

Abstract
Indinavir, a human immunodeficiency virus (HIV) protease inhibitor, inhibits the growth of tumor cells in vivo but does not show any cytotoxicity against cancer cells in vitro. To optimize the anticancer activity of indinavir, two novel analogs, CH05‐0 and CH05‐10, were synthesized. CH05‐10 was much more cytotoxic than indinavir and had similar cytotoxicity to nelfinavir, the one with the best anticancer activities among all HIV protease inhibitors examined. For 14 cell lines representing 10 different types of human malignancies, the 50% inhibitory concentration (IC50) values of CH05‐10 are in the range of 4.64–38.87 μM. Further detailed studies using the lung cancer cell line A549 as the model system showed that the effect of CH05‐10 on the A549 cell line is both time‐ and dose‐dependent. The CH05‐10 treatment not only induced cell cycle arrest at G1 and caused caspase‐dependent apoptosis, but also resulted in caspase‐independent death via the induction of endoplasmic reticulum stress and unfolded protein response. These findings demonstrate that CH05‐10, a novel indinavir analog, is a potent anticancer agent with pleiotropic effects. (Cancer Sci 2010; 101: 2644–2651)
Human immunodeficiency virus protease inhibitors (HIV PI) such as nelfinavir, saquinavir, ritonavir, indinavir and atazanavir are a class of antiretroviral drugs.( 1 ) Unexpectedly, several of them have antitumor activity that is independent of their antiviral or immune‐mediated activity.( 1 ) For example, HIV PI have been shown to inhibit Kaposi’s sarcoma,( 2 ) lymphoma,( 3 ) glioma,( 4 ) multiple myeloma,( 5 ) prostate cancer,( 6 ) breast cancer and ovarian cancer.( 7 , 8 , 9 ) Nelfinavir has the most potent antitumor activity among the HIV PI examined and this activity has even been tested in a clinical trial.( 10 ) Nelfinavir inhibits cell growth in a broad range of cell types at low micromolar concentrations by inducing G1 cell cycle arrest and apoptosis.( 5 , 11 , 12 ) Recently, a new antitumor mechanism of action has been described for nelfinavir where treatment induces cancer cell death by triggering the endoplasmic reticulum stress (ERS) response and autophagy.( 13 , 14 ) It has been reported that saquinavir treatment also induces ERS and autophagy in cancer cells.( 4 , 15 , 16 ) Unlike these HIV PI, indinavir does not inhibit proliferation or induce apoptosis in cancer cells in vitro,( 17 , 18 ) although it does inhibit tumor growth by blocking angiogenesis and cell invasion.( 17 ) While indinavir may be useful as a new and alternative antitumor agent, it needs to be optimized to improve its cellular activity.
In this study, two novel analogs of indinavir, CH05‐0 and CH05‐10, were designed and synthesized. CH05‐10 showed a broad spectrum of anticancer activity, and its inhibitory effects and molecular mechanisms were further assessed in detail using the human lung cancer cell line A549 as the model system. Our results showed that the CH05‐10 treatment inhibited the survival of the A549 cells in a time‐ and dose‐dependent manner by both caspase‐dependent apoptotic cell death and caspase‐independent cell death pathway characterized by induction of the ERS response.
Materials and Methods
Reagents. Indinavir and nelfinavir were purchased from Huahai Pharmaceutical Co. (ZheJiang, China). CH05‐0 (yellow solid) and CH05‐10 (yellow solid) were synthesized as described in our patent.( 19 ) All of the compounds were dissolved in dimethyl sulfoxide (DMSO) as a 20 mM stock solution and stored at −20°C. Each working solution was freshly prepared in RPMI‐1640 medium (Gibco BRL, Gaithersburg, MD, USA); the final DMSO concentration was <0.1% (v/v).( 20 ) Primary antibodies specific for cyclin D1, cyclin E, CDK2, CDK4, CDK6, p21WAF1/Cip, caspase‐7, poly‐ADP‐ribose polymerase (PARP), Akt, phospho‐Akt, ubiquitin, BiP/GRP78, CHOP and LC3II were purchased from Cell Signaling Technology (Danvers, MA, USA). The anti‐GAPDH antibody was purchased from Kangchen Biotech (ShangHai, China). The enhanced chemiluminescent kit was purchased from Amersham Biosciences (Little Chalfont, UK). The annexin V‐PE apoptosis detection kit was purchased from BD Biosciences (San Jose, CA, USA). All other reagents were obtained from Sigma–Aldrich (St Louis, MO, USA).
Cell culture. The human cancer cell lines, MCF‐7 (breast), U251 (central nervous system), HT‐29 (colon), NCI‐H460 and A549 (lung), K562, MOLT‐4 and HL60 (leukemia), A375 (melanoma), OVCAR‐3 (ovarian), and DU‐145 and PC‐3 (prostate) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA); OS‐RC‐2 (kidney) and Hela (cervix) cell lines were obtained from the CAS Cell Bank (ShangHai, China). All cell lines were cultured at 37°C and 5% CO2 in RPMI 1640 medium (Gibco BRL) supplemented with 10% fetal bovine serum (Gibco BRL).
Cytotoxicity analysis. Cells (1 × 104 cells/well) were cultured in 96‐well plates with increasing concentrations of each compound for the indicated period of time. Cell viability was measured using the MTT assay.( 21 ) Briefly, cells were treated with 20 μL MTT (5 mg/mL) at 37°C for 4 h. All media was aspirated and 100 μL DMSO was added to each well to dissolve the formazan product generated by viable cells. The absorbance was taken at 570 nm using a microplate reader (Bio‐Rad, Hercules, CA, USA). The 50% inhibitory concentration (IC50) was calculated using GraphPad Prism software (GraphPad, La Jolla, CA, USA).
Cell counting analysis. A549 cells (1 × 105 cells/well) were cultured in six‐well plates with DMSO or CH05‐10 for 24, 48 or 72 h. Adherent cells were then trypsinized and resuspended in culture medium containing 0.2% (w/v) trypan blue. The number of total cells and viable cells (i.e. those that exclude trypan blue) were counted using a hemocytometer.( 22 ) Triplicate wells were counted for each condition. These data are presented as relative cell viability and proliferation.
