

Abstract
Sunitinib malate (Sutent, SU11248) is a small‐molecule multitargeted tyrosine kinase inhibitor (TKI) used for the treatment of renal cell carcinoma and imatinib‐resistant gastrointestinal stromal tumors. Some TKIs can overcome multidrug resistance conferred by ATP‐binding cassette transporter, P‐glycoprotein (P‐gp)/ABCB1, multidrug resistance‐associated protein 1 (MRP1)/ABCC1, and breast cancer resistance protein (BCRP)/ABCG2. Here, we analyzed the effects of sunitinib on P‐gp and on wild‐type and germ‐line mutant BCRPs. Sunitinib remarkably reversed BCRP‐mediated and partially reversed P‐gp‐mediated drug resistance in the respective transfectants. The in vitro vesicle transport assay indicated that sunitinib competitively inhibited BCRP‐mediated estrone 3‐sulfate transport and P‐gp‐mediated vincristine transport. These inhibitory effects of sunitinib were further analyzed in Q141K‐, R482G‐, R482S‐, and F431L‐variant BCRPs. Intriguingly, the F431L‐variant BCRP, which is expressed by a germ‐line mutant allele 1291T>C, was almost insensitive to both sunitinib‐ and fumitremorgin C (FTC)‐mediated inhibition in a cell proliferation assay. Sunitinib and FTC did not inhibit 125I‐iodoarylazidoprazosin‐binding to F431L‐BCRP. Thus, residue Phe‐431 of BCRP is important for the pharmacological interaction with sunitinib and FTC. Collectively, this is the first report showing a differential effect of a germ‐line variation of the BCRP/ABCG2 gene on the pharmacological interaction between small‐molecule TKIs and BCRP. These findings would be useful for improving our understanding of the pharmaceutical effects of sunitinib in personalized chemotherapy. (Cancer Sci 2010; 00: 000–000)
The ATP‐binding cassette (ABC) transporter proteins, particularly P‐glycoprotein (P‐gp/ABCB1), multidrug resistance‐associated protein 1 (MRP1/ABCC1), and breast cancer resistance protein (BCRP/MXR/ABCP/ABCG2) have been extensively studied as key molecules that are involved in the multidrug‐resistant phenotype of cancer cells.( 1 , 2 ) P‐gp effluxes various anticancer agents including vincristine (VCR), paclitaxel (PTX), doxorubicin (DOX), and mitoxantrone (MXR).( 1 , 3 ) BCRP is referred to as a half‐type ABC transporter that functions as a homodimer and transports anticancer agents such as topotecan, irinotecan, SN‐38 (7‐ethyl‐10‐hydroxycamptothecin), methotrexate, and MXR out of cells.( 4 )
Many compounds have been tested for their ability to overcome ABC transporter‐mediated drug resistance. Verapamil, cyclosporine A (CsA), and other compounds have been identified as inhibitors of P‐gp,( 5 , 6 ) while fumitremorgin C (FTC), tamoxifen derivatives, and certain flavonoids inhibit BCRP.( 7 , 8 , 9 , 10 ) Verapamil, for example, directly interacts with P‐gp and competitively interferes with transporter–substrate binding.( 11 ) Co‐administration of inhibitory compounds would be expected to overcome unwanted anticancer drug resistance during chemotherapy, but is also suspected to affect the pharmacokinetics and pharmacodynamics of substrate anticancer drugs.
Recent genetic analyses of the multidrug resistance gene 1 (MDR1) and BCRP genes have revealed that some germ‐line mutations, including single nucleotide polymorphisms (SNP), affect the pharmacological activities of these ABC transporters.( 3 , 12 , 13 ) We previously reported that the germ‐line mutant allele 3587T>G in MDR1 expresses a nonfunctional P‐gp.( 14 ) We also reported variant BCRP SNP cDNAs harboring 421C>A (amino acid substitution Q141K),( 15 ) and this SNP is physiologically important because the pharmacokinetics of diflomotecan, a new camptothecin‐derivative anticancer agent, and the risk of adverse reactions, such as gefitinib‐induced diarrhea, was affected in patients heterozygous for the A421 allele.( 16 ) In addition, we reported that a germ‐line mutant allele 1291T>C expresses the F431L variant of BCRP with lower functional resistance to SN‐38.( 17 ) This suggests that amino acid substitution F431L may affect substrate recognition of SN‐38.
The small‐molecule TKIs, most of which are competitive inhibitors for ATP, are currently used in various clinical settings.( 18 ) Sunitinib malate (Sutent) is an unique ATP‐competitive multitargeted TKI that inhibits platelet‐derived growth factor receptor (PDGFR) α and β, vascular endothelial cell growth factor receptor (VEGFR) types 1 and 2, stem cell factor receptor c‐KIT, FMS‐like TK‐3 receptor, and the glial cell‐line‐derived neutrophic factor receptor.( 19 ) Sunitinib was approved by the Food and Drug Administration in the USA in 2006 and in Japan in 2008 for the treatment of advanced renal cell carcinoma and imatinib‐resistant gastrointestinal stromal tumor.
Recent studies have shown pharmacological interaction between several clinically important TKIs with the ABC transporters P‐gp and BCRP.( 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 ) Regarding pharmacological properties, imatinib, gefitinib, erlotinib, and sunitinib can inhibit the function of ABC transporters and might cause unexpected adverse effects during novel combination chemotherapy with these TKIs and other drugs in early clinical trials.( 28 , 29 , 30 , 31 ) Unfortunately, as in our recent report, the pharmacological inhibitory effects of TKIs on ABC transporters are dependent upon the pairings between the transporter protein, the substrate drug, and TKIs.( 32 ) Moreover, genetic polymorphisms of the ABC transporters are associated with modulations in functional transporter activity.( 4 , 33 ) Therefore, it is difficult to predict the possible pharmacological interactions between TKIs, anticancer drugs, and ABC transporters in individual patient based on the current insufficient evidence.
Sunitinib is expected to be examined for use in combination with conventional chemotherapies in various tumor settings. Most recently, sunitinib was shown to antagonize P‐gp‐ and BCRP‐mediated drug resistance through direct inhibition of their efflux activities.( 26 , 27 ) In the present study, we found that the inhibitory effects of sunitinib on P‐gp and BCRP are mediated by a competitive mechanism. Moreover, we show for the first time that this inhibitory effect of sunitinib on BCRP is cancelled by a germ‐line mutation of BCRP gene (1291T>C) that causes a single amino acid substitution of F431L.
