

Abstract
Phosphoglycerate kinase 1 (PGK‐1) is a multifunctional protein that is involved in the glycolytic pathway and the generation of the angiogenesis inhibitor angiostatin. In a previous study, we showed that the overexpression of full‐length PGK‐1 in Lewis lung carcinoma (LLC‐1) can reduce tumor growth in vivo by downregulation of COX‐2 expression. Phosphoglycerate kinase 1 has two functional domains: a catalytic domain (CD); and a nucleotide‐binding domain (NBD). To identify the functional domain of PGK‐1 responsible for its antitumor effects, we evaluated the tumorigenicity of LLC‐1 cells overexpressing full‐length PGK‐1 (LLC‐1/PGK), CD (LLC‐1/CD), and NBD (LLC‐1/NBD). Although no difference in tumor cell growth was observed in vitro, the tumor invasiveness was reduced in the LLC‐1/PGK, LLC‐1/CD, and LLC‐1/NBD cells compared to parental LLC‐1 cells in vivo. In addition, in vivo tumor growth retardation by LLC‐1/CD and LLC‐1/NBD cells was observed, similar to that by LLC‐1/PGK cells. However, the reduced stability of COX‐2 mRNA and downregulation of the COX‐2 protein and its metabolite, prostaglandin E2, was only found in LLC‐1/PGK and LLC‐1/NBD cells. Low levels of COX‐2 were also observed in the tumor mass formed by the modified cells when injected into mice. The results indicate that COX‐2 suppression by PGK‐1 is independent of its catalytic activity. COX‐2 targeting by PGK‐1 can be attributed to its NBD and is probably a result of the destabilization of COX‐2 gene transcripts brought about by the mRNA‐binding property of PGK‐1. (Cancer Sci 2010; 101: 2411–2416)
Lung cancer is the leading cause of carcinoma‐related deaths worldwide, and non‐small cell lung carcinoma (NSCLC) constitutes 85% of all lung cancers. Despite advances in lung cancer treatment, poor survival rates are commonly seen in patients with late‐stage disease.( 1 ) Therefore, early diagnosis and novel therapeutic approaches are urgently required. Previous studies indicated that the blocking of oncogenic signaling cascades that targeted the epidermal growth factor receptor‐ and COX‐2‐regulated pathways yielded promising results in specific subsets of lung cancers.( 1 , 2 )
Phosphoglycerate kinase 1 (PGK‐1), an enzyme comprising a common nucleotide‐binding domain (NBD) and two unique catalytic domains (CDI and CDII), is an interesting multifunctional protein.( 3 , 4 , 5 , 6 , 7 , 8 , 9 ) In addition to being an essential enzyme in the glycolytic pathway,( 3 ) PGK‐1 is well known to play an inhibitory role in tumor angiogenesis by facilitating the formation of angiostatin from plasmin.( 8 ) Moreover, PGK‐1 can influence DNA replication and repair in the mammalian nucleus and stimulate viral mRNA synthesis in the cytosol.( 5 , 6 ) The enzyme also serves as an mRNA‐binding protein and is implicated in the negative regulation of the stability of urokinase‐type plasminogen activator receptor (uPAR) mRNA.( 7 ) The reduction in uPAR expression and cellular motility by overexpression of PGK‐1 has been shown in human lung cancer cells.( 7 ) Peptides of PGK‐1 isolated from human colon cancer can stimulate γ‐interferon secretion from infiltrating T lymphocytes, resulting in enhanced T‐cell cytotoxicity against tumor cells.( 9 )
The COX family comprises key prostaglandin biosynthetic enzymes with clinical significance. Cyclooxygenase‐2, the inducible isoform of COX, is overexpressed in early and advanced lung cancer and is associated with a poor prognosis.( 2 , 10 , 11 , 12 , 13 ) Elevated levels of tumor COX‐2 and its metabolite prostaglandin E2 (PGE2) contribute to increasing angiogenesis,( 11 , 14 ) augmentation of tumor invasiveness,( 11 , 15 ) resistance to apoptosis,( 11 , 16 ) and suppression of the immune response.( 17 , 18 , 19 ) It has been shown that inhibition of COX‐2 in lung cancers can reduce tumorigenicity.( 14 , 19 ) Thus, COX‐2 and PGE2 are considered to play pivotal roles in lung tumorigenesis.
In our previous study, we showed that the COX concentration in PGK‐1‐overexpressing Lewis lung carcinoma cells (LLC‐1) is decreased and the generation of Th1 immunity is enhanced, whereby tumor growth is eventually retarded in vivo.( 20 ) In the current study, we further determine the importance of the different PGK‐1 domains, that is, CDI and NBD, in the reduction of COX‐2 levels and regulation of tumor progression. We established LLC‐1 cell lines overexpressing CDI and NBD using the inducible Tet‐Off system (BD Clontech, Palo Alto, CA, USA). The expression levels of the CDI and NBD genes in LLC‐1 could be negatively modulated by the addition of doxycycline (Dox), which in turns refines the effects of CDI and NBD on tumor cells. Similar to full‐length PGK‐1, both the CDI and NBD domains could downregulate the invasive ability of the tumor and reduce in vivo lung cancer cell growth. Moreover, further analyses showed that tumor growth reduced under conditions of NBD overexpression because of the decrease in the levels of COX‐2 and PGE2. The NBD of PGK‐1 lowers the stability of COX‐2 mRNA, whereby it suppresses COX‐2 in lung cancer cells.
