

Abstract
The extracellular signal‐regulated kinase/mitogen‐activated protein kinase (ERK‐MAPK) is critical in human malignancies. It remained to be established whether DNA methyltransferases (Dnmt) and proliferating cell nuclear antigen (PCNA) involved in DNA methylation during RAF‐transformed cell proliferation. The plasmid of constitutively active RAF was used to transfect gastric cell GES‐1 and cancer cell AGS. RAF promoted cell proliferation, growth in soft agar and induced cell cycle progress faster than empty plasmid by accelerating G1/S transition in both cell lines, a massive induction of cyclin D1 and PCNA expression was observed, along with reduced expression of p16INK4A , p21WAF1 and p27KIP1. Methylation‐specific polymerase chain reaction and bisulfite sequencing showed that the promoter of p16INK4A was methylated in RAF‐transformed cells, treatment with 5‐aza‐dC or PD98059 restored the expression of p16INK4A, increased p21WAF1 and p27KIP1 partially, associated with upregulation of the activity of Dnmt in RAF‐transformed cell GES‐1, and also decreased the hypermethylation status of p16INK4A , but not all CpG islands of p21WAF1 and p27KIP1 . These data suggest that RAF may induce cell proliferation through hypermethylation of tumor suppressor gene p16INK4A , while the epigenetic inactivation of p21WAF1 and p27KIP1 may be not a key factor in RAF‐transformed cells. (Cancer Sci 2009; 100: 117–125)
The human gastric epithelium is in a constant state of self‐renewal, proper development and tissue homeostasis, which is regulated through balance between cell proliferation, differentiation and apoptosis. Any disruption in this balance can lead to cancer.( 1 ) Gastric cancer is one of the most common malignancies worldwide, particularly in East Asian countries such as China, Japan and Korea.
The extracellular‐regulated kinase (ERK) cascade is one of mitogen‐activated protein kinases (MAPK) cascades, it consists of RAS, RAF, MEK1/2 and ERK1/2, mediates diverse extracellular signals from cell cytoplasm into nuclear, by activating the cascades phosphorylation to control cell proliferation and differentiation.( 2 ) B‐RAF mutations have been reported in human malignancies, and B‐RAF protein plays a central role in the RAS/RAF/MEK/ERK pathway. Recently, B‐RAF mutations were detected in 12% of tumors in young patients and in 8% of tumors in older patients.( 3 ) The percentages were high compared to those reported previously (0–2.2%).( 4 ) Alterations to the ERK‐MAPK pathway caused by both RAS and B‐RAF mutations may contribute to the pathogenesis of stomach cancer.
Aberrant methylation of 5′‐CpG islands of tumor suppressor genes and elevated levels of the DNA‐(cytosine‐5) methyltransferases (Dnmt) are considered important epigenetic alterations that are intimately involved in the initiation and development of gastric cancer.( 5 , 6 , 7 ) In particular, hypermethylation in the promoters of tumor suppressor genes correlates with the loss of expression of these genes. Previous work has suggested that activation of oncogenic RAS can silence genes through DNA methylation; in addition, elevated Dnmt levels are required to maintain the phenotype of these RAS‐transformed cells.( 8 , 9 , 10 ) The ERK‐MAPK inhibitor treatment or small interfering RNA‐mediated knockdown of ERK‐MAPK decreases DNA methylation in SW1116 colon cancer cells.( 11 )
Carcinogenesis is a multistep process with morphological progression involving multiple genetic and epigenetic events. Genetic abnormalities of the several tumor suppressor genes, including p15INK4B , p16INK4A /p14ARF , p53, p73, and Rb genes have been reported.( 4 ) The deletions and point mutations of these genes are infrequently detected in gastric cancer, while the epigenetic silence of tumor suppressor genes is now recognized as the common molecular defect in immortalization and cancer. We initiated studies to determine the mechanism by which RAF causes downregulation of tumor suppressor genes, and to assess the effects on cell proliferation and methylation status of DNA.
Materials and Methods
Cell cultures and treatments. Non‐malignant human gastric epithelial immortalized cell line (GES‐1) and gastric cancer cell line (AGS) were cultured. Cells were grown to low density 24 h before treatment and switched to culture medium supplemented with 5‐aza‐2‐deoxycytidine (5‐aza‐dC, Sigma Chemical, St Louis, MO, USA; diluted in acidic phosphate‐buffered saline [PBS], at a concentration of 5 µM), or MEK1/2 inhibitor PD98059 (Promega, Madison, WI, USA; diluted in dimethylsulfoxide [DMSO]), cells were treated with PBS or DMSO as a vehicle control according to the observations of others.( 12 , 13 )
Plasmids and cell transfection. The constitutively active mutant of pCMV‐B‐RAF (membrane‐targeted B‐Raf‐CAAX and N‐terminally truncated Raf‐22 W, constitutively active RAF [caRAF]) (kindly provided by Dr Guan KL, University of Michigan) and empty vector pCMV, were used as a vehicle control to transfect cell, with Superfect Transfection Reagent according to the manufacturer's protocol (Qiagen); then, 800 ng/mL G418 was added to the medium. The clones with upregulated RAF were detected by western blotting.
Protein expression and immunoblotting. Protein concentrations were measured with the BCA method (Pierce, Rockford, IL, USA). Equal amounts of proteins were separated by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS‐PAGE). Proteins were detected immunologically following electro‐transfer onto PVDF membrane (Invitrogen, San Diego, CA, USA). The following antibodies were used: MEK1/2, ERK1/2, phospho‐B‐RAF (Ser445), phospho‐MEK1/2, phosphor‐ERK1/2 (Cell Signaling Technology); Dnmt1, Dnmt3a, Dnmt3b (BD Biosciences); β‐actin (Sigma Chemical); cyclin D1, p27KIP1 p16INK4A (Santa Cruz Biotechnology, Santa Cruz, CA, USA); or p21WAF1 (Calbiochem, Germany). Blots were visualized using the enhanced chemiluminescence (ECL; Amersham Biosciences, Amersham, Buckinghamshire, UK).
DNA methyltransferase activity assay. Cells were grown to subconfluence in 60‐mm dishes and were serum‐starved for 24 h. Nuclear extracts from cells were prepared using EpiQuik Nuclear Extraction Kit I (Epigentek, New York, NY, USA) according to the manufacturer's instructions. Dnmt activity was determined by using an EpiQuik DNA Methyltransferase Activity Assay Kit (Epigentek) according to the manufacturer's instructions. The data are represented as the percentage of the values found for cells without stimulation by mean ± standard deviation (SD) of independent experiments. In each experiment, the basal Dnmt activity was set to 100%.