Materials and Methods
Reagents. Sunitinib was kindly provided by Pfizer (Groton, CT, USA). SN‐38 was provided by Yakult Honsha (Tokyo, Japan). Other chemicals were commercially available. The anti‐BCRP polyclonal antibody 3488 was generated as described elsewhere.( 34 )
Cells and drug sensitivity assay. PA317 mouse fibroblast cells, K562 human myelogenous leukemia cells, BCRP‐expressing PA317 cells, BCRP‐expressing K562 cells (K562/BCRP), and P‐gp‐expressing K562 cells (K562/MDR) were established and cultured as previously.( 17 , 21 ) To establish K562/F431L cells, parental K562 cells were transduced with a HaBCRP retrovirus‐harboring Myc‐tagged human BCRP (F431L) cDNA in the Ha retrovirus vector as previously described.( 17 ) After limiting dilution without any selective drugs and screening of over 700 clones, we selected two F431L‐BCRP‐expressing K562 cell clones K562/F431L‐1 and ‐3.
To enrich BCRP‐expressing PA317 cells, cells were labeled with biotin‐labeled anti‐BCRP antibody, and antibody‐attached BCRP‐expressing cells were purified by a magnetic beads system using a MACS streptavidin kit (Miltenyi Biotec, Bergisch Gladbach, Germany). This purification procedure was repeated three or four times, and the cell surface expression of BCRP was confirmed by fluorescence analysis.
The chemosensitivity of the PA317 and K562 cell lines in the presence or absence of sunitinib was evaluated by a cell growth assay in which cell numbers were counted using a Coulter counter or by MTT assay after incubation of the cells for 5 days. The IC50 values (dose of drug achieving 50% inhibition) and the reversal indices (RI50) (concentration of inhibitors sunitinib, CsA, or FTC) that caused a twofold reduction in the IC50 values for anticancer drugs in each resistant cell line) were defined as previously described.( 10 )
Intravesicular transport assay. The vesicular transport assay was done using a rapid centrifugation technique with 3H‐labeled VCR and estrone 3‐sufate (E1S) (Perkin‐Elmer Life Sciences, Boston, MA, USA), essentially as described before.( 21 , 32 ) For Lineweaver–Burk plot analysis, the concentrations of 3H‐labeled VCR were 100, 200, and 400 nmol/L for K562/MDR vesicles, and concentrations of E1S were 50, 100, and 200 nmol/L for K562/BCRP vesicles.
Intracellular accumulation of mitoxantrone and VCR. The effect of sunitinib on cellular accumulation of MXR was determined by flow cytometry as described before.( 32 ) In brief, 5 × 105 K562, K562/BCRP or K562/MDR cells were incubated with 1 μmol/L of MXR for 40 min at 37°C in the absence or presence of sunitinib (1, 3, and 10 μmol/L), CsA (1, 3, and 10 μmol/L), or FTC (1, 3, and 10 μmol/L). MXR fluorescence was measured using a BD LSR II system (Becton Dickinson, San Jose, CA, USA). The cellular uptake of VCR was determined by intracellular accumulation of 3H‐labeled VCR (American Radiolabelled Chemicals, St. Louis, MO, USA). In brief, cells were pre‐incubated with 10 μmol/L sunitinib or CsA for 5 min, and then cultured in the presence of 100 nmol/L of 3H‐labeled VCR for 20 min and washed three times with ice‐cold PBS. The cell pellets were then solubilized and radioactivity was measured using a liquid scintillation counter.
Cellular efflux assay. Cellular efflux assay was done as described before.( 32 ) In brief, cells were incubated with 0.2 μmol/L of 3H‐labeled MXR or VCR for 30 min at 37°C, and washed twice with ice‐cold PBS, and suspended in ice‐cold 3H‐free fresh normal growth medium. At the indicated times, supernatants were collected to measure efflux of [3H] radioactivity levels using a liquid scintillation counter.( 32 )
Cell surface BCRP expression. The cell surface expression of BCRP was determined by fluorescence analysis. Cells were incubated with or without a biotinylated human‐specific monoclonal antibody raised against BCRP (5D3) (100 μg/mL). The cells were then washed and incubated with R‐phycoerythrin‐conjugated streptavidin (400 μg/mL; Becton Dickinson) and fluorescence levels were detected using a BD LSR II system (Becton Dickinson).
Western blot analysis and photoaffinity labeling with iodoarylazidoprazosin (IAAP). Western blotting was performed as previously reported.( 17 ) The photoaffinity labeling assay was done, essentially as previously described.( 26 ) In brief, membrane vesicle fractions from BCRP‐expressing K562 cells (90 μg protein/sample) were pre‐incubated with 0 or 10 μmol/L sunitinib or FTC for 5 min at room temperature in 50 mmol/L Tris‐HCl (pH 7.5). Then, 10 nmol/L [125I]IAAP (2200 Ci/mmol) (Perkin‐Elmer Life Sciences) was added and incubated for an additional 10 min. The sample plate was kept on ice and illuminated with a UV lamp (365 nm, UVP LLC, model B‐100AP; Upland, CA, USA) for 30 min at room temperature. The labeled BCRP protein was solubilized in a buffer containing 1% NP‐40, 0.1% sodium deoxycholate, 20 mmol/L Tris‐HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L EDTA, and immunoprecipitated with the anti‐BCRP antibody BXP‐21 (Millipore, Billerica, MA, USA). Samples were separated by 5–20% SDS–PAGE, and the gels were dried. The binding of [125I]IAAP with BCRP was quantified using the FLA7000 Bioimage analyzer (Fujifilm, Tokyo, Japan) with the software Multi‐Gauge (Fujifilm).
Results
Suppressive effects of sunitinib on BCRP‐ and P‐gp‐mediated drug resistance. The sunitinib dose‐dependently overcame the relative resistance to SN‐38 and MXR in K562/BCRP cells (Fig. 1a and Fig. S1a, Supporting information). The RI50 value for sunitinib, the concentration that caused a 2‐fold reduction in the IC50 for SN‐38 and MXR in K562/BCRP cells, was calculated as shown in Table 1. The RI50 values of sunitinib for SN‐38 and MXR were comparable with those of FTC (0.09 and 0.19 mmol/L) and erlotinib (0.05 and 0.1 mmol/L) as shown in our recent report.( 32 ) Collectively, sunitinib and FTC appeared to exhibit equivalent inhibitory activities against BCRP‐mediated drug resistance in K562/BCRP cells.