Materials and Methods
Cells, mice, and reagents. Lewis lung carcinoma (H‐2b; ATCC CRL‐1642) cells were purchased from the Bioresource Collection and Research Center (Hsinchu, Taiwan). Male C57BL/6 (H‐2b) mice were obtained from the National Laboratory Animal Center (Taipei, Taiwan), and the regulations of the Animal Care Committee of National Yang‐Ming University (Taipei, Taiwan) were followed. Recombinant transforming growth factor‐β1 (TGF‐β1) was obtained from eBioscience (San Diego, CA, USA). Recombinant PGE2 and yeast (Saccharomyces cerevisiae) PGK proteins, anti‐β‐actin (AC‐15) antibody, and actinomycin D were purchased from Sigma‐Aldrich (St. Louis, MO, USA).
Nucleotide‐binding domain and CDI constructs. Full‐length human PGK‐1 cDNA was amplified and cloned into the pRevTRE vector (Clontech Laboratories, Mountain View, CA, USA) as described previously.( 20 ) The CDI and NBD domains of PGK‐1 were amplified from pRevTRE‐PGK using specific primers (Table 1). The insert sequences in pRevTRE‐PGK, pRevTRE‐CDI, and pRevTRE‐NBD were confirmed by automated DNA sequencing.
Table 1.
Sequences of primer pairs used in RT‐PCR or RT–quanti‐tative (q)PCR
| Forward primer/reverse primer | |
|---|---|
| PGK‐1 | 5′‐CGCGGATCCATGTCGCTTTCTAACAAGCTG‐3′ |
| 5′‐CATCTGATGGTTCTCTAGAAACTG‐3′ | |
| PGK‐1/CDI | 5′‐CGGGATCCATGTCGCTTTCTA‐ACAAGCTGACG‐3′ |
| 5′‐CATCTGATGGTTCTCTAGAAACTG‐3′ | |
| PGK‐1/NBD | 5′‐CGGGATCCATGTTGATGAAGAA‐GGAGCTGAAC‐3′ |
| 5′‐CATCTGATGGTTCTCTAGAAACTG‐3′ | |
| COX‐2 | 5′‐TACCAGTCTCTCAATGAGTACC‐3′ |
| 5′‐TGGTAGGCTGTGGATCTTGCACATTG‐3′ | |
| VEGF | 5′‐GTACCTCCACCATGCCAAGT‐3′ |
| 5′‐TCACATCTGCAAGTACGTTCG‐3′ | |
| TGF‐β1 | 5′‐CCTGTCCAAACTAAGGC‐3′ |
| 5′‐GGTTTTCTCATAGATGGCG‐3′ | |
| HIF‐1α | 5′‐GGAATGGCCCAGTGAGAAAA‐3′ |
| 5′‐CCAGCAGAGTGAGAGCATCA‐3′ | |
| ZEB‐1 | 5′‐GTTCGTGATTGTTTGCG‐3′ |
| 5′‐CCAATAGCGTATCCATGC‐3′ | |
| G3PDH | 5′‐ACCACAGTCCATGCCATCAC‐3′ |
| 5′‐TCCACCACCCTGTTGCTGTA‐3′ | |
| COX‐2 (RT‐qPCR) | 5′‐GCCTACTACAAGTGTTTCTTTTTGCA ‐3′ |
| 5′‐CATTTTGTTTGATTGTTCACACCAT ‐3′ | |
| Beta‐2 microglobulin (RT‐qPCR) | 5′‐CCGGAGAATGGGAAGC ‐3′ |
| 5′‐GTAGACGGTCTTGGGC ‐3′ |
CDI, catalytic domain; HIF‐1α, hypoxia‐inducible factor‐1α; NBD, nucleotide‐binding domain; PGK‐1, phosphoglycerate kinase 1; TGF‐β1, transforming growth factor‐β1; VEGF, vascular endothelial growth factor; ZEB1, zinc finger E box‐binding homeobox1.
Preparation of LLC‐1 transfectants. Stable clones were established for LLC‐1/PGK (clone 30), LLC‐1/CDI (clone 14; C14), and LLC‐1/NBD (clone 3; N3) (Fig. 1) as described previously.( 20 ) To analyze tumor growth in vitro, the transfectants (LLC‐1/PGK, LLC‐1/CDI, and LLC‐1/NBD, 5 × 104/mL) were cultured in 10% FBS‐DMEM for 7 days. The cell number was measured daily using the CellTiter96 Aqueous Non‐Radioactive Cell Proliferation Assay (Promega, Madison, WI, USA).
Figure 1.
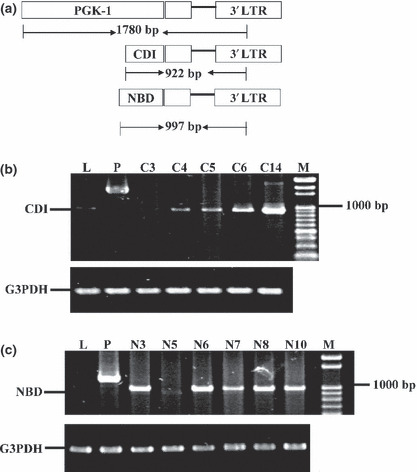
Expression of catalytic domain (CDI) and nucleotide‐binding domain (NBD) of phosphoglycerate kinase 1 (PGK‐1) in transfected lung cancer cells. (a) Schematic diagram of PGK‐1, CDI, and NBD RT‐PCR products. (b,c) Expression of PGK‐1, CDI, NBD, and G3PDH were detected in different clones of Lewis lung carcinoma (LLC‐1) transfectants. C, LLC‐1/CDI clones; L, LLC‐1; M, marker; N, LLC‐1/NBD clones; P, LLC‐1/PGK.