Cell proliferation and growth assay. Cells were seeded in triplicate in 12‐well plates at a density of 5 × 104 cells/well for normal gastric and 2.5 × 104 cells/well for cancer cells. Cell proliferation was assessed daily by the methyl thiazolyl blue tetrazolium bromide (MTT, 3‐[4,5‐dimethylthiazol‐2‐yl]‐2, 5‐diphenytetra‐zolium; Calbiochem, San Diego, CA, USA) colorimetric method in triplicate; cells were incubated with 0.3 mL MTT dye for 2 h at 37°C; the color absorbance was monitored at 570 nm. Cell growth was performed starting with cell population at 3 weeks postselection and subsequently plated in 12‐well plates at a concentration. Cell growth was measured 7 days for cell population using a cell particle counter. Anchorage‐independent growth was evaluated as described previously.( 14 ) Images of colonies were also taken after staining with MTT (0.5 mg/mL) for 3 h at 37°C.
Cell cycle analysis. Approximately 1 × 108 cells were trypsinized, washed twice with PBS, and fixed in 80% ice‐cold ethanol for 1 h. The samples were then concentrated by removing the ethanol and exposed to 1% (v/v) Triton X‐100 (Sigma Chemical) and 100 µg/mL RNaseA (Sigma Chemical) for 10 min at 37°C. Cellular DNA were stained with propidium iodide. Cell cycle distributions were determined using a flow cytometer (Model FACSCALIBAR, Becton‐Dickinson, Lincoln Park, NJ, USA). Data analysis was performed using the MultiCycle software package (Phoenix, San Diego, CA, USA).
Real‐time reverse transcription polymerase chain reaction (RT‐PCR). Total cell RNA was harvested using the commercial kit (Trizol) according to the manufacturer's instructions (Invitrogen/Gibco BRL, Carlsbad, CA, USA). RT and real‐time PCR were performed using the protocols and reagents from Applied Biosystems Taqman RT reagent kit and SYBR Green PCR master mix as described.( 15 ) Relative quantitation data was obtained using the comparative Ct method with the ABI PRISM 7700 Sequence Detection System (software ver. 1.6) according to the manufacturer's protocol. Primers were provided by Shenyou (Shanghai, China). Sequences of PCR primers are shown in Table 1. Real‐time PCR were also performed with the primers for β‐actin to normalize each of the extracts for amplifiable human DNA. Significant alteration of mRNA expression was defined as a threefold difference in the expression level after treatment relative to that before treatment.( 16 )
Table 1.
Sequences of primers and probes for real‐time polymerase chain reaction
| Gene | Primer (forward) (5′→3′) | Primer (reverse) (5′→3′) | GenBank number |
|---|---|---|---|
| p16INK4A | CATAGATGCCGCGGAAGGT | CAGAGCCTCTCTGGTTCTTTCAA | NM_058197 |
| p21WAF1 | CTGGAGACTCTCAGGGTCGAA | GGATTAGGGCTTCCTCTTGGA | NM_078467 |
| p27KIP1 | GCAGTGTCCAGGGATGAGGA | TCTGTTCTGTTGGCCCTTTTGT | NM_004064 |
| Dnmt1 | GCACCTCATTTGCCGAATACA | TCTCCTGCATCAGCCCAAATA | NM_001379 |
| Dnmt3a | GCCCAAGGTCAAGGAGATTATTG | TCTGCCGCACCTCGTACAC | NM_022552 |
| Dnmt3b | CGACAAGAGGGACATCTCACG | CAGAAACTTTGATGGCATCAATCA | XM_009449 |
| β‐actin | CTGGCACCCAGCACAATG | GGACAGCGAGGCCAGGAT | BC016045 |
Methylation‐specific polymerase chain reaction (MSP) and bisulfite sequencing. Cells were incubated in growth medium supplemented with 2 µM 5‐aza‐dC for 6 days, with 25 µM PD98059 for 24 h. Cells treated with DMSO were used as vehicle controls. Methylation patterns in the CpG island of these genes were assessed by chemical bisulfite modification as described previously.( 17 ) MSP was performed with primers specific for either methylated or the bisulfite modified unmethylated DNA (Table 2). The primers were designed without CpG dinucleotides to enable both methylated and unmethylated alleles to be amplified. DNA treated in vitro with Sss I methyltransferase (New England Biolabs) was used as positive control for methylated DNA and that from normal lymphocytes as negative control unmethylated DNA for PCR with the methylation‐specific primers. Each PCR reaction product was directly loaded onto 3% agarose gels and electrophoresed. To further identify the methylation pattern within the CpG islands of the p16INK4A and p21WAF1 in GES‐1 cells, bisulfite sequencing was performed. Primer sequences of p16INK4A and p21WAF1 annealing temperatures, and expected PCR product sizes are summarized in Table 2. PCR products were sequenced using ABI PRISM 3730 sequencer.
Table 2.
Sequences of primers for methylation‐specific polymerase chain reaction and bisulfite genomic sequence
| Primer | Primer (forward) (5′→3′) | Primer (reverse) (5′→3′) | Product size and Tm | GenBank accession number |
|---|---|---|---|---|
| MSP | ||||
| p16INK4A M‐MSP | TTATTAGAGGGTGGGGCGGATCGC | GACCCCGAACCGCGACCGTAA | 150 bp 65°C | X 94154 |
| p16INK4A U‐MSP | TTATTAGAGGGTGGGGTGGATTGT | CAACCCCAAACCACAACCATAA | 151 bp 60°C | |
| p21WAF1 M‐MSP | TGTAGTACGCGAGGTTTCG | TCAACTAACGCAACTCAACG | 202 bp 53°C | NT_007592 |
| p21WAF1 U‐MSP | TTTTTGTAGTATGTGAGGTTTTGG | AACACAACTCAACACAACCCTA | 200 bp 56°C | |
| p27KIP1 M‐MSP | AAGAGGCGAGTTAGCGT | AAA ACGCCGCCGAACGA | 177 bp 65°C | S 76986 |
| p27KIP1 U‐MSP | ATGGAAGAGGTGAGTTAGT | AAAACCCCA ATTAAAACA | 177 bp 65°C | |
| Bisulfite genomic sequencing | ||||
| p16INK4A (first PCR) | ATTTTAGGGGTGTTATAT | CTACCTAATTCCAATTCCCCTACA | 50°C | |
| p16INK4A (second PCR) | TTTTTAGAGGATTTGAGGGATAGG | TTCCAATTCCCCTACAAA | 384 bp, 50°C | |
| p21WAF1 (first PCR) | AATAGTGTTGTGTTTTTTTGGAGAGT | CAAAAATTCCTATACTTATAATCCC | 57°C | |
| p21WAF1 (second PCR) | GGGAGGAGGGAAGTGTTTTT | ACAACTACTCACACCTCAACT | 251 bp, 52°C | |
Tm, annealing temperature; M‐MSP, methylation‐specific polymerase chain reaction for the methylated allele; U‐MSP, MSP for the unmethylated allele.