Cell cycle analysis. A549 cells (1 × 105 cells/well) were cultured in six‐well plates and synchronized at the G0/G1 phase by serum deprivation for 24 h. After treatment with DMSO or CH05‐10 for 12, 24 or 48 h, the cells were fixed with 70% ethanol and stained with propidium iodide solution (10 μg/mL; Qiagen, Hilden, Germany) containing RNase A (50 μg/mL; Qiagen).( 23 ) Cell cycle analysis was performed using a FACScan flow cytometer (Becton Dickinson, Bedford, MA, USA) and cell cycle data were processed with ModFit LT cell cycle analysis software (Verity Software House, Topsham, ME, USA).
Apoptosis assay. Morphological changes in the nuclear chromatin of A549 cells were detected by staining with 4′,6‐diamidino‐2‐phenylindole (DAPI).( 24 ) After treatment with DMSO or CH05‐10 for 24 h, the cells were incubated with 1 μg/mL DAPI solution at 37°C for 10 min. After being washed with PBS, the cells were visualized using a fluorescence microscope (Olympus, Tokyo, Japan). Apoptosis of A549 cells was also quantified using the annexin V‐PE apoptosis detection kit (BD Biosciences). Briefly, A549 cells (1 × 105 cells/mL) were cultured in six‐well plates and treated with DMSO or CH05‐10 for 24, 48 or 72 h. After treatment, cells were washed twice with cold PBS, resuspended in 100 μL of cold binding buffer plus 5 μL PE‐Annexin V and 5 μL 7‐aminoactinomycin D (7‐AAD) and incubated for 15 min at room temperature in the dark. Apoptosis was assessed by dual‐color flow cytometry on a FACScan cytofluorometer (Becton Dickinson) using the Cell Quest software package from Becton Dickinson.
Western blot analysis. A549 cells (1 × 105 cells/well) were cultured in six‐well plates with DMSO or CH05‐10 for 48 h. Cells were collected and lysed in ice‐cold lysis buffer (100 mM Tris–HCl, pH 6.8, 4% [m/v] SDS, 20% [v/v] glycerol, 200 mM β‐mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride and 1 μg/mL aprotinin) for 30 min. Lysates were then centrifuged at 12 000 g for 15 min at 4°C to remove insoluble cell debris. Protein content was quantified using the Bio‐Rad protein assay (Bio‐Rad, Hercules, CA, USA) and 15 μg of protein was separated by SDS–PAGE and transferred onto polyvinylidene difluoride membranes. The membrane was blocked for 1 h in buffer (1 × TBS, 5% [w/v] milk, 0.1% [v/v] Tween 20) and then incubated with the primary antibody in dilution buffer (1 × TBS, 5% [v/v] bovine serum albumin, 0.1% [v/v] Tween 20) overnight at 4°C. The membrane was then washed three times with wash buffer (1 × TBS, 0.1% [v/v] Tween 20), incubated with the appropriate horseradish peroxidase‐linked secondary antibody and visualized with the enhanced chemiluminescent detection system (Amersham Biosciences).
RT‐PCR. Cells were harvested and total RNA was isolated using a high pure RNA isolation kit (Roche, Mannheim, Germany) according to the manufacturer’s instruction. Reverse transcription (RT) was carried out with 2 μg RNA and Oligo dT (Takara, Otsu, Japan) using Sensiscript Reverse Transcriptase (Qiagen). For ATF4, the forward and reverse primers were 5′‐AGGAGTTCGCCTTGGATGCCCTG‐3′ and 5′‐AGTGATATCCACTTCACTGCCCAG‐3′, respectively.( 25 ) The PCR reaction was performed in a final reaction volume of 20 μL under the following conditions: a preheating cycle at 95°C for 5 min, then 33 cycles at 95°C for 20 s, 58°C for 30 s, and 72°C for 30 s, the final elongation step was performed for 5 min at 72°C.
The PCR of XBP1 was essentially performed as described previously.( 26 ) For XBP1, the forward and reverse primers were 5′‐CGCGGATCCGAATGAAGTGAGGCCAGT GG‐3′ and 5′‐GGGGCTTGGTATATATGTGG‐3′, respectively. Amplified fragments covering a 26‐nt intron (nt 531–556) and the flanking exons were digested with Pst I (a unique PstI site existed at nt 556; Takara) and separated on 2.5% polyacrylamide gels. The ERS‐dependent splicing of transcript from this gene should produce an mRNA species with 26 bp deletions in comparison with the normal transcript. The RT‐PCR product of spliced XBP1 could not digested by Pst I.
Statistical analysis. Results were expressed as the mean ± SD. Analysis was performed using a two‐tail Student’s t‐test. P values <0.05 were considered statistically significant.
Results
Indinavir analogs showed broad‐spectrum anticancer activity.
To optimize the biological profile of indinavir, two analogs, CH05‐0 and CH05‐10, were synthesized (Fig. 1). The cytotoxicity of indinavir, CH05‐0, CH05‐10 and nelfinavir to 14 cell lines representing 10 different types of human malignancies was evaluated using the MTT assay. Table 1 shows the relative cellular cytotoxicity of these compounds (nelfinavir ≈ CH05‐10 > CH05‐0 > indinavir), which demonstrates that both analogs have greater antitumor activity than indinavir. In particular, CH05‐10 was 10‐ to 50‐fold more cytotoxic than indinavir, and had a similar cytotoxicity to nelfinavir.
Figure 1.
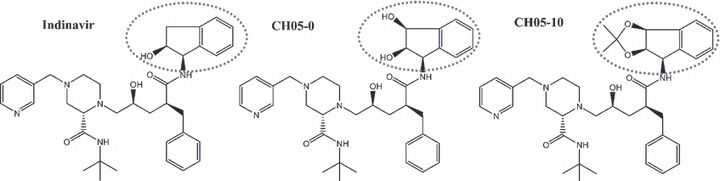
Chemical structures of indinavir, CH05‐0 and CH05‐10. The different moieties of these compounds are indicated.
Table 1.