In vivo tumor model. Determination of the in vivo tumor growth was carried out as described previously.( 20 )
Examination of invasive ability. The invasive ability of the tumor cells was assessed as described previously.( 20 )
Detection of gene expression. Total RNA preparation and RT‐PCR were carried out as described previously.( 20 ) RT‐PCR images were quantified by ImageQuant 5.2 software (Amersham Bioscience, Piscataway, NJ, USA). The levels of COX‐2 mRNA in the tumor cells were further quantified using RT–quantitative (q)PCR and SYBR Green I (Bio‐Rad, Hercules, CA, USA) with an ABI7700 System (Applied Biosystems, Foster City, CA, USA). All qPCR values were normalized against β‐2 microglobulin mRNA. The forward and reverse primers were synthesized by Mission Biotech (Taipei, Taiwan) (Table 1).
Western blotting for COX‐2. An immunoblot assay was carried out as described previously,( 20 ) and we used anti‐COX‐2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti‐β‐actin (Sigma) primary antibodies.
Enzyme‐linked immunosorbent assay for PGE2 and TGF‐β1. The levels of PGE2 and TGF‐β1 in the culture‐conditioned medium were measured using commercially available PGE2 enzyme immunoassay (EIA) (Cayman Chemical, Ann Arbor, MI, USA) and TGF‐β1 ELISA kits (BioSource, Camarillo, CA, USA).
Statistical analysis. Data were expressed as the mean ± SD, and statistical significance was assessed by Student’s t‐test.
Results
Overexpression of PGK‐1, CDI, and NBD in lung cancer cells inhibited tumor growth in vivo but not in vitro. A previous study showed that PGK‐1 overexpression in LLC‐1 inhibited tumor growth in vivo.( 20 ) To determine whether the overexpression of a functional domain of PGK‐1 alone (CDI or NBD) can reduce tumor growth, as full‐length PGK‐1 can, the tumor size was determined at different time points in mice s.c. injected with LLC‐1 and LLC‐1 transfectants overexpressing full‐length PGK‐1 (LLC‐1/PGK), CDI (LLC‐1/CDI), and NBD (LLC‐1/NBD) as well as a vector control (LLC‐1/EV). The LLC‐1/PGK‐, LLC‐1/CDI‐, and LLC‐1/NBD‐injected mice (0.1 cm3/21 days) all showed significantly smaller tumor volumes than the control mice (injected with parental LLC‐1 or LLC‐1/EV; 1.25 cm3/21 days) (Fig. 2a). In addition, the tumor growth rates of the LLC‐1/PGK, LLC‐1/CDI, and LLC‐1/NBD cells were restored when the mice were treated with Dox( 20 ) (Fig. 2b) to further prove the tumor‐suppressive effects of PGK, CDI, and NBD. The survival rate of mice injected with LLC‐1/PGK, LLC‐1/CDI, and LLC‐1/NBD cells remained at 100% on day 27, but none of the mice injected with LLC‐1 and LLC‐1/EV cells survived up to day 27 (data not shown). To rule out the possible cytotoxic effects of overexpressed PGK‐1, CDI, and NBD on lung cancer cells, the proliferation rates of the transduced LLC‐1 cells and control cells were compared in vitro; the growth rates of the transduced cell types were found to be similar (Fig. 2c) and comparable with those of the control cells (LLC‐1 and LLC‐1/EV). Thus, the in vivo antitumor responses are a result of PGK‐1, CDI, and NBD expression by LLC‐1 and not the toxic effects of the overexpressed proteins.
Figure 2.
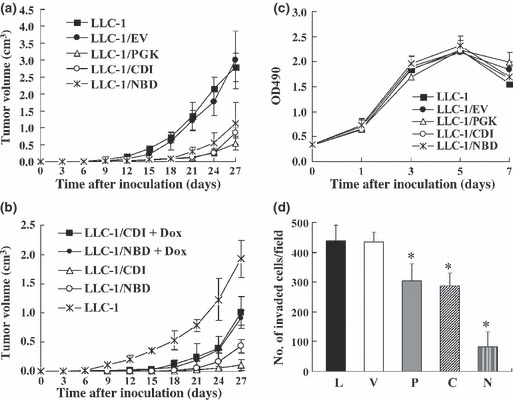
Overexpression of catalytic domain (CDI), nucleotide‐binding domain (NBD), and phospho‐glycerate kinase 1 (PGK‐1) in Lewis lung carcinoma (LLC‐1) cells inhibited in vivo tumor growth and invasive ability. (a,b) C57BL/6 mice (n = 10) were pretreated with or without doxycycline (Dox; 20 μg/mL) for 3 days, and 5 × 105 cells were then s.c. injected into them. (c) The number of tumor cells was measured on different culture days. (d) The invasive ability of the tumor cells (5 × 104) was determined. Ten fields were counted per filter in each group. Each experiment was repeated in triplicate. C, LLC‐1/CDI; EV, LLC‐1/EV (vector control); L, parental LLC‐1; P, LLC‐1/PGK (full‐length PGK‐1); N, LLC‐1/NBD; V, vector. *P < 0.05.