Statistics. Data were representative of at least three independent experiments performed in triplicate, and are presented as means ± standard deviation. Comparisons between groups were made using the Student's paired t‐test. Relationships were analyzed by Fisher's exact test using SAS ver. 6.12 software. Asterisks were used to graphically denote statistical significance (P < 0.05) in the figures.
Results
caRAF plasmid transfection increase ERK‐MAPK phosphorylation in GES‐1 and AGS cells. In the absence of serum, high basal levels of ERK1/2 phosphorylation were observed in AGS only; 24 h of serum stimulation slightly increased ERK1/2 phosphorylated in GES‐1, however, the level of ERK1/2 never reached that observation in cancer cells; consistent with this finding, the phosphorylation of MEK1/2 was strong and sustained in AGS relative to GES‐1 (Fig. 1a). Therefore, we concentrated our attention on the ERK‐MAPK signaling cascade, a major mediator of proliferation in many cell types.
Figure 1.
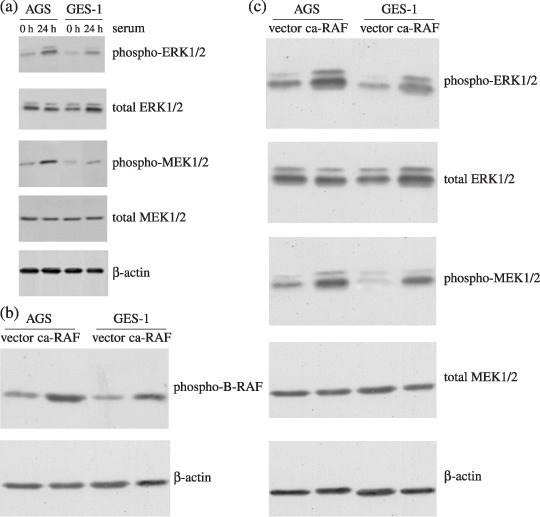
Basal levels of extracellular signal‐regulated kinase/mitogen‐activated protein kinase (ERK‐MAPK) phosphorylation in human gastric epithelial cells and constitutively active RAF (caRAF) transfected cells. (a) The expression of MEK/ERK phosphorylation of cancer cell AGS and epithelial cell GES‐1 after stimulation with serum for 24 h and western blot analysis with antibodies specific for phospho‐MEK1/2 and phospho‐ERK1/2. To control for loading, the membrane was stripped and reblotted with antitotal‐MEK1/2 or antitotal‐ERK1/2 serum. (b) The expression of phospho‐B‐RAF increased in caRAF‐AGS and caRAF‐GES‐1 compared with vehicle control, β‐actin served as a control for loading. (c) The expression of phosphorylated MEK and phosphorylated ERK in caRAF expressing cells.
As shown in Fig. 1(b), the selected populations expressed a high level of phospho‐B‐RAF as determined by western blot analysis. The effect of caRAF on the activities of ERK‐MAPK signaling pathway was assessed using antibody specifically recognizing phosphorylation, the form of kinase activities of MEK1/2 or ERK1/2. Fig. 1(c) shows that caRAF‐AGS and caRAF‐GES‐1 cells exhibited more phosphorylated MEK1/2 or ERK1/2 compared with empty‐expressing cells after normalization of expression levels.
caRAF transfection induces cell proliferation in AGS and GES‐1 cell lines. The control of AGS cell populations grew steadily and reached confluence by day 6 post‐seeding, and then stopped accumulating; caRAF‐AGS cells continued to proliferate and accumulate after confluence day 6 post‐seeding. While the GES‐1 with empty vector grew slowly, they did not reach confluence day 7 post‐seeding, the caRAF‐GES‐1 reached confluence day 6 post‐seeding and then stopped accumulation (Fig. 2a). The OD values of caRAF‐expressing cells were significantly higher than the control cell in both AGS and GES‐1 (Fig. 2b). The range of concentrations effective for PD98059 in the inhibition of phosphorylated MEK was 50 µM (caRAF‐AGS) to 25 µM (caRAF‐GES‐1). Both of them led to a striking inhibition of cell proliferation with a significant reduction of cell viability. GES‐1 with caRAF increased both the number and size of the colonies compared with the empty vector; PD98059 caused proliferation arrest and even a loss of cells. The quantification of transformed GES‐1 cells as foci after 14 days in culture from triplicate plates is shown in Fig. 2(c).
Figure 2.
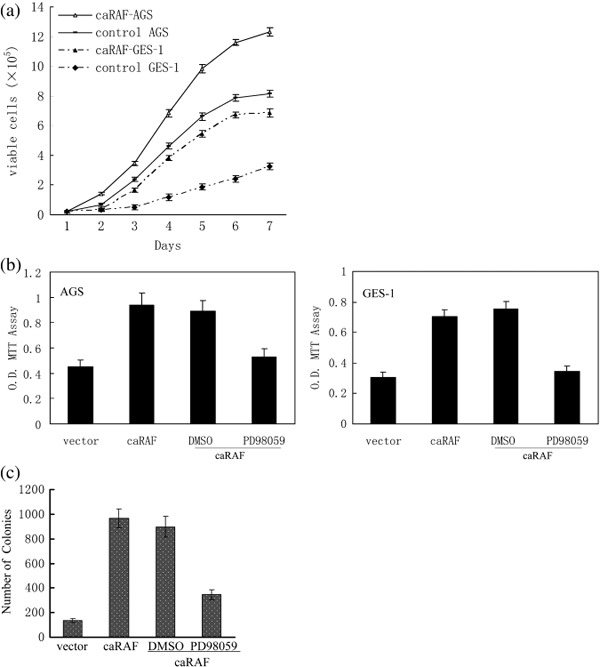
Effects of constitutively active RAF (caRAF) or PD98059 on cell proliferation. Cell proliferation was measured by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenytetra‐zolium) (MTT) assay. caRAF‐GES‐1 and caRAF‐AGS were seeded in 12‐well plates and cultured in complete media. (a) Relative growth rates of these cells were compared for 7 days. (b) The vitality of caRAF‐GES‐1 and caRAF‐AGS, as well as the OD value of both cells treated with PD98059 or dimethylsulfoxide (DMSO) for 48 h. The effective inhibited concentrations of PD98059 for caRAF‐GES‐1 was 25 µM and caRAF‐AGS was 50 µM. (c) Quantification of transformed caRAF‐GES‐1 cells as foci after 14 days in culture from triplicate plates, the data from soft agar assay.