Growth inhibitory activity of four compounds (nelfinavir, indinavir, CH05‐0 and CH05‐10) in human cancer cells
| Cancer type | Cell line | IC50(μM) | |||
|---|---|---|---|---|---|
| Nelfinavir | Indinavir | CH05‐0 | CH05‐10 | ||
| Breast | MCF‐7 | 15.8 ± 3.0 | >1000 | 611.0 ± 49.3 | 18.3 ± 2.8 |
| CNS | U251 | 17.6 ± 4.0 | >1000 | 395.8 ± 13.8 | 24.8 ± 3.6 |
| Colon | HT‐29 | 15.5 ± 0.4 | 543.6 ± 0.5 | 549.4 ± 67.1 | 17.6 ± 1.4 |
| Lung | NCI‐H460 | 17.2 ± 3.3 | >1000 | 654.7 ± 45.2 | 16.8 ± 0.7 |
| Lung | A549 | 12.7 ± 1.4 | >1000 | 607.8 ± 27.4 | 22.2 ± 0.2 |
| Leukemia | K562 | 34.6 ± 0.0 | >1000 | 957.2 ± 56.3 | 25.5 ± 1.4 |
| Leukemia | MOLT‐4 | 10.5 ± 0.7 | 385.8 ± 0.3 | 330 ± 28.4 | 4.6 ± 1.6 |
| Leukemia | HL60 | 9.7 ± 2.6 | 610.5 ± 0.2 | 395.9 ± 34.9 | 6.9 ± 0.9 |
| Melanoma | A375 | 25.0 ± 7.1 | >1000 | 689.6 ± 60.1 | 19.3 ± 6.3 |
| Ovarian | OVCAR‐3 | 25.5 ± 9.7 | >1000 | ∼1000 | 18.1 ± 4.7 |
| Prostate | DU‐145 | 33.5 ± 1.6 | >1000 | 347.4 ± 18.8 | 20.0 ± 3.0 |
| Prostate | PC‐3 | 18.8 ± 2.8 | >1000 | 495.3 ± 51.2 | 17.7 ± 1.7 |
| Renal | OS‐RC‐2 | 31.8 ± 0.6 | >1000 | >1000 | 38.9 ± 3.5 |
| Cervical | Hela | 34.6 ± 5.1 | >1000 | >1000 | 20.5 ± 0.4 |
CH05‐10 inhibited the survival of A549 cells in a time‐ and dose‐dependent manner. Further experiments were performed using the human lung cancer cell line A549. After treatment with indinavir, CH05‐0 or CH05‐10, the cells were visualized under light microscopy. Only treatment with CH05‐10 resulted in morphological changes to the cell, including cellular rounding and detachment from the surface of culture dishes (Fig. 2A). After exposure to increasing concentrations of CH05‐10 for 6, 48 or 72 h, cell viability was measured using the MTT assay. CH05‐10 treatment inhibited the survival of A549 cells in a time‐ and dose‐dependent manner (Fig. 2B). Although treatment with a low concentration of CH05‐10 (20 μM) for 48 h did not cause the cellular morphological changes, the cell size was increased (data not show). These data suggest that CH05‐10 treatment inhibits the proliferation of A549 cells, which was confirmed by cell‐counting analysis. After treatment with CH05‐10 for 48 h, few cells had died (Fig. 2C), but the total number of viable cells was decreased compared with the controls (Fig. 2D).
Figure 2.
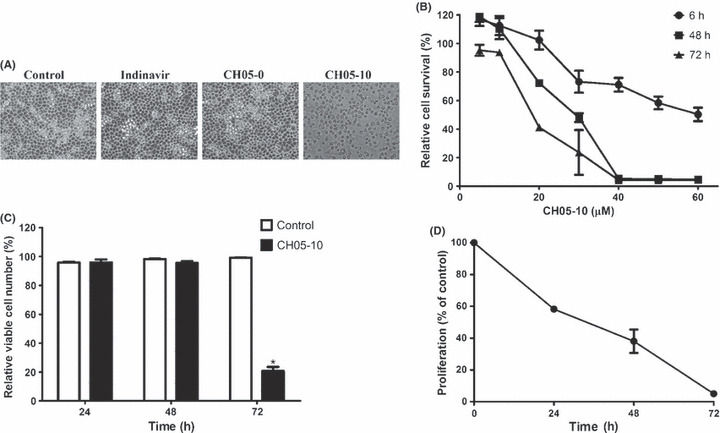
CH05‐10 inhibited cell growth of A549 cancer cells. (A) Morphological changes induced by CH05‐10. A549 cells were treated with DMSO, 40 μM indinavir, CH05‐0 and CH05‐10 for 48 h. Cell morphology was visualized by optical microscopy (magnification, ×200). (B) A549 cells were treated with various concentrations (5–60 μM) of CH05‐10 for 6, 48 and 72 h. Cell survival was measured using the MTT assay. The percentage of viable cells was calculated as the ratio of treated cells to control cells. (C) A low concentration of CH05‐10 did not induce A549 cell death. A549 cells were treated with 20 μM CH05‐10 for 24, 48 and 72 h, and the dead and living cells were counted. The percentage of viable cells was the ratio of living cells to the total cell population. *Different from untreated cells (P < 0.05). (D) A low concentration of CH05‐10 inhibited the proliferation of A549 cells. The A549 cells were treated with 20 μM CH05‐10 for 24, 48 and 72 h, and the living cells were counted. The percentage of proliferating cells was calculated by the ratio of treated cells to control cells.
CH05‐10 induced G1 phase cell cycle arrest in A549 cells. CH05‐10 treatment caused a marked G1 arrest in A549 cells as indicated by a higher percentage of cells in the G1 phase and a lower percentage of cells in the S and G2‐M phase (Fig. 3A). Upon CH05‐10 treatment, the G1 arrest was evident after as short as 12 h. In addition, the CH05‐10‐induced cell cycle arrest is not dose‐ and time‐dependent (Fig. 3B). As such, the regulatory effect of CH05‐10 treatment on the protein level of cyclins and cyclin‐dependent kinases (CDK), which play important roles in controlling cell cycle regulation, was investigated. CH05‐10 treatment downregulated the protein levels of cyclin D1 and cyclin E but had no effect on the protein levels of CDK2, CDK4 and CDK6 at 48 h (Fig. 3C). P21WAF1/Cip is a potent cyclin‐dependent kinase inhibitor (CKI). The p21WAF1/Cip protein binds to and inhibits the activity of cyclin‐CDK2 or ‐CDK4 complexes, and thus functions as a regulator of cell cycle progression at G1. After CH05‐10 treatment, the p21WAF1/Cip protein level declined at 12 h and returned to a normal level at 24 h (Fig. 3C).