Figure 1.
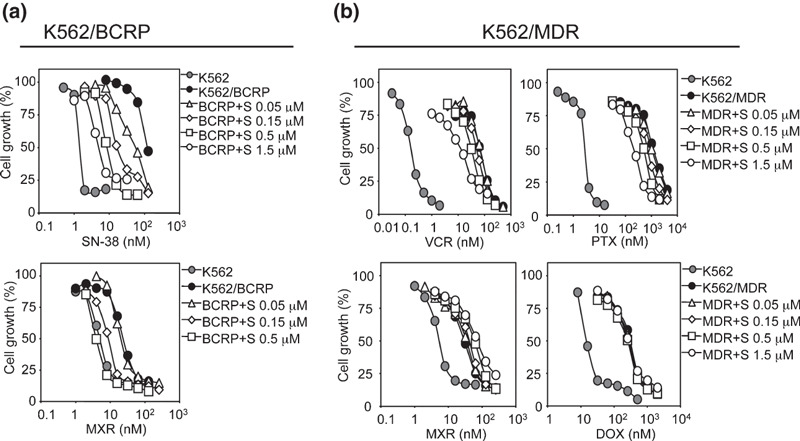
Overcoming drug resistance using sunitinib. The chemosensitivity of K562/breast cancer resistance protein (BCRP) (a) and K562/multidrug resistance protein (MDR) cells (b) to anticancer drugs were determined without (filled circles) or with sunitinib (0.05 μmol/L, open triangles; 0.15 μmol/L, open diamonds; 0.5 μmol/L, open squares; 1.5 μmol/L, open circles). Chemosensitivity of parental K562 cells is shown in gray circles. Cell growth inhibition after 5 days of culture was determined using the MTT assay. The growth inhibition curves were established from the means ± SD from triplicate determinations.
Table 1.
Reversal of BCRP‐mediated drug resistances
| RI50 values (μmol/L) for K562/BCRP | |
|---|---|
| Sunitinib | |
| MXR | 0.10 ± 0.003 |
| SN‐38 | 0.064 ± 0.004 |
Results are means ± SD of triplicate experiments. BCRP, breast cancer resistance protein; MXR, mitoxantrone; RI50, 50% reversal index; SN‐38, 7‐ethyl‐10‐hydroxycamptotecin.
We next tested the effects of sunitinib on P‐gp‐mediated drug resistance in K562/MDR cells. K562/MDR cells were resistant to VCR (relative resistance: ∼390‐fold), PTX (∼450‐fold), DOX (∼20‐fold), and MXR (∼6‐fold), and the effects of sunitinib on overcoming the resistance of K562/MDR cells to these drugs were weak (Fig. 1b). The dose‐dependent effect of sunitinib on P‐gp‐mediated drug resistance was analyzed (Fig. 1b) and the RI50 value of sunitinib on K562/MDR cells was determined as shown in Table 2. These experiments indicated that P‐gp‐mediated resistance to DOX and MXR was not inhibited by sunitinib although sunitinib partially reversed P‐gp‐mediated resistance to VCR and PTX, with RI50 values for sunitinib of 0.44 and 0.3 mmol/L, respectively. As a typical inhibitor for P‐gp, CsA was subjected to the same experiments (Fig. S1c, Supporting information), and the results showed that CsA reversed P‐gp‐mediated drug resistance to VCR, PTX, DOX, and MXR in K562/MDR cells equivalently (Table 2 and Fig. S1c, Supporting information).
Table 2.
Reversal of P‐gp‐mediated drug resistances
| RI50 values (μmol/L) for K562/MDR | ||
|---|---|---|
| Sunitinib | CsA | |
| VCR | 0.44 ± 0.02 | 0.17 ± 0.004 |
| PTX | 0.30 ± 0.06 | 0.20 ± 0.02 |
| DOX | Not determined | 0.29 ± 0.02 |
| MXR | Not determined | 0.24 ± 0.01 |
Results are means ± SD of triplicate experiments. CsA, cyclosporin A; DOX, doxorubicin; MXR, mitoxantrone; PTX, paclitaxel; RI50, 50% reversal index; VCR, vincristine.
Sunitinib co‐treatment consistently increased the intracellular accumulation of MXR and suppressed MXR efflux through BCRP with an efficacy that was similar to FTC (Fig. 2a–c). In contrast, sunitinib did not in K562/MDR cells (Fig. 2d–f). However, sunitinib partially suppressed P‐gp‐mediated VCR efflux and increased intracellular VCR accumulation in K562/MDR cells (Fig. 2g,h). Therefore, the inhibitory effect of sunitinib on P‐gp appeared to be different from that of CsA, and our observations indicate that the inhibitory effects of sunitinib were dependent on the specific substrate involved in the P‐gp‐mediated resistance phenotype.
Figure 2.