Expression of exogenous caRAF promotes cell cycle progression by shortening of G1 phase. We synchronized AGS and GES‐1 cells transfected caRAF or empty vector with most of cells in the G1 phase (90%); RAF significantly increased in the S‐phase population and decreased in the G0–G1 population, as illustrated in Fig. 3(a,b). Before and after PD98059 treatment, in caRAF‐AGS the percentage of G1/G0 population increased from 48.2% to 60.1%, and the S phase decreased from 41.2% to 27.9%; in the caRAF‐GES‐1 cell, G1/G0 increased from 58.1% to 65.8%, and S phase decreased from 34.8% to 24.2%; the cell cycle distribution was similar to the empty vector without PD98059 (Fig. 3c,d).
Figure 3.
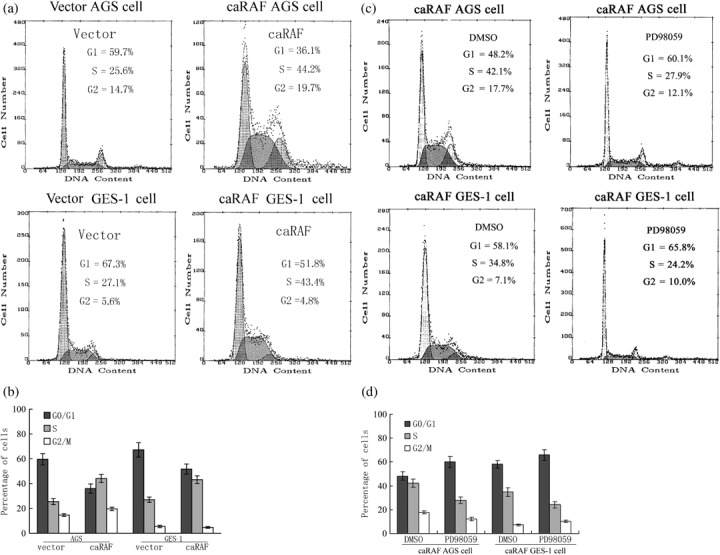
Constitutively active RAF (caRAF) promotes cell cycle progression, and MEK inhibitor causes cell cycle to delay in phases G1 to S. The number in the frame represents the mean ± standard deviation of three independent experiments, Fisher's exact test. (a) Flow cytometry analysis of the distribution of cell cycle phases in GES‐1 and AGS, expressing caRAF or vector. (b) The percentage of cells in each cell cycle phase was quantitated using CellQuest software. (c) Flow cytometry analysis cell cycle distribution of caRAF‐GES‐1 or caRAF‐AGS cells treated with PD98059 or dimethylsulfoxide (DMSO) for 24 h. (d) The percentage of each cell cycle phase in caRAF‐expressing cells with PD98059 or DMSO.
Link between the RAF/MEK/ERK and cell cycle machinery in cell proliferation. Western blot analysis indicated that cyclin D1 was higher expressed in the two cell lines with caRAF than without caRAF; the checkpoint proteins such as proliferating cell nuclear antigen (PCNA) increased in caRAF‐AGS and caRAF‐GES‐1. This suggested that RAF/MEK/ERK pathway control cell proliferation was partly driving cyclin D1 and PCNA expression in gastric cells. The increase of phospho‐MEK1/2 and phospho‐ERK1/2 were detected during conversion of a normal gastric cell into a transformed cell (Fig. 4a). The tumor suppressor genes such as p16INK4A , p21WAF1 and p27KIP1 , the regulators of cell cycle, were higher expressed in gastric normal cells GES‐1, and lower expressd in gastric cancer cells AGS. The expression of p16INK4A, p21WAF1 and p27KIP1 were decreased in caRAF‐GES‐1 cells compared with the vector of GES‐1, in agreement with the levels of mRNA of these CDKI. A similar comparative study was performed for 24 h using caRAF‐GES‐1 cell with PD98059, the protein of CDKI, were upregulated (Fig. 4b,c).
Figure 4.
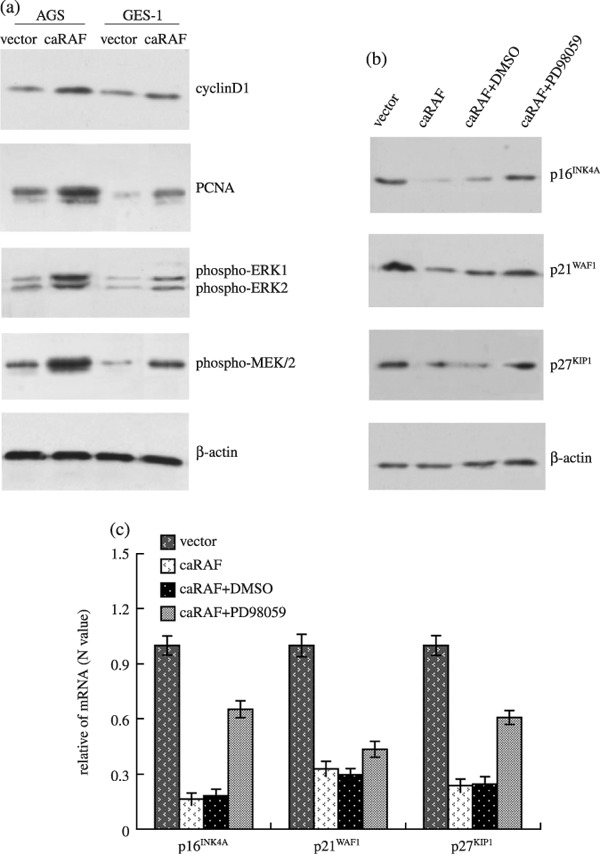
Effects of constitutively active RAF (caRAF) or PD98059 on the expression of tumor associated genes. Equal amount of total protein prepared from caRAF‐GES‐1 and caRAF‐AGS cells, and also from caRAF‐GES‐1 that treated with 25 µM PD98059 for 24 h. (a) caRAF activated MEK and extracellular signal‐regulated kinase (ERK) and increased their phosphorylation levels, as well as proliferative cell nuclear antigen (PCNA), and increased cyclin D1. (b) PD98059 restored the expression of p16INK4A, p21WAF1 and p27KIP1 proteins to a certain degree, which was inhibited by caRAF in GES‐1 cells. (c) The data of p16INK4A , p21WAF1 and p27KIP1 expression in mRNA levels were representative of real‐time reverse transcription polymerase chain reaction experiments. DMSO, dimethylsulfoxide.