Figure 3.
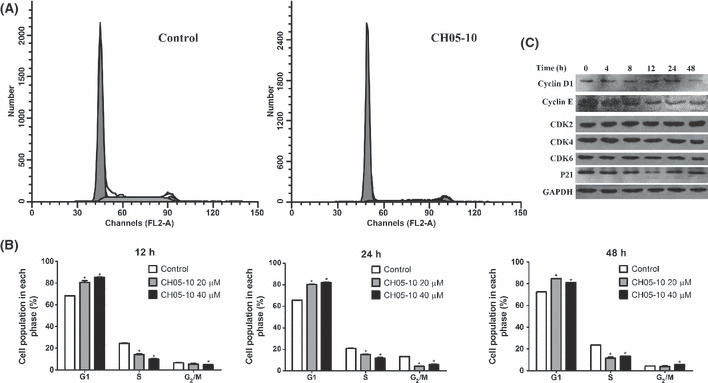
Cell cycle analysis of CH05‐10‐treated A549 cells. (A) A549 cells were treated with DMSO or 40 μM CH05‐10 for 48 h. The cells were stained with propidium iodide (PI) and analyzed by flow cytometry. (B) The graphs show the cell distributions at each phase of the cell cycle. A549 cells were treated with DMSO, 20 or 40 μM CH05‐10 for the indicated times. The cells were stained with PI and analyzed by flow cytometry. *Different from untreated cells (P < 0.05). (C) Western blot analysis of cyclin D1, cyclin E, CDK2, CDK4, CDK6 and p21 WAF1/Cip in A549 cells treated with 40 μM CH05‐10 for the indicated times. The GAPDH protein levels were used as the protein loading control.
CH05‐10 induces caspase‐dependent apoptosis. Next, we determined whether CH05‐10 treatment induces apoptosis in A549 cells. The nuclear morphology of dying cells was examined using the fluorescent DNA‐binding agent DAPI. After treatment with 40 μM CH05‐10 for 24 h, the condensed chromosome in the nucleus was observed, which is a typical morphological feature of apoptotic cells (Fig. 4A). To further confirm the induction of apoptosis, cells were stained with annexin V and 7‐AAD after treatment with 40 μM CH05‐10 for 24, 48 or 72 h. The proportion of annexin V+/7‐AAD+ cells in CH05‐10‐treated cells increased in a dose‐ and time‐dependent manner (Fig. 4B). Because caspases often play an important role in mediating various apoptotic responses, we examined whether CH05‐10 might activate caspase‐7 during the induction of apoptosis. The cleaved form of caspase‐7 was observed in CH05‐10‐treated cells after 48 h (Fig. 4C). In general, PARP is cleaved by caspases during apoptosis. In this study, the protein levels of full‐length PARP were decreased and levels of the cleaved form were increased in the CH05‐10‐treated cells (Fig. 4C), which provides further evidence supporting CH05‐10‐induced apoptosis in A549 cells. To assess whether caspase activation was required for CH05‐10‐induced death, we further introduced a pan‐caspase inhibitor (z‐VAD‐fmk) to CH05‐10 containing A549 cell culture medium. After those treatments, cell viability was measured by MTT assay. Treatment with z‐VAD‐fmk can only partially block CH05‐10‐mediated cell death in A549 cells (Fig. 4D). CH05‐10 induced cellular vacuolization with or without z‐VAD‐fmk. Akt, a serine/threonine protein kinase, is often involved in cellular survival pathways through the inhibition of apoptotic processes and promotion of cell survival signals by phosphorylation and inactivation of several proapoptotic proteins.( 27 , 28 ) Constitutive activation of the Akt pathway is very common in human cancers.( 14 , 29 , 30 ) Therefore, inhibition of the PI3K/Akt pathway was investigated by assessing the expression and activation of Akt in the presence of CH05‐10. Indeed, CH05‐10 treatment decreased the protein level of both total Akt and phospho‐Akt (Fig. 4C).
Figure 4.
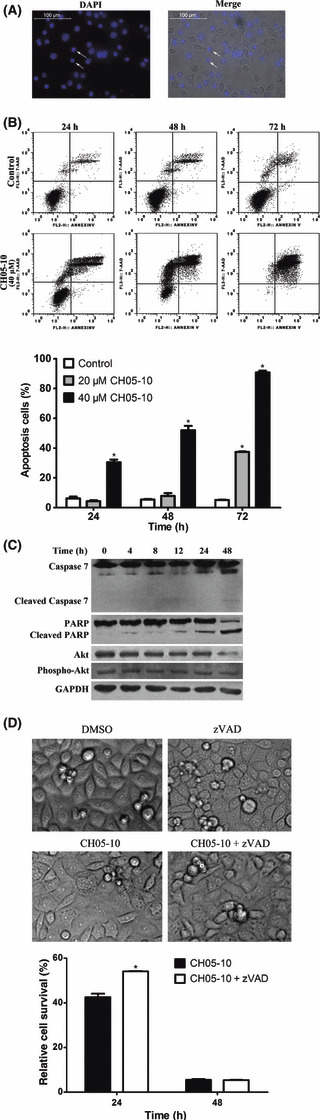
CH05‐10 induces apoptosis in A549 cells. (A) Cells were treated with 40 μM CH05‐10 for 24 h, stained with DAPI, and visualized with a fluorescence microscope (magnification, ×200). Scale bar, 100 μm. Arrows indicate apoptotic nuclei with condensed chromatin. (B) Quantitative analysis of CH05‐10‐induced apoptotic cell death by flow cytometry. Cells were treated with DMSO, 20 or 40 μM CH05‐10 for the indicated times. *Different from untreated cells (P < 0.05). (C) Western blot analysis of caspase‐7, poly‐ADP‐ribose polymerase (PARP), Akt and phospho‐Akt in A549 cells treated with 40 μM CH05‐10 for the indicated times. The GAPDH protein levels were used as the protein loading control. (D) Treatment with z‐VAD‐fmk only partially blocked CH05‐10‐mediated cell death in the A549 cells. The A549 cells were divided into four groups: control (treated with DMSO); z‐VAD‐fmk (treated with 40 mM z‐VAD‐fmk); CH05‐10 (treated with 40 μM CH05‐10); and CH05‐10 plus z‐VAD‐fmk (treated with both 40 μM CH05‐10 and 40 mM z‐VAD‐fmk) for 24 or 48 h. Cell viability was measured with an MTT assay. Phase‐contrast micrographs were taken 24 h after CH05‐10 treatment. *Different from CH05‐10‐treated cells (P < 0.05).