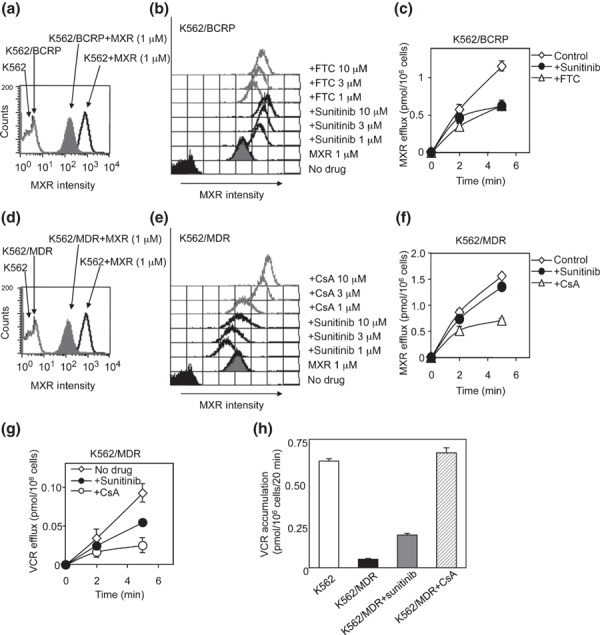
Effects of sunitinib on the uptake and efflux of mitoxantrone (MXR). Intracellular accumulation of MXR in K562 as a control, K562/breast cancer resistance protein (BCRP) (a) and K562/multidrug resistance protein (MDR) (d) cells was determined as the fluorescence intensity of MXR measured by flow cytometry. The histogram for MXR fluorescence intensity shows the intracellular accumulation level of MXR. The cellular uptake of MXR in the presence of sunitinib or fumitremorgin C (FTC) (b, gray lines) or cyclosporine A (CsA) (e, gray lines) in K562/BCRP (b, bold lines), and K562/MDR (e, bold lines) cells was measured as above. The fluorescence intensity patterns without MXR (control) are shown as filled histograms, and those without inhibitors as gray histograms. To measure the cellular efflux of MXR, K562/BCRP (c) and K562/MDR (f) cells were pre‐treated with 200 nmol/L of 3H‐labeled MXR for 30 min and then incubated in fresh growth medium without 3H‐labeled MXR. The efflux of 3H‐labeled MXR was determined by measuring the level of radioactivity exported into the culture medium. The time‐dependent efflux of MXR in the absence of inhibitors is shown as open diamond symbols (c,f). The effects of sunitinib (filled circles), FTC (c, open triangles), and cyclosporine A (CsA) (f, open triangles) on MXR efflux were determined. Results are means ± SD of triplicate determinations. (g) To measure cellular efflux of VCR, K562/MDR cells were pre‐treated with 100 nmol/L of 3H‐labeled VCR for 30 min and then incubated in fresh growth medium without 3H‐labeled VCR. The efflux of 3H‐labeled VCR was determined as above. The time‐dependent efflux of VCR in the presence of sunitinib (filled circles), CsA (open circles) or absence of inhibitor (open diamond symbols) is shown. (h) Cellular uptake of VCR in K562/MDR cells was determined in the absence or presence of 10 μmol/L of inhibitors. The accumulation of VCR is shown for parental K562 (white column), K562/MDR (black column), K562/MDR cells with sunitinib (gray column), and K562/MDR cells with CsA (hatched column). Results are means ± SD of triplicate determinations.
Effects of sunitinib on BCRP‐ and P‐gp‐mediated transports. An intravesicular transport assay was performed to analyze the kinetics of sunitinib inhibition on BCRP‐mediated transport in vitro using membrane vesicles from K562/BCRP cells. As shown in Figure 3(a), ATP‐dependent [3H]E1S transport was dose‐dependently inhibited by sunitinib, similar to that with FTC (IC50 values for sunitinib and FTC: 0.24 and 0.28 μmol/L, respectively). Moreover, the Lineweaver–Burk plot analysis showed that sunitinib acted as a competitive inhibitor for BCRP‐mediated E1S transport (Fig. 3b). The calculated Vmax (pmol/mg/min) was 8.8 for the control condition, 6.1 for sunitinib (at 1 μmol/L), and 8.5 for FTC (at 1 μmol/L), and the calculated Ki values for sunitinib and FTC were both 0.32 μmol/L. Therefore, sunitinib acts as a competitive inhibitor for BCRP‐mediated E1S transport, and our analysis revealed that sunitinib and FTC have equivalent inhibitory activity on BCRP.
Figure 3.
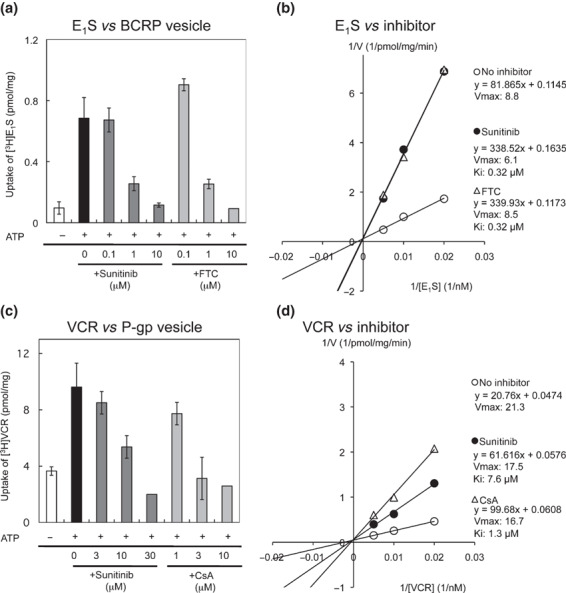
The intravesicular transport of estrone 3‐sufate (E1S) by breast cancer resistance protein (BCRP) and of vincristine (VCR) by P‐glycoprotein (P‐gp). (a) The transport of [3H]E1S was determined by measuring the radioactivity incorporated into the membrane vesicles from K562/BCRP cells, as described in the Materials and Methods. (b) Lineweaver–Burk plot analysis was used to determine the mode of sunitinib inhibition of BCRP in the intravesicular transport assay. The concentration of 3H‐labeled E1S was 50 nmol/L in experiments (a) and 50, 100, and 200 nmol/L, respectively in (b). Sunitinib at 1 μmol/L and FTC at 1 μmol/L were tested in experiment (b). (c) Effects of sunitinib on VCR transport by P‐gp. The transport of [3H]VCR was determined using membrane vesicles from K562/MDR cells as above. (d) Lineweaver–Burk plot analysis was performed to determine the mode of sunitinib inhibition of P‐gp. The concentration of 3H‐labeled VCR was 100 nmol/L in the experiments (c) and 100, 200, and 400 nmol/L, respectively, in (d). Sunitinib at 15 μmol/L and CsA at 5 μmol/L were tested in experiment (d). Results shown in (a) and (c) are means ± SD of triplicate determinations, and results in (b) and (d) are means of duplicate determinations.