RAF downregulates tumor suppressor genes through promoter methylation. Promoter methylation represents one common mechanism of epigenetic gene silencing in human carcinomas.( 18 ) To determine whether loss of tumor suppressor genes p16INK4A , p21WAF1 and p27KIP1 expression are due to DNA methylation, we first treated caRAF‐GES‐1 cells with the azadeoxycytidine demethylating agent, 5‐aza‐dC. As shown in Fig. 5(a), 5‐aza‐dC increased the expression of p16INK4A , p21WAF1 and p27KIP1 mRNA levels compared with those in untreated of caRAF‐GES‐1 cells, and p16INK4A increased significantly (P < 0.05). Next, we examined DNA methylation of these tumor suppressor genes by MSP and DNA sequencing (Fig. 5b); primers that specifically amplified either the methylated or unmethylated form of p16INK4A , p21WAF1 and p27KIP1 promoter produced different methylated bands from mock‐treated GES‐1 cells, indicating that p16INK4A , p21WAF1 and p27KIP1 DNA were present in the unmethylated forms in GES‐1 cells, the methylated forms were observed in caRAF‐GES‐1 cells. We also inhibited RAF signal pathway by PD98059, as shown in Fig. 5(b), there was more unmethylated product than the corresponding methylated product in 5‐aza‐dC or PD98059 treated cells. The methylated forms of p16INK4A promoter were abolished, but the methylated forms of p21WAF1 and p27KIP1 were not abolished with PD98059.
Figure 5.
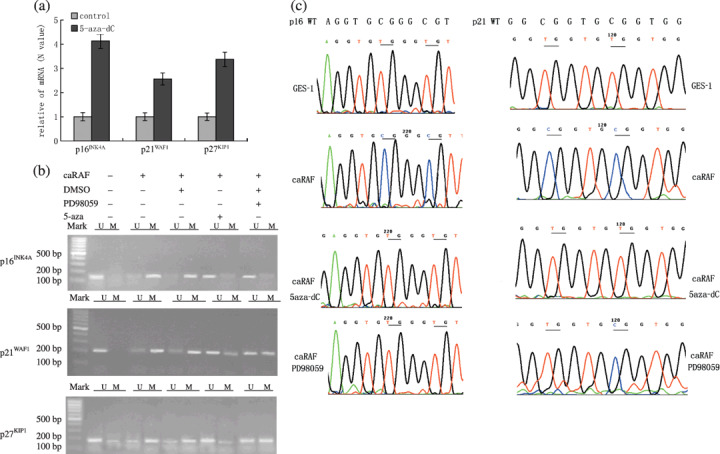
The expressions of tumor suppressor genes are decreased through DNA methylation by constitutively active RAF (caRAF). (a) 5‐aza‐dC restores the mRNA transcription of p16INK4A , p21WAF1 and p27KIP1 in caRAF‐GES‐1. (b) The promoters of p16INK4A , p21WAF1 and p27KIP1 were methylated in caRAF‐GES‐1 cells. Primers of specific for unmethylated (U) or methylation (M) DNA were used to amplify DNA from caRAF‐GES‐1, or treated with 25 µM PD98059, or with 5‐aza‐dC (2 µM). (c) Bisulfite sequencing chromatogram of p16INK4A and p21WAF1 CpGs of p16INK4A in caRAF‐GES‐1 cells show numerous thymidines due to sodium bisulfite conversion of unmethylated cytosines to uracil. Whereas GES‐1 cells (with empty plasmid transfection) or the caRAF‐GES‐1 cells (treatment with 5‐aza‐dC or PD98059) show multiple cytosines due to the resistance of methylated cytosines within CpG dinucleotides to bisulfite conversion. The sequencing of p21WAF1 shows that not all methylated cytosines within CpG dinucleotides are abolished by PD98059. Both sequences are compared to the wild‐type (WT) genomic DNA sequence. DMSO, dimethylsulfoxide.
Next, we used sequencing analysis to confirm the methylation change of p16INK4A and p21WAF1 . CpGs of p16INK4A promoters in GES‐1 cells showed numerous thymidines due to sodium bisulfite conversion of unmethylated cytosines to uracil. caRAF‐GES‐1, however, showed multiple cytosines, due to the resistance of methylated cytosines within CpG dinucleotides to bisulfite conversion. However, caRAF‐GES‐1 cells treated with 5‐aza‐dC or PD98059 showed numerous thymidines, the same as that in GES‐1. However, the sequencing map of p21WAF1 showed that not all methylated cytosines within CpG dinucleotides of p21WAF1 were abolished by PD98059 (Fig. 5c). Both the methylation and demethylation status of p27KIP1 were found in the control cells and caRAF‐GES‐1 cells. PD98059 and 5‐aza‐dC failed to demethylate p27KIP1 (data not shown).
Taken together, these data show that caRAF results in ERK‐MAPK‐dependent downregulation of p16INK4A , which is significantly associated with CpG methylation of promoter.
RAF causes the change of Dnmt activity involved in CpG hypermethylation. Because the hypermethylation of tumor suppressor gene p16INK4A was restored upon treatment with 5‐aza‐dC or PD98059, we assessed whether RAF causes a change of three Dnmt involved in CpG hypermethylation. As shown in Fig. 6(a), caRAF‐GES‐1 cells showed significant increase of mRNA levels in both Dnmt1 and Dnmt3b, but not Dnmt3a. Western blot supported this finding (Fig. 6b). The activity of total Dnmt was elevated by transfection with RAF (216 ± 13% of vector transfection control) in GES‐1 cells, 193 ± 32% in caRAF‐AGS (Fig. 6c). Inhibition of the ERK‐MAPK signal pathway by PD98059 decreased the protein expression and transcript levels of Dnmt1 (Fig. 6a,b). Therefore, caRAF activation might cause an increase in Dnmt responsible for genomic DNA hypermethylation, resulting in silencing of the tumor suppressor gene p16INK4A .