Overexpression of PGK, CDI, and NBD reduced the invasive ability of the tumor cells. The influence of PGK‐1, CDI, and NBD on the invasive ability of the tumor cells was also studied. Compared to the number of LLC‐1 and LLC‐1/EV control cells, the number of LLC‐1/PGK, LLC‐1/CDI and LLC‐1/NBD cells that invaded the Matrigel‐coated filter (Fig. 2d) was significantly lower. A 41%, 40%, and 77% reduction in invasiveness was observed for the LLC‐1/PGK, LLC‐1/CDI, and LLC‐1/NBD cells, respectively, compared to the invasiveness of the controls (Fig. 2d). In addition, overexpression of PGK‐1, CDI, or NBD reduced the invasive ability of human A549 lung cancer cells (data not shown). Thus, the overexpression of PGK‐1, CDI, or NBD can curb primary tumor growth as well as tumor cell invasion.
Cyclooxygenase‐2 transcription was downregulated in LLC‐1/PGK and LLC‐1/NBD cells. To investigate the mechanisms by which cellular invasion and tumor growth were inhibited in vivo by PGK‐1, CDI, and NBD, the transcript levels of COX‐2, TGF‐β1, angiogenic factor (vascular endothelial growth factor [VEGF]), hypoxia‐inducible factor‐1α, and transcriptional repressor of E‐cadherin (zinc finger E box‐binding homeobox1) in the controls and LLC‐1 transfectants were analyzed (Fig. 3). The results showed that the COX‐2 and TGF‐β1 transcripts were downregulated in LLC‐1/PGK cells as compared with the control LLC‐1 and LLC‐1/EV cells (Fig. 3a). In addition, the mRNA level of COX‐2, but not that of TGF‐β1, significantly decreased in the LLC‐1/NBD cells (Fig. 3b,c). However, the overexpression of PGK‐1 and NBD in the LLC‐1 cells did not significantly affect the levels of VEGF, hypoxia‐inducible factor‐1α, or zinc finger E box‐binding homeobox1. Furthermore, LLC‐1/CDI cells displayed the same levels of all the tested genes as the LLC‐1 and LLC‐1/EV control cells (Fig. 3b,c).
Figure 3.
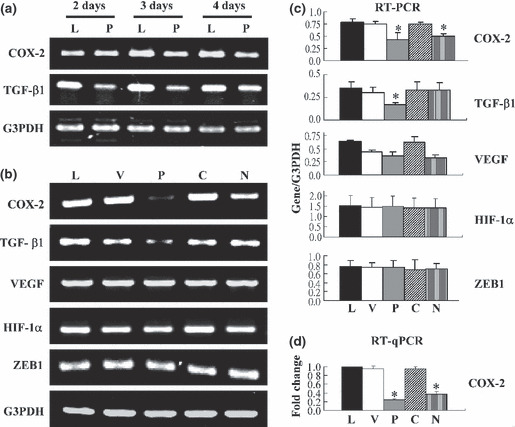
Expression of COX‐2 was downregulated in Lewis lung carcinoma (LLC‐1) cells overexpressing phosphoglycerate kinase 1 (PGK) and nucleotide‐binding domain (NBD). (a) Cells (8 × 104/mL) were cultured for 2, 3, and 4 days. Gene expression was detected by RT‐PCR. (b) Gene expression profiles were surveyed in the 3‐day culture of tumor cells by RT‐PCR. (c) RT‐PCR images were quantified using ImageQuant software and normalized against those of G3PDH. (d) All values from the RT‐qPCR analysis of COX‐2 were normalized, and the gene expression was plotted as fold change relative to the level in parental LLC‐1 cells. L, parental LLC‐1; V, LLC‐1/EV (vector control); P, LLC‐1/PGK; C, LLC‐1/CDI; N, LLC‐1/NBD; TGF‐β1, transforming growth factor‐β1; VEGF, vascular endothelial growth factor; HIF‐1α, hypoxia‐inducible factor‐1α; ZEB1, zinc finger E box‐binding homeobox1. The means of three independent experiments were calculated. *P < 0.05.
Levels of COX‐2 and PGE2 were reduced in the LLC‐1/NBD and LLC‐1/PGK cells. The mRNA and protein levels of COX‐2 in the control cells and transfectants were determined by using RT‐qPCR (Fig. 3d) and Western blot analysis (Fig. 4a). Compared with the control cells, the LLC‐1/PGK and LLC‐1/NBD cells, but not the LLC‐1/CDI cells, contained lower amounts of COX‐2 mRNA and protein. The levels of COX‐2 were also reduced in the A549 cells transfected with PGK‐1 or NBD (data not shown). In addition, Dox‐treated LLC‐1/PGK cells had repressed expression of the PGK transgene, whereby COX‐2 protein expression was recovered (Fig. 4b), further confirming the effects of PGK‐1 on tumor cells.
Figure 4.