Although sunitinib did not inhibit P‐gp‐mediated MXR efflux (as shown in 1, 2 and Fig. S1, Supporting information), sunitinib partially overcame P‐gp‐mediated VCR resistance in K562/MDR cells (1, 2 and Fig. S1b, Supporting information). Therefore, we examined the effect of sunitinib on P‐gp‐mediated VCR transport by a transport assay using membrane vesicles from K562/MDR cells and [3H]VCR as a transporter substrate. These experiments showed that both sunitinib and CsA inhibited P‐gp‐mediated VCR transport (Fig. 3c). However, while 10 mmol/L CsA completely suppressed VCR transport, a higher concentration (30 mmol/L) of sunitinib was required for complete inhibition of P‐gp‐mediated VCR transport, indicating that the inhibitory activity of sunitinib was weaker than that of CsA, with IC50 values of 16.2 and 2.2 μmol/L, respectively. The Lineweaver–Burk plot analysis (Fig. 3d) indicated that the calculated Vmax values for control, sunitinib (at 15 μmol/L)‐treated and CsA (at 5 μmol/L)‐treated samples were similar (21.3, 17.5, and 16.7 pmol/mg/min, respectively). The calculated Ki value for sunitinib was about 7.6 μmol/L and that for CsA was about 1.3 μmol/L for P‐gp‐mediated VCR transport. Therefore, the inhibitory mode of sunitinib for P‐gp‐mediated VCR transport appeared to involve competition, similar to that of CsA. Our analysis also suggested that the inhibitory activity of sunitinib for P‐gp is weaker than that of CsA. Overall, these results indicate that sunitinib acts as a competitive inhibitor on the transporter function of BCRP and P‐gp, and that sunitinib shows better activity against BCRP than against P‐gp.
Effects of sunitinib on BCRP variants. Previous molecular cloning studies of BCRP cDNAs from drug‐selected cells and normal tissues have uncovered functional variants of BCRP with amino acid substitutions and their substrate preferences.( 13 , 35 , 36 , 37 ) The Q141K variant, a widespread SNP in Japanese individuals, is associated with the low protein expression of BCRP,( 15 ) and the F431L variant, also a germ‐line mutation of BCRP, shows a low level of resistance to SN‐38. The R482T and R482G are BCRP variants identified after in vitro selection of culture cells and these variants confer DOX‐ and MXR‐resistances.( 34 ) Before examining the suppressive effect of sunitinib on these BCRP variant‐expressing murine fibroblast PA317 cells, cell populations with high BCRP expression were selectively enriched using immunomagnetic beads. The BCRP protein expression levels were confirmed to be comparable between each enriched variant BCRP‐expressing PA317 cells (Fig. S2a–c, Supporting information). Using these cells, we determined the drug resistance of BCRP‐expressing cells to SN‐38 with or without various concentrations of sunitinib (Fig. 4a), and sunitinib‐mediated reversal of the relative resistance to SN‐38 was calculated (Fig. 4b). To our surprise, sunitinib showed only marginal ability to overcome SN‐38 resistance in the F431L‐BCRP variant, even though the other BCRP variants were all sensitive to sunitinib with comparable efficacy.
Figure 4.
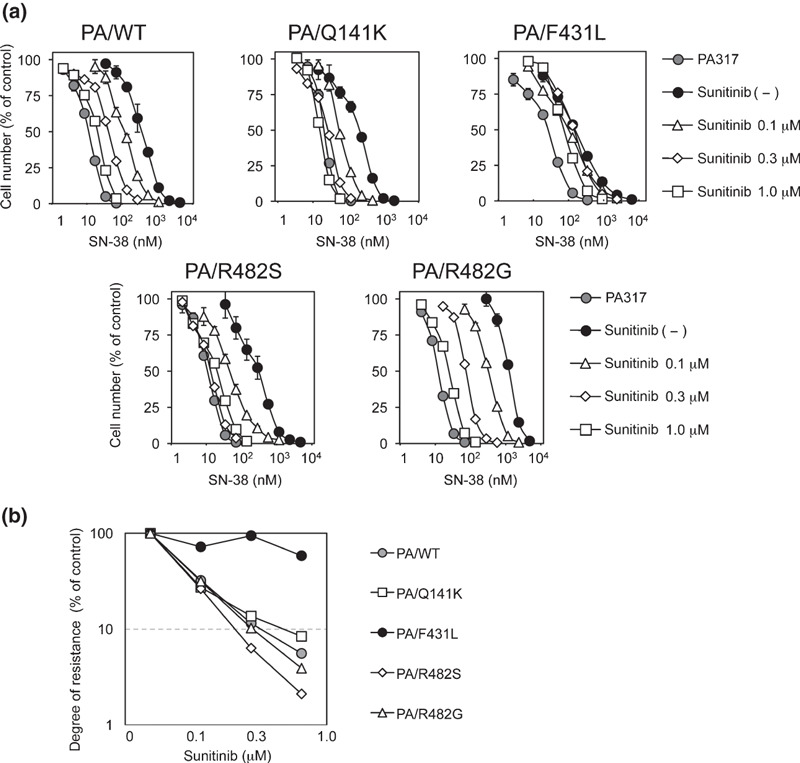
Sunitinib overcomes variant breast cancer resistance protein (BCRP)‐mediated drug resistance. (a) Variant BCRP‐expressing PA317 cell lines were cultured for 5 days with SN‐38 and sunitinib. Cell numbers were determined with a Coulter counter and the cell growth inhibition curves (% of control) are shown. Results are means ± SD of triplicate determinations. (b) BCRP‐mediated resistance to SN‐38 in the presence of sunitinib was determined as IC50 values and relative resistance was calculated as the ratio of the IC50 value in the presence of sunitinib divided by the IC50 value in the absence of sunitinib. Reversal ratios are shown for wild‐type (gray circles), and Q141K (open squares), F431L (filled circles), R482S (open diamonds), and R482G (open triangles) BCRP variant‐expressing cells.
To confirm these observations, we also tested the effect of sunitinib on F431L‐BCRP in different cell lines. The F431L BCRP‐expressing K562 cell clones K562/F431L‐1 and ‐3 were established without the drug selection process. FACS analysis showed that the protein expression of F431L‐BCRP in K562 cells was relatively low (1/8–1/4‐fold compared with wild‐type BCRP‐expressing K562 cells) (Fig. 5a). The reason for the lower protein expression of F431L‐BCRP in K562 cells is unknown, but we could not isolate K562/F431L clones expressing high levels of F431L‐BCRP protein, even after the screening of over 700 clones. These additional experiments also revealed that sunitinib could not overcome SN‐38 resistance conferred by F431L‐BCRP (Fig. 5b). We also examined the inhibition of the F431L‐BCRP variant by the typical BCRP inhibitor FTC and found that the F431L‐BCRP variant was also resistant to FTC‐mediated inhibition (Fig. 5c). These data suggested that the residue Phe‐431 of BCRP would be important for the interaction with sunitinib and FTC.
Figure 5.
