMed] [Google Scholar]
- 5. Bardi G, Johansson B, Pandis N et al. Karyotypic abnormalities in tumours of the pancreas. Br J Cancer 1993; 67: 1106–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hahn SA, Seymour AB, Hoque AT et al. Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res 1995; 55: 4670–5. [PubMed] [Google Scholar]
- 7. Armengol G, Knuutila S, Lluis F, Capella G, Miro R, Caballin MR. DNA copy number changes and evaluation of MYC, IGF1R, and FES amplification in xenografts of pancreatic adenocarcinoma. Cancer Genet Cytogenet 2000; 116: 133–41. [DOI] [PubMed] [Google Scholar]
- 8. Griffin CA, Hruban RH, Morsberger LA et al. Consistent chromosome abnormalities in adenocarcinoma of the pancreas. Cancer Res 1995; 55: 2394–9. [PubMed] [Google Scholar]
- 9. Mahlamaki EH, Hoglund M, Gorunova L et al. Comparative genomic hybridization reveals frequent gains of 20q, 8q, 11q, 12p, and 17q, and losses of 18q, 9p, and 15q in pancreatic cancer. Genes Chromosomes Cancer 1997; 20: 383–91. [DOI] [PubMed] [Google Scholar]
- 10. Schleger C, Arens N, Zentgraf H, Bleyl U, Verbeke C. Identification of frequent chromosomal aberrations in ductal adenocarcinoma of the pancreas by comparative genomic hybridization (CGH). J Pathol 2000; 191: 27–32. [DOI] [PubMed] [Google Scholar]
- 11. Heidenblad M, Jonson T, Mahlamaki EH et al. Detailed genomic mapping and expression analyses of 12p amplifications in pancreatic carcinomas reveal a 3.5‐Mb target region for amplification. Genes Chromosomes Cancer 2002; 34: 211–23. [DOI] [PubMed] [Google Scholar]
- 12. Gorunova L, Hoglund M, Andren‐Sandberg A et al. Cytogenetic analysis of pancreatic carcinomas: intratumor heterogeneity and nonrandom pattern of chromosome aberrations. Genes Chromosomes Cancer 1998; 23: 81–99. [DOI] [PubMed] [Google Scholar]
- 13. Heidenblad M, Schoenmakers EF, Jonson T et al. Genome‐wide array‐based comparative genomic hybridization reveals multiple amplification targets and novel homozygous deletions in pancreatic carcinoma cell lines. Cancer Res 2004; 64: 3052–9. [DOI] [PubMed] [Google Scholar]
- 14. Aguirre AJ, Brennan C, Bailey G et al. High‐resolution characterization of the pancreatic adenocarcinoma genome. Proc Natl Acad Sci USA 2004; 101: 9067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holzmann K, Kohlhammer H, Schwaenen C et al. Genomic DNA‐chip hybridization reveals a higher incidence of genomic amplifications in pancreatic cancer than conventional comparative genomic hybridization and leads to the identification of novel candidate genes. Cancer Res 2004; 64: 4428–33. [DOI] [PubMed] [Google Scholar]
- 16. Bashyam MD, Bair R, Kim YH et al. Array‐based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia 2005; 7: 556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gysin S, Rickert P, Kastury K, McMahon M. Analysis of genomic DNA alterations and mRNA expression patterns in a panel of human pancreatic cancer cell lines. Genes Chromosomes Cancer 2005; 44: 37–51. [DOI] [PubMed] [Google Scholar]
- 18. Mahlamaki EH, Kauraniemi P, Monni O, Wolf M, Hautaniemi S, Kallioniemi A. High‐resolution genomic and expression profiling reveals 105 putative amplification target genes in pancreatic cancer. Neoplasia 2004; 6: 432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nowak NJ, Gaile D, Conroy JM et al. Genome‐wide aberrations in pancreatic adenocarcinoma. Cancer Genet Cytogenet 2005; 161: 36–50. [DOI] [PubMed] [Google Scholar]
- 20. Loukopoulos P, Kanetaka K, Takamura M, Shibata T, Sakamoto M, Hirohashi S. Orthotopic transplantation models of pancreatic adenocarcinoma derived from cell lines and primary tumors and displaying varying metastatic activity. Pancreas 2004; 29: 193–203. [DOI] [PubMed] [Google Scholar]
- 21. Japan Pancreas Society. Classification of Pancreatic Carcinoma, 2nd English edn. Tokyo: Kanehara and Co., 2003. [Google Scholar]
- 22. Tanabe C, Aoyagi K, Sakiyama T et al. Evaluation of a whole‐genome amplification method based on adaptor‐ligation PCR of randomly sheared genomic DNA. Genes Chromosomes Cancer 2003; 38: 168–76. [DOI] [PubMed] [Google Scholar]
- 23. Sonoda I, Imoto I, Inoue J et al. Frequent silencing of low density lipoprotein receptor‐related protein 1B (LRP1B) expression by genetic and epigenetic mechanisms in esophageal squamous cell carcinoma. Cancer Res 2004; 64: 3741–7. [DOI] [PubMed] [Google Scholar]