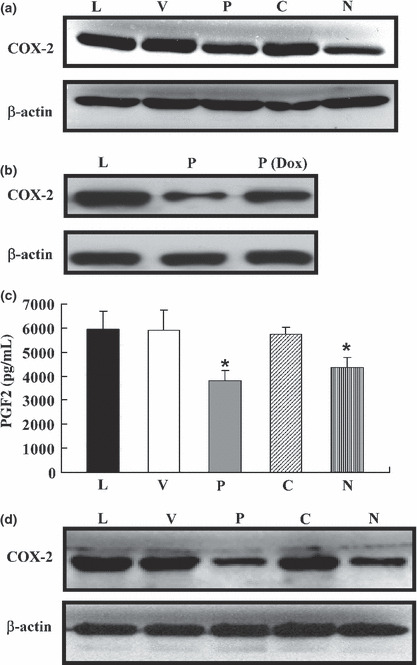
Expression levels of COX‐2 and prostaglandin E2 (PGE2) in Lewis lung carcinoma (LLC‐1) cells overexpressing nucleotide‐binding domain (NBD) and phosphoglycerate kinase 1 (PGK) were markedly downregulated in vitro and in vivo. (a,b) Both COX‐2 and β‐actin proteins were detected. (c) Tumor cells (8 × 104/mL) were cultured for 2 days. The PGE2 concentrations in the culture‐conditioned media were measured. (d) C57BL/6 mice were s.c. injected with 5 × 105 tumor cells. After 36 days, COX‐2 protein expression in the tumors was detected. L, parental LLC‐1; V, LLC‐1/EV (vector control); P, LLC‐1/PGK; C, LLC‐1/CDI; N, LLC‐1/NBD; Dox, doxycycline. Each experiment was repeated in triplicate. *P < 0.05.
We further analyzed the levels of PGE2, a downstream product of COX‐2 that has been known to upregulate cellular migration and invasion as well as angiogenesis.( 2 , 10 , 11 ) The level of PGE2 was significantly reduced in the conditioned medium in which the LLC‐1/PGK (3900 pg/mL) and LLC‐1/NBD (4800 pg/mL) cells were cultured in comparison with the levels in the conditioned medium in which the control cells were cultured (6000 pg/mL) (Fig. 4c). Additionally, mice injected with LLC‐1/PGK and LLC‐1/NBD cells showed reduced COX‐2 protein expression in the primary lung tumor mass (Fig. 4d). These results indicated that the levels of COX‐2 and PGE2 were decreased by the overexpression of PGK‐1 or NBD in LLC‐1 cells.
Prostaglandin E2 promoted invasion of LLC‐1 cells. The variations in the TGF‐β1 mRNA levels (Fig. 3) in the LLC‐1 transfectants were confirmed by ELISA. In accordance with the RT‐PCR results, only the LLC‐1/PGK cells, and not the LLC‐1/CDI and LLC‐1/NBD cells, displayed a decrease in the amount of secreted TGF‐β1 (500 pg/mL) compared to the control LLC‐1/EV cells (1100 pg/mL) (Fig. 5a). The role of TGF‐β1 and PGE2 in the invasion of lung cancer cells was examined by treating LLC‐1 cells with TGF‐β1 and PGE2. When the LLC‐1 cells were stimulated with PGE2, their invasive ability increased, whereas LLC‐1 stimulation with TGF‐β1 did not enhance invasiveness (Fig. 5b). These data indicate that the decrease in the level of PGE2 induced by PGK‐1 and NBD may reduce the invasive ability of LLC‐1 cells.
Figure 5.
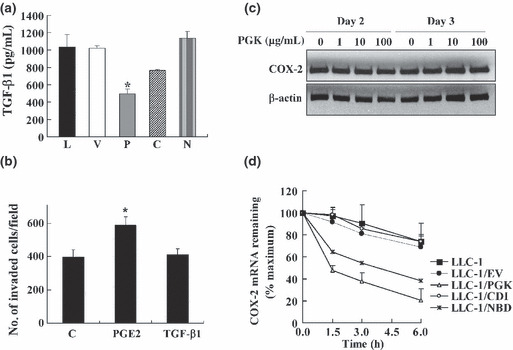
Expression of COX‐2 in Lewis lung carcinoma (LLC‐1) cells overexpressing nucleotide‐binding domain (NBD) and phosphoglycerate kinase 1 (PGK) cells was reduced because of decreased COX‐2 mRNA stability. (a) Transfected tumor cells (8 × 104/mL) were cultured for 4 days. The transforming growth factor‐β1 (TGF‐β1) concentrations in the culture supernatants were measured. (b) LLC‐1 cells (2 × 105/mL) were stimulated with prostaglandin E2 (PGE2; 10 μg/mL) or TGF‐β1 (6 ng/mL) for 2 days. The invasive ability of the treated cells was assessed. (c) LLC‐1 cells were treated with yeast PGK at different concentrations for 2 or 3 days. The level of COX‐2 protein in the treated cells was detected. (d) LLC‐1 and LLC‐1 transfectants were treated with actinomycin D (5 μg/mL) for different time periods. The COX‐2 mRNA levels were determined by RT–quantitative PCR. C, LLC‐1/catalytic domain (CDI); EV, LLC‐1/EV (vector control); L, parental LLC‐1; V, LLC‐1/EV (vector control); P, LLC‐1/PGK; C, LLC‐1/CDI; N, LLC‐1/NBD; EV, vector control. The means of three independent experiments were calculated. *P < 0.05.