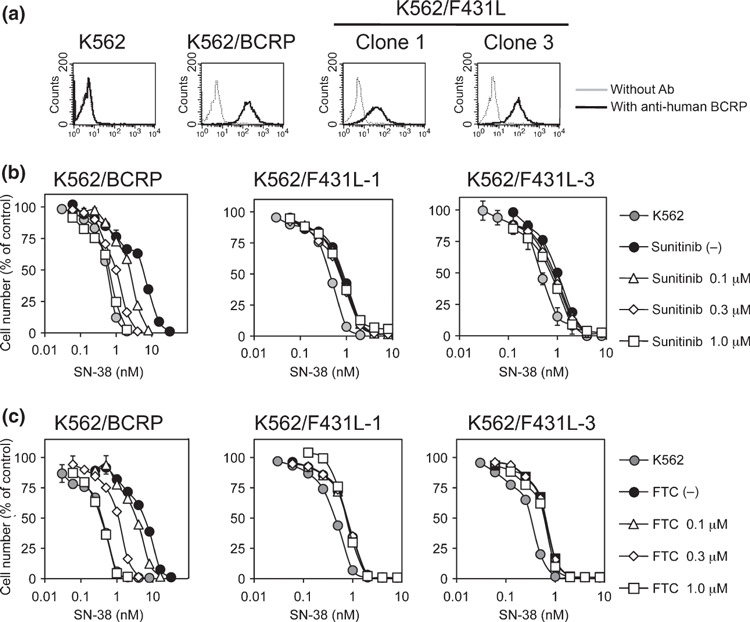
Sunitinib failed to overcome F431L‐breast cancer resistance protein (BCRP)‐mediated drug resistance in K562 cells. Protein expression of BCRP in K562/F431L cells was analyzed by flow cytometry (a). The F431L‐BCRP‐expressing K562 cell lines K562/F431L‐1 and ‐3 were cultured for 5 days with SN‐38 and sunitinib (b) or fumitremorgin C (FTC) (c). Cell growth was determined with a Coulter counter and cell growth inhibition curves (% of control) are shown. Results are means ± SD of triplicate determinations.
Effects of sunitinib on IAAP binding to F431L‐BCRP. We next examined the effect of sunitinib on photoaffinity labeling of wild‐type and F431L‐BCRP with [I125]IAAP‐binding to investigate the direct competition between the substrate IAAP and sunitinib on the F431L variant. Because F431L‐BCRP protein expression was lower than wild‐type BCRP in K562 cell lines (Fig. 6a), [I125]IAAP‐binding to the membrane vesicles prepared from these K562/F431L cells was weaker than that from wild‐type BCRP‐expressing membrane vesicles. Consistent with other reports, sunitinib (10 μmol/L) and FTC (10 μmol/L) inhibited [I125]IAAP‐binding to wild‐type BCRP, whereas [I125]IAAP‐binding to the F431L variant was apparently resistant to sunitinib‐ and FTC‐mediated inhibition (Fig. 6b,c). These data clearly showed that the F431L variant has decreased affinity for physical interactions with sunitinib and FTC.
Figure 6.
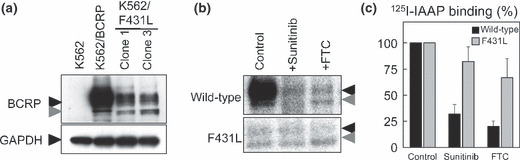
Effect of sunitinib on photoaffinity labeling of wild‐type and F431L‐breast cancer resistance protein (BCRP) with [125I]IAAP. Protein expression of BCRP was analyzed by western blotting (a) in F431L‐BCRP‐transduced K562 cells. Membrane vesicles (90 μg/mL) from K562/BCRP and K562/F431L‐3 cells were pre‐incubated for 5 min with sunitinib or fumitremorgin C (FTC) (10 μmol/L each) and then 10 nmol/L of [125I]IAAP was added for a further 10 min. After cross‐linking by UV irradiation, BCRP proteins were immunoprecipitated and resolved by SDS–PAGE. The binding of [125I]IAAP to BCRPs was visualized (b) and quantified (c) with an FLA7000 instrument. Black and gray arrowheads are BCRP protein as described in reference 17. A representative result of three independent experiments is shown in (b) and the means and SD from three independent experiments are shown in (c).
Overall, our results indicate that sunitinib can inhibit the transporter function of both BCRP and P‐gp, albeit less efficiently for P‐gp. Moreover, the germ‐line BCRP variant F431L showed decreased affinity for sunitinib and therefore would be irrelevant to the pharmacological and physical interaction between BCRP and sunitinib.
Discussion
Various small‐molecule TKIs can modulate the functional activity of ABC transporters such as P‐gp, BCRP, and MRP1.( 38 , 39 , 40 ) We have studied the mode of action of TKIs and ABC transporters, and previously reported the effects of gefitinib and erlotinib on BCRP and P‐gp. We showed that gefitinib and erlotinib are competitive inhibitors for BCRP, while erlotinib is a complex‐type inhibitor for P‐gp.( 21 , 32 , 41 ) In this study, we first examined the activity of sunitinib as a competitive inhibitor for both BCRP and P‐gp, and found that BCRP appeared to be a better target for sunitinib than P‐gp. Second, we investigated the effect of sunitinib on mutants of BCRP, and found that the F431L variant conferred resistance to sunitinib‐mediated suppression. Thus, this is the first report showing that the germ‐line mutation of BCRP/ABCG2 affects the pharmacological interaction with TKIs.
A study by Shukla et al. demonstrated that inhibitory effect of sunitinib on P‐gp‐mediated drug resistance appeared to be partial compared with that on BCRP, and that the ATPase activity of P‐gp was stimulated by higher concentrations of sunitinib than were required for BCRP.( 26 ) Dai et al. reported no significant effect of sunitinib on P‐gp‐mediated drug resistance.( 27 ) In our experiments, sunitinib partially suppressed P‐gp‐mediated resistance to VCR and PTX at sub‐micromolar concentrations, but did not to DOX and MXR. The P‐gp drug interaction sites are thought to be localized to the transmembrane domains and the presence of multiple drug‐binding sites has been suggested.( 42 ) The physical interaction sites for MXR and DOX are not well defined for P‐gp, and the interaction mode of vinca alkaloids with P‐gp seems to be different from that of DOX.( 42 ) Our data suggest that the putative region on P‐gp that interacts with sunitinib may be associated with the VCR‐binding site, but is distinct from the DOX interaction site. Further molecular studies are required to elucidate the modes of interaction between sunitinib and P‐gp.