CH05‐10 induced caspase‐independent death, ERS and autophagy. CH05‐10 treatment inhibited the growth of A549 cells as early as 6 h (Fig. 2B); however, the indicators of apoptosis were not detected until 24 h (Fig. 4C), and the majority of apoptotic cells were in the late apoptotic stages of cell death (Annexin V+/7‐AAD+) (Fig. 4B). Overall, these findings suggested that, in addition to a caspase‐dependent mechanism of CH05‐10‐mediated cell death, CH05‐10 might also trigger a caspase‐independent, non‐apoptotic cell death pathway in A549 cells. Interestingly, it has been reported that nelfinavir and saquinavir impair tumor cell growth via induction of the ERS response.( 13 , 16 ) As such, we sought to determine if CH05‐10 induced a similar caspase‐independent mechanism of cell death in A549 cells. The A549 cells were treated with CH05‐10 and cytoplasmic vacuolization, which is a morphological change consistent with induction of ERS, was observed under the phase contrast microscope (Fig. 5A). Endoplasmic reticulum stress triggers the degradation of misfolded endoplasmic reticulum (ER) proteins via upregulation of ubiquitination enzymes.( 30 ) A marked increase in polyubiquitinated proteins of various sizes, represented by a smear on the blot, was observed in the CH05‐10‐treated cells (Fig. 5B). Binding immunoglobulin protein (BiP, also known as GRP78, glucose‐regulated protein) is a molecular chaperone that senses ER homeostasis and initiates the ERS response by transiently binding to newly synthesized, misfolded or unassembled proteins in the ER lumen. C/EBP homologous protein (CHOP) is a member of the C/EBP family of bZIP transcription factors that induces apoptosis through a Bcl‐2–inhibitable mechanism during ERS. Expression of BiP/GRP78 and CHOP increases to high levels following ERS( 4 ) and is considered a marker of the ERS response. As such, changes in the protein levels of BiP/GRP78 and CHOP were investigated by western blot analysis following CH05‐10 treatment and were uniformly increased after 8 and 12 h of treatment (Fig. 5B), which suggests that CH05‐10 treatment induces ERS. The chop gene promoter contains binding sites for all of the major inducers of unfolded protein response (UPR), including activating transcription factor 4 (ATF4), activating transcription factor 6 (ATF6) and X‐box binding protein 1 (XBP1), and these transcription factors play causative roles in inducing chop gene transcription.( 31 ) Transcribed XBP‐1 mRNA is converted to its active form by unconventional cytoplasmic splicing mediated by inositol‐requiring enzyme‐1 (IRE‐1) upon ERS.( 32 ) After CH05‐10 treatment, spliced XBP1 mRNA was observed (Fig. 5C) and the mRNA level of ATF4 was upregulated (Fig. 5D).
Figure 5.
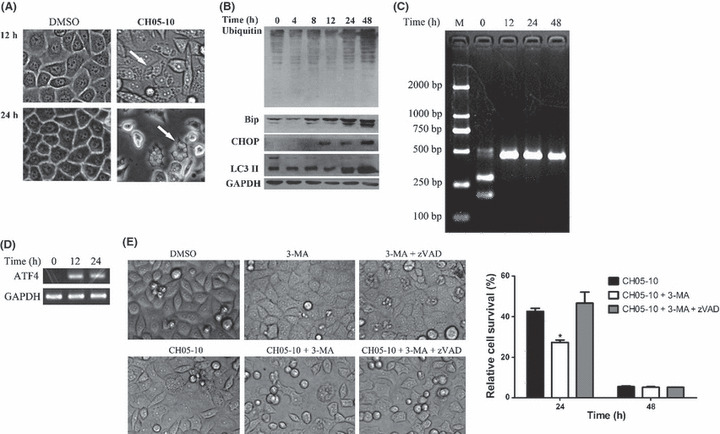
CH05‐10 induced endoplasmic reticulum stress (ERS) and autophagy. (A) A549 cells were treated with 40 μM CH05‐10 for 12 or 24 h and then visualized using a phase contrast microscope (magnification, ×400). Arrows pointed to dilated ER cisternae. (B) Western blot analysis of BiP/GRP78, CHOP and LC3 II in the A549 cells treated with 40 μM CH05‐10 for the indicated times. The GAPDH protein levels were used as the protein loading control. (C) The A549 cells were treated with 40 μM CH05‐10 for the indicated times and the splicing of X‐box binding protein 1 (XBP1) was detected by RT‐PCR and Pst I digestion. (D) The A549 cells were treated with 40 μM CH05‐10 for the indicated times and the transcription of ATF4 was detected by RT‐PCR. The GAPDH levels were used as the RT‐PCR control. (E) 3‐MA enhanced CH05‐10‐mediated cell death in A549 cells. The A549 cells were divided into six groups: control (treated with DMSO); 3‐MA (treated with 10 mM 3‐MA); CH05‐10 (treated with 40 μM CH05‐10); CH05‐10 plus 3‐MA (treated with 40 μM CH05‐10 and 10 mM 3‐MA); 3‐MA plus z‐VAD‐fmk (treated with 10 mM 3‐MA and 40 mM z‐VAD‐fmk) and CH05‐10 plus 3‐MA plus z‐VAD‐fmk (treated with 40 μM CH05‐10, 10 mM 3‐MA and 40 mM z‐VAD‐fmk) for 24 or 48 h. Cell viability was measured with an MTT assay. Phase‐contrast micrographs were taken 24 h after CH05‐10 treatment. *Different from CH05‐10‐treated cells (P < 0.05).