Reduction in COX‐2 level due to overexpression of PGK‐1 and NBD was through reduction in Cox‐2 mRNA stability. In addition to being an intracellular protein, PGK‐1 is also secreted into medium by cells, in which the catalytic formation of angiostatin is produced.( 8 ) We investigated whether extracellular PGK downregulates expression of COX‐2 in LLC‐1 cells. Two and three days after adding the recombinant PGK‐1 protein, the PGK‐treated cells were tested for protein expression levels of COX‐2. We found that there was no difference in COX‐2 levels in the PGK‐treated LLC‐1 and non‐treated LLC‐1 cells (Fig. 5c). The data suggest that regulation of COX‐2 can be attributed to the intracellular effects of PGK‐1. Previous studies have shown that PGK‐1 is an mRNA‐binding protein and that it downregulated uPAR, interleukin‐6 (IL‐6), IL‐8, and VEGF in cancer cells overexpressing it.( 7 , 21 ) We inferred that PGK‐1 may be directly involved in the post‐transcriptional regulation of COX‐2 mRNA in LLC‐1/PGK cells. Therefore, we measured the post‐transcriptional level of COX‐2 expression in the PGK‐ and NBD‐overexpressing LLC‐1 cells. Transfectants were treated with actinomycin D to inhibit transcription and analyzed for the COX‐2 mRNA level by RT‐qPCR. The results indicated that the level of COX‐2 mRNA was significantly reduced in the LLC‐1/PGK and LLC‐1/NBD cells (Fig. 5d), but not the LLC‐1/CDI cells, compared to the control cells. Therefore, it is likely that the overexpression of PGK‐1 or NBD led to the destabilization of COX‐2 mRNA in the LLC‐1 cells.
Discussion
Despite recent improvements in chemotherapy and radiotherapy for lung cancer management, novel multimodality treatments are still required to further benefit patients.( 1 , 2 ) The inhibition of COX‐2 represents one such treatment method.( 2 ) In this study, we showed that the inhibition of COX‐2 by overexpression of the NBD of PGK‐1 in lung cancer cells could retard tumor growth in vivo. In addition to reducing COX‐2 expression, PGK‐1 can further limit tumorigenicity by downregulating uPAR,( 7 ) generating angiostatin,( 8 ) and promoting immune responses against cancer cells.( 9 ) The tumor vascularity and growth rate were found to be reduced in fibrosarcoma‐ and pancreatic tumor‐bearing mice injected with recombinant PGK‐1 protein.( 8 ) In prostate cancer, enhanced expression of PGK‐1 was found to lead to the arrest of angiogenesis as a result of angiostatin generation and reduced secretion of pro‐angiogenic factors (VEGF, IL‐6, and IL‐8). However, it has been shown that when prostate cancers metastasize to tissues with high chemokine (C‐X‐C motif) ligand (CXCL)‐12 production, the expression of PGK‐1 is inhibited by CXCL‐12 signaling through chemokine[C‐X‐C motif] receptor 4 (CXCR4), which results in angiogenic switch and metastatic growth.( 21 ) In NSCLC, PGK‐1 overexpression not only activated peripheral immune cells to trigger specific antitumor immunity, but also inhibited the expression of oncogenic factors (COX‐2, PGE2, and TGF‐β1), tumor invasion, and angiogenesis, resulting in the retardation of tumor growth in both immunocompetent and immunodeficient mice.( 20 ) In the current study, we first showed that the overexpression of the functional domains CDI and NBD of PGK‐1 could also lead to reduced tumor invasive ability (Fig. 2d) and in vivo lung cancer growth (Fig. 2a). Thus, the CDI and NBD domains of PGK‐1 play an important role in the regulation of tumor development.
Constitutive COX‐2 overexpression in tumors is responsible for the dysregulation of post‐transcriptional and translational mechanisms, whereby tumorigenesis is stimulated.( 22 ) AU‐rich sequence element (ARE)‐binding proteins bind to the ARE of COX‐2 3′‐untranslated region and influence the fate of COX‐2 mRNA by regulating mRNA degradation, stabilization, or translation.( 22 , 23 ) In serous cancer, the enhanced binding of HuR, an RNA‐binding protein, to COX‐2 ARE was found to contribute to an increase in COX‐2 mRNA stability.( 22 , 24 , 25 ) An mRNA‐binding protein, PGK‐1 has been shown to interact directly with the mRNA coding region sequence of uPAR, resulting in reduced uPAR mRNA stability in H157 human lung carcinoma cells.( 7 ) Despite lacking enzyme activity, mutant PGK‐1 overexpressed in lung carcinoma cells can downregulate uPAR expression and the invasive ability of the tumor cells.( 7 ) We also showed that the overexpression of wild‐type PGK‐1 accelerated COX‐2 mRNA degradation (Fig. 5d), leading to reduced levels of COX‐2 protein and tumorigenicity attenuation (2, 4). Furthermore, even though it lacks catalytic activity, the NBD of PGK‐1 destabilized COX‐2 mRNA (Fig. 5d), which in turn contributed to hampered tumor cell invasive ability and tumor growth (Fig. 2a,d). As PGK‐1 is an mRNA binding protein, the NBD domain of PGK‐1 may interact directly with the mRNA coding region sequence of COX‐2, resulting in reduced COX‐2 mRNA stability. Therefore, the NBD is essential for the PGK‐1‐mediated reduction in COX‐2 mRNA expression at the post‐transcriptional level, and the downregulation of COX‐2 mRNA is independent of the catalytic activity of PGK‐1. The catalytic domain of PGK‐1 (CDI) contains many cysteine residues that may be involved in thiol reductase activity of PGK‐1 on plasminogen for the generation of angiostatin, leading to reduction of tumor vascularity in vivo.( 8 ) Therefore, the CDI domain may be important for PGK‐1‐mediated tumor suppression (Fig. 2a) due to the generation of angiostatin.