The germ‐line mutant F431L‐BCRP was previously shown to have low ability to confer drug‐resistance to SN‐38.( 34 ) Our present study also showed that the F431L mutation in BCRP compromised the pharmacological and physical interactions with sunitinib and FTC. Regarding the physical interaction between TKIs and BCRP, gefitinib binds to ATP‐bound BCRP at an as yet unknown binding site,( 43 ) while erlotinib stimulates the ATPase activity of BCRP but does not compete with IAAP‐binding to BCRP.( 23 ) By contrast, a study by Shukla et al. and our data showed a direct interaction between sunitinib and IAAP‐binding sites on BCRP and P‐gp.( 26 ) Thus F431L substitution may reduce substrate/inhibitor‐recognition efficacy or may be an important amino acid residue involved in the functional transporter activity of BCRP.
Phe‐431 is thought to be located near the boundary region between the extracellular side and the second transmembrane domain of BCRP,( 44 , 45 ) but it is still unclear whether Phe‐431 is associated with the substrate‐binding pocket. Li et al. reported that another mutant, F431S‐BCRP, is still functional because pheophorbide A transport by F431S‐BCRP was completely suppressed by FTC.( 44 ) Therefore, they excluded the possible importance of the Phe‐431 residue at the BCRP dimer interface and proposed that Phe‐431 faced the lipid bilayer. Our observations also indicate that the F431L‐BCRP protein forms a dimer (data not shown), but our data appeared to be inconsistent with their conclusion because the F431L‐BCRP variant showed reduced sensitivity to FTC. Unfortunately, we failed to confirm the inhibitory effects of sunitinib and FTC on F431L‐BCRP‐mediated drug transport using the in vitro vesicle transport assay because the membrane vesicles prepared from K562/F431L cells did not show good transport activity in vitro (data not shown). However, we also monitored anti‐BCRP antibody 5D3 reactivity to F431L‐BCRP in the presence or absence of FTC because the direct interaction between FTC and BCRP is thought to stimulate the binding efficacy of the anti‐BCRP antibody 5D3 by inducing a conformational change in BCRP.( 46 ) In this 5D3 reactivity test, the fluorescence intensity associated with 5D3 antibody binding was changed by FTC treatment in K562/BCRP cells, but not in K562/F431L cells (Fig. S3, Supporting information). This experiment suggested that F431L‐BCRP is resistant to the FTC‐induced conformational change required for 5D3 antibody‐binding to BCRP. Overall, we suspect that the F431L variant shows compromised physical interaction with FTC and sunitinib, or altered conformational dynamics that are required for substrate recognition and transport cycling by BCRP.
It is likely that sunitinib administration not only modulates the normal functions of BCRP expressed in digestive organs, the kidney, and the blood–brain barrier, but is also likely to influence the efficacy of anticancer drugs during combination chemotherapy.( 31 ) Although F431L‐BCRP had lower transporter activity than wild‐type BCRP, this mutant BCRP still conferred significant drug‐resistance in both PA317 and K562 cells, so that we should pay attention to functional relevance between drug–drug interaction and this mutant BCRP. Importantly, our findings demonstrate that germ‐line mutations of the BCRP/ABCG2 gene 1291T>C (F431L), affect its pharmacological interaction with sunitinib. Recent pharmacogenetic analyses revealed that two polymorphisms (−15622C/T and 1143C/T) of the BCRP/ABCG2 gene that result in reduced BCRP expression are strongly associated with erlotinib‐ and sunitinib‐induced cytotoxicity in patients.( 30 , 31 ) Taken together, the results of studies investigating the pharmacological interaction between TKIs, ABC transporters, and their substrates should contribute to the understanding of the molecular basis of the pharmacokinetics of sunitinib and other drugs used in chemotherapy. In future personalized medicine, functional analysis of germ‐line mutation affecting efficacy of drug–drug interactions such as F431L‐BCRP with sunitinib would contribute to design for the evidence‐based optimized chemotherapy regimen in each patient.
Supporting information
Fig. S1. Reversal of drug resistance by sunitinib. (a) Breast cancer resistance protein (BCRP)‐mediated resistance to SN‐38 (open circles) or mitoxantrone (MXR) (filled circles) in the presence of sunitinib was determined as IC50 values, and the relative resistance was calculated as the ratio of the IC50 value in the presence of sunitinib divided by the IC50 value in the absence of sunitinib. Similar analyses were performed for P‐gp‐mediated resistance to MXR (filled circles), doxorubicin (DOX) (filled triangles), vincristine (VCR) (open circles), and paclitaxel (PTX) (open diamonds) to investigate the effects of sunitinib (b) and cyclosporine A (CsA) (c).
Fig. S2. Positions of the substituted amino acids in the breast cancer resistance protein (BCRP) protein (a). Variant BCRP protein expression levels were analyzed by western blotting (b) and flow cytometry (c) in selected BCRP‐transduced PA317 cells. For western blotting, cell lysates (10 μg/lane) were resolved by SDS‐PAGE and the expression of BCRP and GAPDH was detected using anti‐BCRP (3488) or anti‐GAPDH antibodies, respectively. For FACS analysis, cells were incubated with or without a biotinylated anti‐BCRP 5D3 antibody, and labeled with R‐phycoerythrin‐conjugated streptavidin.
Fig. S3. Binding of the 5D3 antibody to F431L‐breast cancer resistance protein (BCRP). Interactions between sunitinib and BCRP were monitored with the 5D3 reactivity change, as described in the Materials and Methods. The cells were incubated in the presence or absence of 1, 3, and 10 μmol/L FTC for 5 min at 37°C followed by incubation with the 5D3 antibody (2 μg/mL) for 1 h at 37°C. After incubation with the secondary antibody, the cells were washed and fluorescence intensity was measured using a BDTM LSR II system.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
This work was supported by Grants‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology, and from the Ministry of Health, Labor and Welfare of Japan. We thank Yuka Shimomura for performing the initial experiments in this study, and other laboratory members for their helpful discussions.