Figure 6.
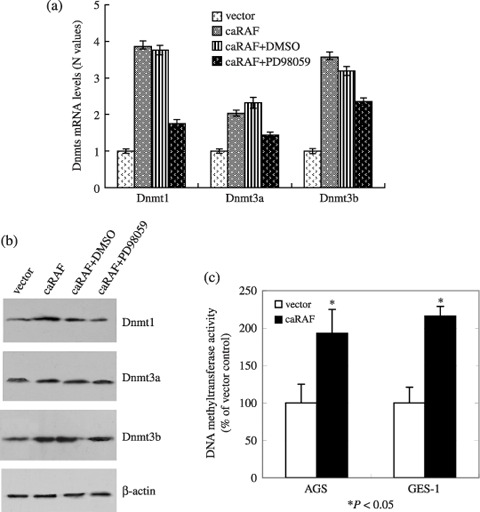
The expressions of Dnmt1 and Dnmt3b in mRNA or protein levels were upregulated in constitutively active RAF (caRAF)‐GES‐1. PD98059 reversed the upregulation of Dnmt1. Total RNA and protein from GES‐1 expressing caRAF or vector only, along with caRAF‐GES‐1 cells treated with 25 µM PD98059. (a) The graphical representation of the Dnmt1, Dnmt3a and Dnmt3b mRNA, data are mean ± standard deviation of triplicates of a representative experiment. Similar results were obtained in two experiments. (b) Cell lysates were used for analyzed by western blot with antibody specific for Dnmt1, Dnmt3a and Dnmt3b, β‐actin serving as a loading control. (c) caRAF can increase the activity of DNA methyltransferase in GES‐1 (216 ± 13%) and AGS (193 ± 32%) cell lines compared with each control. DMSO, dimethylsulfoxide.
Discussion
The ERK‐MAPK (RAS/RAF/MEK/ERK) kinase pathway mediates cellular responses to growth signals. Inappropriate activation of ERK‐MAPK is a common feature in human cancers.( 19 ) The three RAF genes code for cytoplasmic serine/threonine kinases that are regulated by binding RAS. B‐RAF is commonly activated by somatic mutations in human cancer; mutated RAS and RAF proteins have elevated kinase activity and show equivalent abilities to cause transformation of NIH 3T3 and other rodent fibroblast cell lines.( 20 ) Therefore, caRAF was used to transfect gastric cell lines.
We found that the expression of caRAF was sufficient to mimic RAS‐mediated cell proliferation and cell cycle progression in gastric epithelial cells. The RAF‐induced transformation of gastric epithelial cell was associated with downregulated tumor suppressor gene expression, such as p16INK4A , p21WAF1 and p27KIP1 , which contributed to soft agar growth, cell cycle and proliferation. Finally, RAF activation of ERK‐MAPK along with a yet unidentified pathway(s) results in downregulation of p16INK4A by causing an increase in the expression of CpG methyltransferases. The methylation of promoters of key genes such as p16INK4A may then result in downregulation of its expression and thus contribute to the process of transformation, while the epigenetic inactivation of p21WAF1 and p27KIP1 may not play the import role in RAF‐transformed cells.
Proliferating cell nuclear antigen, an index of proliferative activity, increased in caRAF‐AGS and caRAF‐GES‐1 compared with non‐transfected cells. RAF can induce the upregulation of cyclin D1 and cell proliferation in different cell types.( 21 ) The amount of DNA at S phase was increased approximately threefold by RAF, indicating that RAF was a strong promoter of G1 to S phase progression in gastric cell lines. This correlated with cooperative effects on the transcriptional upregulation of cell cycle regulatory protein cyclin D1 and downregulation of the CIP/KIP family, p21CIP1/WAF1 p27KIP1 and INK4 family, p16INK4A, which not only regulate the transition from G1 to S phases of the cell cycle, but also affect anchorage dependence and loss of density‐dependent growth. MEK inhibitor PD98059 reduced cell proliferation. The inactivation of tumor suppressor genes, such as p53, Rb, p16INK4A , p21CIP1/WAF1 and p27KIP1 and the activation of oncogenes genes, such as RAS and B‐RAF, constitute the basis leading to the lost control of normal gastric epithelia cell proliferation and adenocarcinoma transformation, associated with human gastric cancer progression.( 18 ) Our studies found that tumor suppressor genes, p16INK4A , p21CIP1/WAF1 and p27KIP1 , had lower expression in cancer cell line, AGS, and higher expression in normal cell line, GES‐1. These results are consistent with and expand upon the work of others who demonstrated that multiple RAS effector pathways contribute to G1 cell cycle progression and oncogenic transformation; p21WAF1 and p27KIP1 may act as assembly factors to sustain G1 arrest that is ensured by p27KIP1 expression and by cyclin D1 loss in pancreatic cancer cells and RD cells.( 22 , 23 ) Some cell studies have shown that ERK pathway can increase concentrations of p21WAF1 protein and induces S‐phase arrest,( 24 ) but on the contrary, some studies have found that MAPK suppress expression of p21WAF1 and overexpress cyclin D1 and shorten the G1 phase in colonic cancer HT‐29 and gastric adenocarcinoma cell AGS.( 25 ) These findings suggest that the ERK signaling pathway may have the opposite effects in different cell lines.