In the early phases of tumorigenesis, TGF‐β1 inhibits the proliferation of epithelial cells. Owing to changes in the differentiation of tumors and their sensitivity to TGF‐β1, an increase in tumor TGF‐β1 production enhances cancer progression in the late phase.( 26 ) Elevated TGF‐β1 secretion by many types of tumors is positively related to poor clinical outcome.( 27 ) Overproduction of TGF‐β1 influences proliferation, differentiation, migration, invasion, angiogenesis, and immune surveillance in tumor cells.( 28 ) We showed that the overexpression of full‐length PGK‐1 in lung carcinoma cells reduced the level of TGF‐β1, but the overexpression of NBD or CDI did not (3, 5). The regulation of TGF‐β1 may require coordination between NBD and CDI domains of PGK‐1. However, TGF‐β1 treatment did not enhance the migratory or invasive abilities of LLC‐1 cells (Fig. 5b). Our previous result found that overexpression of full‐length PGK‐1 in LLC‐1 may trigger an effective antitumor immune response in vivo.( 20 ) In the LLC study, Young et al. found that LLC cells can induce immune suppressive activity of immature myeloid cells through production of TGF‐β1.( 28 , 29 ) When anti‐TGF‐β antibodies were added to the TGF‐β1‐containing supernatants, the suppressive activity of bone myeloid cells was diminished. This effect promoted CD4 T cell proliferation in an LLC tumor cell–dentritic cell coculture system.( 30 ) Hence tumor‐derived TGF‐β1 is able to regulate differentiation of myeloid dendritic cells and function of T cells, which can provide a tumor‐supporting microenvironment to accelerate tumor growth. Based on previous studies and our experimental data, the reduction in the levels of tumor‐derived TGF‐β1 by wild‐type PGK‐1 may be involved in the activation of antitumor immunity.
In addition to the downregulation of uPAR and the generation of angiostatin,( 7 , 8 ) PGK‐1 expressed in lung cancer cells also limits the tumor levels of COX‐2 and TGF‐β1, the tumor invasive ability, angiogenesis as well as tumor immunosurveillance, leading to suppression of tumor growth.( 20 ) Interestingly, both CDI and NBD of PGK‐1 possess the ability to inhibit tumor cell invasion and delay tumor growth. The antitumor effects of NBD are evident by decreasing COX‐2 and PGE2 expression. The targeting of COX‐2 by PGK‐1 is independent of its catalytic activity. Taken together, our findings show the detailed mechanism of action of PGK‐1, which may be an effective target for pharmacological treatment or gene therapy intervention for NSCLC.
Acknowledgments
This work was supported by grants from the National Science Council (NSC 98‐2320‐B‐010‐001‐MY3), the Veterans General Hospital University System of Taiwan Joint Research Program, Tsou’s Foundation (VGHUST 99‐P6‐30), and Taipei City Hospital, Taipei, Taiwan.
References
- 1. Sun S, Schiller JH, Spinola M, Minna JD. New molecularly targeted therapies for lung cancer. J Clin Invest 2007; 117: 2740–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee JM, Yanagawa J, Peebles KA, Sharma S, Mao JT, Dubinett SM. Inflammation in lung carcinogenesis: new targets for lung cancer chemoprevention and treatment. Crit Rev Oncol Hematol 2008; 66: 208–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. VandeBerg JL. The phosphoglycerate kinase isozyme system in mammals: biochemical, genetic, developmental, and evolutionary aspects. Isozymes Curr Top Biol Med Res 1985; 12: 133–87. [PubMed] [Google Scholar]
- 4. Michelson AM, Blake CC, Evans ST, Orkin SH. Structure of the human phosphoglycerate kinase gene and the intron‐mediated evolution and dispersal of the nucleotide‐binding domain. Proc Natl Acad Sci USA 1985; 82: 6965–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Popanda O, Fox G, Thielmann HW. Modulation of DNA polymerases α, δ and ε by lactate dehydrogenase and 3‐phosphoglycerate kinase. Biochim Biophys Acta 1998; 1397: 102–17. [DOI] [PubMed] [Google Scholar]
- 6. Ogino T, Iwama M, Kinouchi J, Shibagaki Y, Tsukamoto T, Mizumoto K. Involvement of a cellular glycolytic enzyme, phosphoglycerate kinase, in Sendai virus transcription. J Biol Chem 1999; 274: 35999–6008. [DOI] [PubMed] [Google Scholar]
- 7. Shetty S, Ganachari M, Liu MC, Azghani A, Muniyappa H, Idell S. Regulation of urokinase expression by phosphoglycerate kinase is independent of its catalytic activity. Am J Physiol Lung Cell Mol Physiol 2005; 289: L591–8. [DOI] [PubMed] [Google Scholar]
- 8. Lay AJ, Jiang XM, Kisker O et al. Phosphoglycerate kinase acts in tumor angiogenesis as a disulphide reductase. Nature 2000; 408: 869–73. [DOI] [PubMed] [Google Scholar]