Endoplasmic reticulum stress and autophagy are closely related.( 16 , 33 , 34 ) Microtubule‐associated protein 1A/1B‐light chain 3 (LC3) is distributed ubiquitously in mammalian tissues and cultured cells. LC3 exists as a cytoplasmic form (LC3 I) or as a membranous form (LC3 II) that is only associated with autophagosomes( 35 ) and is a marker for autophagy. After treatment with CH05‐10, a significant increase in the expression of LC3‐II was detected (Fig. 5B). Autophagy has been reported to increase as a result of chemotherapy, either leading the cancer cell to autophagic cell death, called programmed cell death (PCD), or mediating its adaptation and prevention from damage by drug cytotoxicity, as in apoptosis.( 36 ) To assess whether CH05‐10‐induced autophagy contributed to cell death, we measured CH05‐10‐induced cell viability in the absence or presence of 10 mM 3‐methyladenine (3‐MA, a popular inhibitor of the autophagic pathway) using MTT assay (Fig. 5E). 3‐MA enhanced CH05‐10‐induced cell death in the A549 cells. It suggests that autophagy is a cellular response that mitigated CH05‐10‐induced death. z‐VAD‐fmk could rescue the CH05‐10 and 3‐MA‐induced cell death in A549 (Fig. 5E). It suggests that 3‐MA enhanced CH05‐10‐induced cell death by triggering apoptosis.
Discussion
The HIV PI have antineoplastic effects via multiple potential mechanisms.( 1 , 9 ) In this study, we showed that CH05‐10, an analog of indinavir, is a novel compound with potent and broad‐spectrum antitumor activity. Compared with indinavir and CH05‐0, a small structural change in CH05‐10 resulted in a significant increase in cytotoxicity (Fig. 1 and Table 1). We demonstrated that CH05‐10 induces both a caspase‐dependent apoptotic cell death (Fig. 4) as well as a caspase‐independent cell death (Fig. 5). Our investigation into the caspase‐independent mechanism of cell death demonstrated that CH05‐10 induces ERS and autophagy in the A549 cells.
The ER is the site of folding of membrane and secreted proteins in the cell. Physiological or pathological processes that disturb protein folding in the endoplasmic reticulum cause ERS and activate a set of signaling pathways termed the unfolded protein response (UPR).( 31 ) The UPR halts general protein synthesis while upregulating ER resident chaperones and other regulatory components of the secretory pathway, giving the cell a chance to correct the environment within the ER. In mammals, the UPR is a complex signaling network mediated by three ER‐resident factors: IRE1; the PKR like ER kinase (PERK); and ATF6. The ER lumenal domains of each of these factors are normally bound to Bip/GRP78. Upon accumulation of unfolded proteins in the lumen of the ER, Bip/GRP78 dissociates from these three sensors permitting their signaling capabilities. Activated IRE1 acts as both a kinase and an endoribonuclease. IRE1 alternatively splices the mRNA of XBP1 removing a 26 nucleotide intron that allows sXBP1 to act as a transcriptional activator for several ER chaperones.( 26 ) Activated PERK phosphorylates the translation initiation factor eIF2a, which halts translation and prevents the continual accumulation of newly synthesized proteins into the ER when protein folding conditions are compromised. Paradoxically, this translational shutdown leads to the selective translation of transcription factor ATF4, a member of the bZIP family of transcription factors. The PERK‐eIF2α‐ATF4 axis regulates the expression of genes involved in amino acid biosynthesis and transport functions, antioxidant stress responses and apoptosis.( 25 ) When the initial cellular responses fail to restore ER homeostasis, sustained ERS causes the UPR to switch from an adaptive to a cell death pathway. The main effector of PERK‐mediated apoptosis is the proapoptotic transcription factor CHOP.( 4 ) Our data showed that CH05‐10 triggers ERS/UPR through at least two different signaling pathways, including IRE1‐XBP1 and PERK‐eIF2α‐ATF4 (Fig. 5C,D).
The time‐dependent expression of ERS markers and autophagy were correlated. Expression of BiP/GRP78 and CHOP increased after 8 and 12 h respectively of CH05‐10 treatment, while the increase in LC3 II expression was observed after 24 h, suggesting that CH05‐10 induces ERS followed by autophagy. These results are similar to those observed with the HIV PI nelfinavir and saquinavir.( 13 , 16 ) Like CH05‐10, both nelfinavir and saquinavir have a proline‐like structure, which suggests that triggering of the ERS response might be related to their chemical structure. However, indinavir also has a proline‐like structure but does not induce ERS and autophagy,( 37 ) which suggests that subtle differences in the structures of indinavir and CH05‐10 play an important role in the induction of ERS.
For survival, the proteolytic degradation of tumor suppressors and cell cycle inhibitors is increased in tumor cells, which mostly requires the ubiquitin/proteasome system. Inhibition of proteasome function has been shown to induce apoptosis in cancer cells. Previous studies for HIV PI revealed that ritonavir,( 38 ) saquinavir( 39 ) and nelfinavir( 14 ) inhibit proteasome activity in cancer cells. The cleavage sites of action for HIV protease were once thought to be unique and distinct from those of mammalian proteases. Based on the chemical structure of the retrovirus‐specific cleavage site, HIV PI have been designed successfully. Almost all the HIV PI in clinical use are peptidomimetics. However, recently, the 20s proteasome has been shown to cleave the same sites. Also, HIV PI have been shown to directly inhibit 20s and 26s proteasome function. (39) It is most likely that CH05‐10, an analogue of HIV PI, induces cell cycle arrest and apoptosis by inhibiting the function of proteasome.