The expression of various tumor suppressor genes, including BRCA1, E‐cadherin, MLH1, p16INK4A , VHL and RB1, is inactivated by hypermethylation in many human cancers.( 26 ) Promoter methylation represents one common mechanism of epigenetic gene silencing in human carcinomas. Therefore, we assessed whether DNA methylation are involved in oncogenic RAF to induce the suppression of p16INK4A, p21WAF1 and p27KIP.1. The study shows that treatment of caRAF‐GES‐1 cells with 5‐aza‐dC (a known inhibitor of CpG methylation) or PD98059 can restore the expression of p16INK4A significantly. MSP and DNA sequencing show that caRAF results in p16INK4A silencing through aberrant cytosine methylation. The loss of hypermethylation of p16INK4A upon PD98059 treatment suggests that RAF utilizes a RAF/MEK/ERK‐dependent pathway leading to DNA methylation. While treatment of caRAF‐GES‐1 cells with 5‐aza‐dC induces cell cycle arrest and the re‐expression of tumor suppressor genes, it restored the expression of p21WAF1 and p27KIP1 only several folds. Also, PD98059 does not change the hypermethylation status of p21WAF1 and p27KIP1 , and the induction of p21WAF1 and p27KIP1 may not only be caused by demethylation of the promoter region, but also by other mechanisms. Recently, a study showed that RAS/RAF also downregulates tropomyosin and Par‐4 through cytosine methylation of the promoter.( 27 ) Thus, the promoter methylation and gene silencing of a variety of tumor suppressor genes may be an important mechanism of RAS/RAF‐mediated transformation.
The association of RAF and DNA methylation is highly intertwined.( 28 , 29 ) Activation of the RAS/RAF/MEK/ERK pathway has been shown to induce DNA methyltransferases, in addition, the elevated activity of Dnmt levels are required to maintain the phenotype of RAS‐transformed cells.( 30 , 31 ) Dnmt1 methylates hemimethylated DNA, and is responsible for maintenance of methylation patterns. Dnmt3a and Dnmt3b are thought to be responsible for de novo methylation of previously unmethylated DNA.( 32 , 33 ) We found that caRAF‐GES‐1 cells showed significant increase of Dnmt1 and Dnmt3b expression, with elevated levels of Dnmt activity; while PD98059 decreases the activity of Dnmt, the promoter methylation status of tumor suppressor gene p16INK4A is associated with Dnmt. One study showed that inhibition of RAS activity in the Y1 mouse adrenocortical tumor cell line, which overexpresses wild‐type K‐RAS by 30‐fold, resulted in a 50‐fold decrease in DNA methyltransferase enzyme activity and mRNA expression.( 8 , 9 ) In addition, injection of Dnmt1 antisense oligodeoxy nucleotides into Y1 mouse tumors inhibits tumor growth.( 34 )
Taken together, these studies suggest a dependence of RAF‐mediated tumor formation on p16INK4A silencing through DNA methylation by a mechanism that involves upregulation of Dnmt methyltransferases, PCNA or along with a yet unidentified pathway(s). Thus, the clinical utility of pharmacological drugs that target the Raf/MEK/ERK pathway may inhibit tumor growth in part by restoring tumor suppressor gene expression and DNA methylation. Additional studies to determine the mechanism by which RAF initiates upregulation of Dnmt will further our understanding.
Supporting information
Fig. S1. RAF activation cause an increase of DNA methyltransferase activity in gastric cell lines GES‐1 transfected with constitutively active RAF (caRAF), while inhibition of extracellular signal‐regulated kinase/mitogen‐activated protein kinase (ERK‐MAPK) signal pathway by PD98059 also decreased the activity of DNA methyltransferases (Dnmt) to some degree. Therefore, we think that the hyperactivity of ERK‐MAPK may affect the DNA methyltransferase to regulate DNA methylation. The increased activity of DNA methyltransferase by caRAF is decreased by MEK inhibitor PD98059 in the GES‐1 cell line. The gastric cell GES‐1 with vector containing the empty plasmid and dimethylsulfoxide (DMSO) was used as a control; the caRAF expressing GES‐1 showed hyperactivity of Dnmt (216 ± 13%), while PD98059 inhibited the activity of Dnmt (153 ± 24%) compared with the control. The difference was significantly (*P < 0.05).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Acknowledgments
Thanks are given to Weiqi Gu and Hong Yin Zhu for performing the real‐time PCR and MSP, Ms Hongyu Luo and Ms Guanfeng Shen for performing the flow cytometry. This work was supported by grants from the National Basic Research Program of China 973 program (no. 2005CB522400), the National Science Fund for Distinguished Young Scholars (no. 30625034), and the National Natural Science Foundation of China (no. 30500235).
References
- 1. Neureiter D, Herold C, Ocker M. Gastrointestinal cancer – only a deregulation of stem cell differentiation? (Review). Int J Mol Med 2006; 17: 483–9. [PubMed] [Google Scholar]
- 2. Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol 2005; 6: 322–7. [DOI] [PubMed] [Google Scholar]
- 3. Sasao S, Hiyama T, Tanaka S, Yoshihara M, Yasui W, Chayama K. Clinicopathologic and genetic characteristics of gastric cancer in young male and female patients. Oncol Rep 2006; 16: 11–15. [PubMed] [Google Scholar]
- 4. Lee SH, Lee JW, Soung YH et al . BRAF and KRAS mutations in stomach cancer. Oncogene 2003; 22: 6942–5. [DOI] [PubMed] [Google Scholar]
- 5. Hirasawa Y, Arai M, Imazeki F et al . Methylation status of genes upregulated by demethylating agent 5‐aza‐2′‐deoxycytidine in hepatocellular carcinoma. Oncology 2006; 71: 77–85. [DOI] [PubMed] [Google Scholar]
- 6. Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet 2000; 1: 11–19. [DOI] [PubMed] [Google Scholar]
- 7. Scarano MI, Strazzullo M, Matarazzo MR, D’Esposito M. DNA methylation 40 years later: its role in human health and disease. J Cell Physiol 2005; 204: 21–35. [DOI] [PubMed] [Google Scholar]
- 8. MacLeod AR, Rouleau J, Szyf M. Regulation of DNA methylation by the Ras signaling pathway. J Biol Chem 1995; 270: 11327–37. [DOI] [PubMed] [Google Scholar]
- 9. Rouleau J, MacLeod AR, Szyf M. Regulation of the DNA methyltransferase by the Ras‐AP‐1 signaling pathway. J Biol Chem 1995; 270: 1595–601. [DOI] [PubMed] [Google Scholar]