- 9. Shichijo S, Azuma K, Komatsu N et al. Two proliferation‐related proteins, TYMS and PGK1, could be new cytotoxic T lymphocyte‐directed tumor‐associated antigens of HLA‐A2+ colon cancer. Clin Cancer Res 2004; 10: 5828–36. [DOI] [PubMed] [Google Scholar]
- 10. Brown JR, DuBois RN. Cyclooxygenase as a target in lung cancer. Clin Cancer Res 2004; 10: 4266s–9s. [DOI] [PubMed] [Google Scholar]
- 11. Castelao JE, Bart RD III, DiPerna CA, Sievers EM, Bremner RM. Lung cancer and cyclooxygenase‐2. Ann Thorac Surg 2003; 76: 1327–35. [DOI] [PubMed] [Google Scholar]
- 12. Hida T, Yatabe Y, Achiwa H et al. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res 1998; 58: 3761–4. [PubMed] [Google Scholar]
- 13. Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimaki A. Expression of cyclooxygenase‐2 in human lung carcinoma. Cancer Res 1998; 58: 4997–5001. [PubMed] [Google Scholar]
- 14. Põld M, Zhu LX, Sharma S et al. Cyclooxygenase‐2‐dependent expression of angiogenic CXC chemokines ENA‐78/CXC Ligand (CXCL) 5 and interleukin‐8/CXCL8 in human non‐small cell lung cancer. Cancer Res 2004; 64: 1853–60. [DOI] [PubMed] [Google Scholar]
- 15. Dohadwala M, Batra RK, Luo J et al. Autocrine/paracrine prostaglandin E2 production by non‐small cell lung cancer cells regulates matrix metalloproteinase‐2 and CD44 in cyclooxygenase‐2‐dependent invasion. J Biol Chem 2002; 277: 50828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hida T, Kozaki K, Muramatsu H et al. Cyclooxygenase‐2 inhibitor induces apoptosis and enhances cytotoxicity of various anticancer agents in non‐small cell lung cancer cell lines. Clin Cancer Res 2000; 6: 2006–11. [PubMed] [Google Scholar]
- 17. Sharma S, Yang SC, Zhu L et al. Tumor cyclooxygenase‐2/prostaglandin E2‐dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res 2005; 65: 5211–20. [DOI] [PubMed] [Google Scholar]
- 18. Sharma S, Zhu L, Yang SC et al. Cyclooxygenase 2 inhibition promotes IFN‐gamma‐dependent enhancement of antitumor responses. J Immunol 2005; 175: 813–9. [DOI] [PubMed] [Google Scholar]
- 19. Stolina M, Sharma S, Lin Y et al. Cyclooxygenase 2 inhibition promotes IFN‐gamma‐dependent enhancement of antitumor response. Specific inhibition of cyclooxygenase 2 restores antitumor reactivity by altering the balance of IL‐10 and IL‐12 synthesis. J Immunol 2000; 164: 361–70. [DOI] [PubMed] [Google Scholar]
- 20. Tang SJ, Ho MY, Cho HC et al. Phosphoglycerate kinase 1‐overexpressing lung cancer cells reduce cyclooxygenase 2 expression and promote anti‐tumor immunity in vivo . Int J Cancer 2008; 123: 2840–8. [DOI] [PubMed] [Google Scholar]
- 21. Wang J, Wang J, Dai J et al. A glycolytic mechanism regulating an angiogenic switch in prostate cancer. Cancer Res 2007; 67: 149–59. [DOI] [PubMed] [Google Scholar]
- 22. Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase‐2 in human neoplasia: rationale and promise. Cancer Cell 2003; 4: 431–6. [DOI] [PubMed] [Google Scholar]
- 23. Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM. Post‐transcriptional control of cyclooxygenase‐2 gene expression The role of the 3′‐untranslated region. J Biol Chem 2000; 275: 11750–7. [DOI] [PubMed] [Google Scholar]
- 24. Dixon DA, Tolley ND, King PH et al. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase‐2 expression in colon cancer cells. J Clin Invest 2001; 108: 1657–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sengupta S, Jang BC, Wu MT, Paik JH, Furneaux H, Hla T. The RNA‐binding protein HuR regulates the expression of cyclooxygenase‐2. J Biol Chem 2003; 278: 25227–33. [DOI] [PubMed] [Google Scholar]
- 26. Kim R, Emi M, Tanabe K, Uchida Y, Toge T. The role of Fas ligand and transforming growth factor beta in tumor progression: molecular mechanisms of immune privilege via Fas‐mediated apoptosis and potential targets for cancer therapy. Cancer 2004; 100: 2281–91. [DOI] [PubMed] [Google Scholar]
- 27. Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor‐beta and the immune response: implications for anticancer therapy. Clin Cancer Res 2007; 13: 5262–70. [DOI] [PubMed] [Google Scholar]
- 28. Jakowlew SB. Transforming growth factor‐beta in cancer and metastasis. Cancer Metastasis Rev 2006; 25: 435–57. [DOI] [PubMed] [Google Scholar]
- 29. Young MR, Wright MA, Coogan M, Young ME, Bagash J. Tumor‐derived cytokines induce bone marrow suppressor cells that mediate immunosuppres‐sion through transforming growth factor beta. Cancer Immunol Immunother 1992; 35: 14–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu Q, Zhang C, Sun A, Zheng Y, Wang L, Cao X. Tumor‐educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J Immunol 2009; 182: 6207–16. [DOI] [PubMed] [Google Scholar]