However, during the UPR, the expression of some nonessential proteins, such as cyclin D1,( 40 ) cyclin E( 40 ) and even Akt( 41 ) is downregulated. Our data shows that CH05‐10 triggered an ERS response in an early event. It seems that the downregulation of cyclin D1, cyclin E (Fig. 3C) and Akt (Fig. 4C) were related to CH05‐10‐induced ERS. And it suggests that CH05‐10‐induced cell cycle arrest is not associated with changes of cyclin D1 and cyclin E. CHOP suppresses cell cycle regulator p21WAF1/Cip expression during ERS facilitating the commitment of cells into a proapoptotic program.( 42 ) In this study, CH05‐10 upregulates CHOP (Fig. 5B) at 12 h, as well as downregulates p21WAF1/Cip (Fig. 3C). Because CH05‐10 also induced caspase‐dependent apoptosis, the protein level of p21WAF1/Cip returned at 24 h when the cleaved form of PARP was increased (Fig. 4C).
Heat shock protein 90 (Hsp90) is a major molecular chaperone that plays an essential role in the maintenance of several signaling molecules, most of which are oncogenic kinases. Hsp90 inhibitors block the activity of such proteins and lead to inhibition of multiple signaling cascades. Due to this, Hsp90 has emerged as an important target for the treatment of cancer. It has been proven that Hsp90 inhibitors can affect CDKs and Cyclin D1 and cause cell cycle arrest.( 43 ) Hsp90 inhibition also induces ERS, which leads to apoptosis and upregulates the ER chaperones, such as BiP/GRP78 and GRP94.( 44 ) Ritonavir, an HIV PI, can bind Hsp90 and partially inhibit its chaperone function.( 45 ) However, it is less likely that CH05‐10 might inhibit Hsp90 because of the lack of CDK4 downregulation and a little bit too strong induction of ERS/UPR.
In summary, this study demonstrated that CH05‐10, a novel indinavir analog, is a broad‐spectrum anticancer agent, which is much more potent than indinavir and is as potent as nelfinavir. CH05‐10 inhibited the growth of human lung cancer A549 cells via multiple mechanisms including inhibition of proliferation, induction of cell cycle arrest and caspase‐dependent apoptosis. CH05‐10 treatment also caused ERS, which led to nonapoptotic cell death. In total, these results indicate that CH05‐10 may be a promising candidate as an antitumor therapeutic.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported in part by a grant from the Guangzhou Science and Technology Programs (2007Z3‐E4131) and the Guangzhou Development District Fund (2008Ss‐P010). The authors thank Yayong Li, Siting Zhao, Guangjie Liu, Benpeng Yin, Nashunbayaer and Haixiang Zhang for their technical assistance.
References
- 1. Monini P, Sgadari C, Toschi E et al. Antitumour effects of antiretroviral therapy. Nat Rev Cancer 2004; 4: 861–75. [DOI] [PubMed] [Google Scholar]
- 2. Monini P, Sgadari C, Grosso MG et al. Clinical course of classic Kaposi’s sarcoma in HIV‐negative patients treated with the HIV protease inhibitor indinavir. AIDS 2009; 23: 534–8. [DOI] [PubMed] [Google Scholar]
- 3. Dewan MZ, Tomita M, Katano H et al. An HIV protease inhibitor, ritonavir targets the nuclear factor‐kappaB and inhibits the tumor growth and infiltration of EBV‐positive lymphoblastoid B cells. Int J Cancer 2009; 124: 622–9. [DOI] [PubMed] [Google Scholar]
- 4. Pyrko P, Kardosh A, Wang W et al. HIV‐1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res 2007; 67: 10920–8. [DOI] [PubMed] [Google Scholar]
- 5. Jiang W, Mikochik PJ, Ra JH et al. HIV protease inhibitor nelfinavir inhibits growth of human melanoma cells by induction of cell cycle arrest. Cancer Res 2007; 67: 1221–7. [DOI] [PubMed] [Google Scholar]
- 6. Yang Y, Ikezoe T, Takeuchi T et al. HIV‐1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signalling. Cancer Sci 2005; 96: 425–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kumar S, Bryant CS, Chamala S et al. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol Cancer 2009; 22: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brüning A, Burger P, Vogel M et al. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol Ther 2009; 8: 226–32. [DOI] [PubMed] [Google Scholar]
- 9. Bernstein WB, Dennis PA. Repositioning HIV protease inhibitors as cancer therapeutics. Curr Opin HIV AIDS 2008; 3: 666–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brunner TB, Geiger M, Grabenbauer GG et al. Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J Clin Oncol 2008; 26: 2699–706. [DOI] [PubMed] [Google Scholar]
- 11. Yang Y, Ikezoe T, Nishioka C et al. NFV, an HIV‐1 protease inhibitor, induces growth arrest, reduced Akt signalling, apoptosis and docetaxel sensitisation in NSCLC cell lines. Br J Cancer 2006; 95: 1653–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chow WA, Guo S, Valdes‐Albini F. Nelfinavir induces liposarcoma apoptosis and cell cycle arrest by upregulating sterol regulatory element binding protein‐1. Anticancer Drugs 2006; 17: 891–903. [DOI] [PubMed] [Google Scholar]
- 13. Gills JJ, Lopiccolo J, Tsurutani J et al. Nelfinavir, A lead HIV protease inhibitor, is a broad‐spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res 2007; 13: 5183–94. [DOI] [PubMed] [Google Scholar]
- 14. Gupta AK, Li B, Cerniglia GJ et al. The HIV protease inhibitor nelfinavir downregulates Akt phosphorylation by inhibiting proteasomal activity and inducing the unfolded protein response. Neoplasia 2007; 9: 271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kraus M, Malenke E, Gogel J et al. Ritonavir induces endoplasmic reticulum stress and sensitizes sarcoma cells toward bortezomib‐induced apoptosis. Mol Cancer Ther 2008; 7: 1940–8. [DOI] [PubMed] [Google Scholar]
- 16. McLean K, VanDeVen NA, Sorenson DR et al. The HIV protease inhibitor saquinavir induces endoplasmic reticulum stress, autophagy, and apoptosis in ovarian cancer cells. Gynecol Oncol 2009; 112: 623–30. [DOI] [PubMed] [Google Scholar]
- 17. Esposito V, Palescandolo E, Spugnini EP et al. Evaluation of antitumoral properties of the protease inhibitor indinavir in a murine model of hepatocarcinoma. Clin Cancer Res 2006; 12: 2634–9. [DOI] [PubMed] [Google Scholar]
- 18. Chow WA, Jiang C, Guan M. Anti‐HIV drugs for cancer therapeutics: back to the future? Lancet Oncol 2009; 10: 61–71. [DOI] [PubMed] [Google Scholar]
- 19. Chen XP, Qiu FY, You JL et al. The synthesis of HIV PI’s analogs and their application in antitumor. Chinese patent 200910037600.4. 2009.