- 10. Esteller M, Toyota M, Sanchez‐Cespedes M et al . Inactivation of the DNA repair gene O6‐methylguanine‐DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K‐ras in colorectal tumorigenesis. Cancer Res 2000; 60: 2368–71. [PubMed] [Google Scholar]
- 11. Lu R, Wang X, Chen ZF, Sun DF, Tian XQ, Fang JY. Inhibition of the extracellular signal‐regulated kinase/mitogen‐activated protein kinase pathway decreases DNA methylation in colon cancer cells. J Biol Chem 2007; 282: 12249–59. [DOI] [PubMed] [Google Scholar]
- 12. Reiners JJ Jr, Lee JY, Clift RE, Dudley DT, Myrand SP. PD98059 is an equipotent antagonist of the aryl hydrocarbon receptor and inhibitor of mitogen‐activated protein kinase kinase. Mol Pharmacol 1998; 53: 438–45. [DOI] [PubMed] [Google Scholar]
- 13. Primeau M, Gagnon J, Momparler RL. Synergistic antineoplastic action of DNA methylation inhibitor 5‐AZA‐2′‐deoxycytidine and histone deacetylase inhibitor depsipeptide on human breast carcinoma cells. Int J Cancer 2003; 103: 177–84. [DOI] [PubMed] [Google Scholar]
- 14. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 15. Benbow U, Tower GB, Wyatt CA, Buttice G, Brinckerhoff CE. High levels of MMP‐1 expression in the absence of the 2G single nucleotide polymorphism is mediated by p38 and ERK1/2 mitogen‐activated protein kinases in VMM5 melanoma cells. J Cell Biochem 2002; 86: 307–19. [DOI] [PubMed] [Google Scholar]
- 16. Scanlan MJ, Gordon CM, Williamson B et al . Identification of cancer/testis genes by database mining and mRNA expression analysis. Int J Cancer 2002; 98: 485–92. [DOI] [PubMed] [Google Scholar]
- 17. Xiong Z, Wu AH, Bender CM et al . Mismatch repair deficiency and CpG island hypermethylation in sporadic colon adenocarcinomas. Cancer Epidemiol Biomarkers Prev 2001; 10: 799–803. [PubMed] [Google Scholar]
- 18. Shames DS, Minna JD, Gazdar AF. DNA methylation in health, disease, and cancer. Curr Mol Med 2007; 7: 85–102. [DOI] [PubMed] [Google Scholar]
- 19. Shaul YD, Seger R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim Biophys Acta 2007; 1773: 1213–26. [DOI] [PubMed] [Google Scholar]
- 20. Davies H, Bignell GR, Cox C et al . Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–54. [DOI] [PubMed] [Google Scholar]
- 21. Naderi S, Wang JY, Chen TT, Gutzkow KB, Blomhoff HK. cAMP‐mediated inhibition of DNA replication and S phase progression: involvement of Rb, p21Cip1, and PCNA. Mol Biol Cell 2005; 16: 1527–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reuven H, Klein S, Levitzki A. The inhibition of Ras farnesylation leads to an increase in p27Kip1 and G1 cell cy i cle arrest. Eur J Biochem 2003; 270: 2759–72. [DOI] [PubMed] [Google Scholar]
- 23. Han J, Tsukada Y, Hara E, Kitamura N, Tanaka T. Hepatocyte growth factor induces redistribution of p21 (CIP1) and p27 (KIP1) through ERK‐dependent p16 (INK4a) up‐regulation, leading to cell cycle arrest at G1 in HepG2 hepatoma cells. J Biol Chem 2005; 280: 31548–56. [DOI] [PubMed] [Google Scholar]
- 24. Zhu H, Zhang L, Wu S et al . Induction of S‐phase arrest and p21 overexpression by a small molecule 2[[3‐(2,3‐dichlorophenoxy) propyl] amino]ethanol in correlation with activation of ERK. Oncogene 2004; 23: 4984–92. [DOI] [PubMed] [Google Scholar]
- 25. McMillan L, Butcher SK, Pongracz J, Lord JM. Opposing effects of butyrate and bile acids on apoptosis of human colon adenoma cells: differential activation of PKC and MAP kinases. Br J Cancer 2003; 88: 748–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 2002; 21: 5427–40. [DOI] [PubMed] [Google Scholar]
- 27. Pruitt K, Ulku AS, Frantz K et al . Ras‐mediated loss of the pro‐apoptotic response protein Par‐4 is mediated by DNA hypermethylation through Raf‐independent and Raf‐dependent signaling cascades in epithelial cells. J Biol Chem 2005; 280: 23363–70. [DOI] [PubMed] [Google Scholar]
- 28. Deng C, Kaplan MJ, Yang J et al . Decreased Ras‐mitogen‐activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum 2001; 44: 397–407. [DOI] [PubMed] [Google Scholar]
- 29. Ramchandani S, Bhattacharya SK, Cervoni N, Szyf M. DNA methylation is a reversible biological signal. Proc Natl Acad Sci USA 1999; 96: 6107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rountree MR, Bachman KE, Herman JG, Baylin S. DNA methylation, chromatin inheritance, and cancer. Oncogene 2001; 20: 3156–65. [DOI] [PubMed] [Google Scholar]
- 31. Detich N, Ramchandani S, Szyf M. A conserved 3′‐untranslated element mediates growth regulation of DNA methyltransferase 1 and inhibits its transforming activity. J Biol Chem 2001; 276: 24881–90. [DOI] [PubMed] [Google Scholar]
- 32. Ordway JM, Williams K, Curran T. Transcription repression in oncogenic transformation: common targets of epigenetic repression in cells transformed by Fos, Ras or Dnmt1. Oncogene 2004; 23: 3737–48. [DOI] [PubMed] [Google Scholar]
- 33. Bachman KE, Rountree MR, Baylin SB. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J Biol Chem 2001; 276: 32282–7. [DOI] [PubMed] [Google Scholar]
- 34. Bigey P, Ramchandani S, Theberge J, Araujo FD, Szyf M. Transcriptional regulation of the human DNA Methyltransferase (dnmt1) gene. Gene 2000; 242: 407–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. RAF activation cause an increase of DNA methyltransferase activity in gastric cell lines GES‐1 transfected with constitutively active RAF (caRAF), while inhibition of extracellular signal‐regulated kinase/mitogen‐activated protein kinase (ERK‐MAPK) signal pathway by PD98059 also decreased the activity of DNA methyltransferases (Dnmt) to some degree. Therefore, we think that the hyperactivity of ERK‐MAPK may affect the DNA methyltransferase to regulate DNA methylation. The increased activity of DNA methyltransferase by caRAF is decreased by MEK inhibitor PD98059 in the GES‐1 cell line. The gastric cell GES‐1 with vector containing the empty plasmid and dimethylsulfoxide (DMSO) was used as a control; the caRAF expressing GES‐1 showed hyperactivity of Dnmt (216 ± 13%), while PD98059 inhibited the activity of Dnmt (153 ± 24%) compared with the control. The difference was significantly (*P < 0.05).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item