- 24. Takada H, Imoto I, Tsuda H et al. Screening of DNA copy‐number aberrations in gastric cancer cell lines by array‐based comparative genomic hybridization. Cancer Sci 2005; 96: 100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peng W‐X, Shibata T, Katoh H et al. Array‐based comparative genomic hybridization analysis of high‐grade neuroendocrine tumors of the lung. Cancer Sci 2005; 96: 661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Katoh H, Shibata T, Kokubu A et al. Genetic profile of hepatocellular carcinoma revealed by array‐based comparative genomic hybridization: Identification of genetic indicators to predict patient outcome. J Hepatol 2005; 43: 863–74. [DOI] [PubMed] [Google Scholar]
- 27. Yamazaki K, Sakamoto M, Ohta T, Kanai Y, Ohki M, Hirohashi S. Overexpression of KIT in chromophobe renal cell carcinoma. Oncogene 2003; 22: 847–52. [DOI] [PubMed] [Google Scholar]
- 28. Tanno S, Mitsuuchi Y, Altomare DA, Xiao GH, Testa JR. AKT activation up‐regulates insulin‐like growth factor I receptor expression and promotes invasiveness of human pancreatic cancer cells. Cancer Res 2001; 61: 589–93. [PubMed] [Google Scholar]
- 29. Kitoh H, Ryozawa S, Harada T et al. Comparative genomic hybridization analysis for pancreatic cancer specimens obtained by endoscopic ultrasonography‐guided fine‐needle aspiration. J Gastroenterol 2005; 40: 511–17. [DOI] [PubMed] [Google Scholar]
- 30. Iacobuzio‐Donahue CA, Van Der Heijden MS, Baumgartner MR et al. Large‐scale allelotype of pancreaticobiliary carcinoma provides quantitative estimates of genome‐wide allelic loss. Cancer Res 2004; 64: 871–5. [DOI] [PubMed] [Google Scholar]
- 31. Iwao K, Watanabe T, Fujiwara Y et al. Isolation of a novel human lung‐specific gene, LUNX, a potential molecular marker for detection of micrometastasis in non‐small‐cell lung cancer. Int J Cancer 2001; 91: 433–7. [DOI] [PubMed] [Google Scholar]
- 32. Phillips AC, Ernst MK, Bates S, Rice NR, Vousden KH. E2F‐1 potentiates cell death by blocking antiapoptotic signaling pathways. Mol Cell 1999; 4: 771–81. [DOI] [PubMed] [Google Scholar]
- 33. Girault I, Tozlu S, Lidereau R, Bieche I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin Cancer Res 2003; 9: 4415–22. [PubMed] [Google Scholar]
- 34. Friess H, Lu Z, Andren‐Sandberg A et al. Moderate activation of the apoptosis inhibitor bcl‐xL worsens the prognosis in pancreatic cancer. Ann Surg 1998; 228: 780–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ghaneh P, Kawesha A, Evans JD, Neoptolemos JP. Molecular prognostic markers in pancreatic cancer. J Hepatobiliary Pancreat Surg 2002; 9: 1–11. [DOI] [PubMed] [Google Scholar]
- 36. Heidenblad M, Lindgren D, Veltman JA et al. Microarray analyses reveal strong influence of DNA copy number alterations on the transcriptional patterns in pancreatic cancer: implications for the interpretation of genomic amplifications. Oncogene 2005; 24: 1794–801. [DOI] [PubMed] [Google Scholar]
- 37. Matsumoto H, Matsuyama H, Fukunaga K, Yoshihiro S, Wada T, Naito K. Allelic imbalance at 1p36 may predict prognosis of chemoradiation therapy for bladder preservation in patients with invasive bladder cancer. Br J Cancer 2004; 91: 1025–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li J, Yang Y, Peng Y et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet 2002; 31: 133–4. [DOI] [PubMed] [Google Scholar]
- 39. Hirasawa A, Saito‐Ohara F, Inoue J et al. Association of 17q21‐q24 gain in ovarian clear cell adenocarcinomas with poor prognosis and identification of PPM1D and APPBP2 as likely amplification targets. Clin Cancer Res 2003; 9: 1995–2004. [PubMed] [Google Scholar]
- 40. Hyman E, Kauraniemi P, Hautaniemi S et al. Impact of DNA amplification on gene expression patterns in breast cancer. Cancer Res 2002; 62: 6240–5. [PubMed] [Google Scholar]
- 41. Pollack JR, Sorlie T, Perou CM et al. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA 2002; 99: 12 963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Platzer P, Upender MB, Wilson K et al. Silence of chromosomal amplifications in colon cancer. Cancer Res 2002; 62: 1134–8. [PubMed] [Google Scholar]
- 43. Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 1999; 400: 687–93. [DOI] [PubMed] [Google Scholar]
- 44. Collins C, Rommens JM, Kowbel D et al. Positional cloning of ZNF217 and NABC1: genes amplified at 20q13.2 and overexpressed in breast carcinoma. Proc Natl Acad Sci USA 1998; 95: 8703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bakolitsa C, Cohen DM, Bankston LA et al. Structural basis for vinculin activation at sites of cell adhesion. Nature 2004; 430: 583–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item