- 20. Zhang XX, Chakrabarti S, Malick AM et al. Effects of different styrene metabolites on cytotoxicity, sister‐chromatid exchanges and cell‐cycle kinetics in human whole blood lymphocytes in vitro. Mutat Res 1993; 302: 213–8. [DOI] [PubMed] [Google Scholar]
- 21. Lui VW, Boehm AL, Koppikar P et al. Antiproliferative mechanisms of a transcription factor decoy targeting signal transducer and activator of transcription (STAT) 3: the role of STAT1. Mol Pharmacol 2007; 71: 1435–43. [DOI] [PubMed] [Google Scholar]
- 22. Tatebe H, Shimizu M, Shirakami Y et al. Synergistic growth inhibition by 9‐cis‐retinoic acid plus trastuzumab in human hepatocellular carcinoma cells. Clin Cancer Res 2008; 14: 2806–12. [DOI] [PubMed] [Google Scholar]
- 23. Shimizu M, Suzui M, Deguchi A et al. Effects of acyclic retinoid on growth, cell cycle control, epidermal growth factor receptor signaling, and gene expression in human squamous cell carcinoma cells. Clin Cancer Res 2004; 10: 1130–40. [DOI] [PubMed] [Google Scholar]
- 24. Park S, Hong SP, Oh TY et al. Paclitaxel augments cytotoxic effect of photodynamic therapy using verteporfin in gastric and bile duct cancer cells. Photochem Photobiol Sci 2008; 7: 769–74. [DOI] [PubMed] [Google Scholar]
- 25. Li B, Li D, Li GG et al. P58(IPK) inhibition of endoplasmic reticulum stress in human retinal capillary endothelial cells in vitro. Mol Vis 2008; 14: 1122–8. [PMC free article] [PubMed] [Google Scholar]
- 26. Yoshida H, Oku M, Suzuki M et al. pXBP1(U) encoded in XBP1 pre‐mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol 2006; 172: 4565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu P, Cheng H, Roberts TM et al. Targeting the phosphoinositide 3‐kinase pathway in cancer. Nat Rev Drug Discov 2009; 8: 627–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009; 9: 550–62. [DOI] [PubMed] [Google Scholar]
- 29. Gupta AK, Cerniglia GJ, Mick R et al. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res 2005; 65: 8256–65. [DOI] [PubMed] [Google Scholar]
- 30. Gaedicke S, Firat‐Geier E, Constantiniu O et al. Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: induction of tumor‐cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res 2002; 62: 6901–8. [PubMed] [Google Scholar]
- 31. Szegezdi E, Logue SE, Gorman AM et al. Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep 2006; 7: 880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yoshida H, Matsui T, Yamamoto A et al. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001; 107: 881–91. [DOI] [PubMed] [Google Scholar]
- 33. Ogata M, Hino S, Saito A et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26: 9220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yorimitsu T, Nair U, Yang Z et al. Endoplasmic reticulum stress triggers autophagy. J Biol Chem 2006; 281: 30299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kabeya Y, Mizushima N, Ueno T et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19: 5720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T and Kuwano H. Inhibition of autophagy by 3‐MA enhances the effect of 5‐FU‐induced apoptosis in colon cancer cells. Ann Surg Oncol 2009; 16: 761–71. [DOI] [PubMed] [Google Scholar]
- 37. Clare M. HIV protease: structure‐based design. Perspect Drug Discovery Des 1993; 1: 49–68. [Google Scholar]
- 38. Laurent N, De Bouard S, Guillamo JS et al. Effects of the proteasome inhibitor ritonavir on glioma growth in vitro and in vivo. Mol Cancer Ther 2004; 3: 129–36. [PubMed] [Google Scholar]
- 39. Pajonk F, Himmelsbach J, Riess K et al. The human immunodeficiency virus (HIV)‐1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radiosensitization in non‐HIV‐associated human cancer cells. Cancer Res 2002; 62: 5230–5. [PubMed] [Google Scholar]
- 40. Bruno RD, Gover TD, Burger AM et al. 17alpha‐Hydroxylase/17,20 lyase inhibitor VN/124‐1 inhibits growth of androgen‐independent prostate cancer cells via induction of the endoplasmic reticulum stress response. Mol Cancer Ther 2008; 7: 2828–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yung H, Korolchuk S, Tolkovsky AM et al. Endoplasmic reticulum stress exacerbates ischemia‐reperfusion‐induced apoptosis through attenuation of Akt protein synthesis in human choriocarcinoma cells. FASEB J 2007; 21: 872–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mihailidou C, Papazian I, Papavassiliou AG et al. CHOP‐dependent regulation of p21/waf1 during ER stress. Cell Physiol Biochem 2010; 25: 761–6. [DOI] [PubMed] [Google Scholar]
- 43. Peng B, Xu L, Cao F et al. HSP90 inhibitor, celastrol, arrests human monocytic leukemia cell U937 at G0/G1 in thiol‐containing agents reversible way. Mol Cancer 2010; 9: 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taiyab A, Sreedhar AS, Rao Ch M. Hsp90 inhibitors, GA and 17AAG, lead to ER stress‐induced apoptosis in rat histiocytoma. Biochem Pharmacol 2009; 78: 142–52. [DOI] [PubMed] [Google Scholar]
- 45. Srirangam A, Mitra R, Wang M et al. Effects of HIV protease inhibitor ritonavir on Akt‐regulated cell proliferation in breast cancer. Clin Cancer Res 2006; 12: 1883–96. [DOI] [PMC free article] [PubMed] [Google Scholar]