References
- 1. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP‐dependent transporters. Nat Rev Cancer 2002; 2: 48–58. [DOI] [PubMed] [Google Scholar]
- 2. Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P‐glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol 2005; 204: 216–37. [DOI] [PubMed] [Google Scholar]
- 3. Ambudkar SV, Kimchi‐Sarfaty C, Sauna ZE, Gottesman MM. P‐glycoprotein: from genomics to mechanism. Oncogene 2003; 22: 7468–85. [DOI] [PubMed] [Google Scholar]
- 4. Sugimoto Y, Tsukahara S, Ishikawa E, Mitsuhashi J. Breast cancer resistance protein: molecular target for anticancer drug resistance and pharmaco‐kinetics/pharmacodynamics. Cancer Sci 2005; 96: 457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res 1981; 41: 1967–72. [PubMed] [Google Scholar]
- 6. Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist 2003; 8: 411–24. [DOI] [PubMed] [Google Scholar]
- 7. Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res 2000; 60: 47–50. [PubMed] [Google Scholar]
- 8. Sugimoto Y, Tsukahara S, Imai Y, Ueda K, Tsuruo T. Reversal of breast cancer resistance protein‐mediated drug resistance by estrogen antagonists and agonists. Mol Cancer Ther 2003; 2: 105–12. [PubMed] [Google Scholar]
- 9. Imai Y, Tsukahara S, Asada S, Sugimoto Y. Phytoestrogens/flavonoids reverse breast cancer resistance protein/ABCG2‐mediated multidrug resistance. Cancer Res 2004; 64: 4346–52. [DOI] [PubMed] [Google Scholar]
- 10. Katayama K, Masuyama K, Yoshioka S, Hasegawa H, Mitsuhashi J, Sugimoto Y. Flavonoids inhibit breast cancer resistance protein‐mediated drug resistance: transporter specificity and structure‐activity relationship. Cancer Chemother Pharmacol 2007; 60: 789–97. [DOI] [PubMed] [Google Scholar]
- 11. Yusa K, Tsuruo T. Reversal mechanism of multidrug resistance by verapamil: direct binding of verapamil to P‐glycoprotein on specific sites and transport of verapamil outward across the plasma membrane of K562/ADM cells. Cancer Res 1989; 49: 5002–6. [PubMed] [Google Scholar]
- 12. Iida A, Saito S, Sekine A et al. Catalog of 605 single‐nucleotide polymorphisms (SNPs) among 13 genes encoding human ATP‐binding cassette transporters: ABCA4, ABCA7, ABCA8, ABCD1, ABCD3, ABCD4, ABCE1, ABCF1, ABCG1, ABCG2, ABCG4, ABCG5, and ABCG8. J Hum Genet 2002; 47: 285–310. [DOI] [PubMed] [Google Scholar]
- 13. Yanase K, Tsukahara S, Mitsuhashi J, Sugimoto Y. Functional SNPs of the breast cancer resistance protein‐therapeutic effects and inhibitor development. Cancer Lett 2006; 234: 73–80. [DOI] [PubMed] [Google Scholar]
- 14. Mutoh K, Mitsuhashi J, Kimura Y et al. A T3587G germ‐line mutation of the MDR1 gene encodes a nonfunctional P‐glycoprotein. Mol Cancer Ther 2006; 5: 877–84. [DOI] [PubMed] [Google Scholar]
- 15. Imai Y, Nakane M, Kage K et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low‐level drug resistance. Mol Cancer Ther 2002; 1: 611–6. [PubMed] [Google Scholar]
- 16. Sparreboom A, Gelderblom H, Marsh S et al. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin Pharmacol Ther 2004; 76: 38–44. [DOI] [PubMed] [Google Scholar]
- 17. Yoshioka S, Katayama K, Okawa C et al. The identification of two germ‐line mutations in the human breast cancer resistance protein gene that result in the expression of a low/non‐functional protein. Pharm Res 2007; 24: 1108–17. [DOI] [PubMed] [Google Scholar]
- 18. Steeghs N, Nortier JW, Gelderblom H. Small molecule tyrosine kinase inhibitors in the treatment of solid tumors: an update of recent developments. Ann Surg Oncol 2007; 14: 942–53. [DOI] [PubMed] [Google Scholar]
- 19. Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol 2007; 25: 884–96. [DOI] [PubMed] [Google Scholar]
- 20. Burger H, Van Tol H, Boersma AW et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004; 104: 2940–2. [DOI] [PubMed] [Google Scholar]
- 21. Yanase K, Tsukahara S, Asada S, Ishikawa E, Imai Y, Sugimoto Y. Gefitinib reverses breast cancer resistance protein‐mediated drug resistance. Mol Cancer Ther 2004; 3: 1119–25. [PubMed] [Google Scholar]
- 22. Elkind NB, Szentpetery Z, Apati A et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib). Cancer Res 2005; 65: 1770–7. [DOI] [PubMed] [Google Scholar]
- 23. Shi Z, Peng XX, Kim IW et al. Erlotinib (Tarceva, OSI‐774) antagonizes ATP‐binding cassette subfamily B member 1 and ATP‐binding cassette subfamily G member 2‐mediated drug resistance. Cancer Res 2007; 67: 11012–20. [DOI] [PubMed] [Google Scholar]
- 24. Brendel C, Scharenberg C, Dohse M et al. Imatinib mesylate and nilotinib (AMN107) exhibit high‐affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia 2007; 21: 1267–75. [DOI] [PubMed] [Google Scholar]
- 25. Polli JW, Humphreys JE, Harmon KA et al. The role of efflux and uptake transporters in N‐{3‐chloro‐4‐[(3‐fluorobenzyl)oxy]phenyl}‐6‐[5‐({[2‐(methylsulfonyl)ethyl]amino} methyl)‐2‐furyl]‐4‐quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos 2008; 36: 695–701. [DOI] [PubMed] [Google Scholar]
- 26. Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib (Sutent, SU11248), a small‐molecule receptor tyrosine kinase inhibitor, blocks function of the ABC transporters, P‐glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos 2009; 37: 359–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dai CL, Liang YJ, Wang YS et al. Sensitization of ABCG2‐overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett 2009; 279: 74–83. [DOI] [PubMed] [Google Scholar]
- 28. Cusatis G, Gregorc V, Li J et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst 2006; 98: 1739–42. [DOI] [PubMed] [Google Scholar]
- 29. McDowell HP, Meco D, Riccardi A et al. Imatinib mesylate potentiates topotecan antitumor activity in rhabdomyosarcoma preclinical models. Int J Cancer 2007; 120: 1141–9. [DOI] [PubMed] [Google Scholar]
- 30. Rudin CM, Liu W, Desai A et al. Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity. J Clin Oncol 2008; 26: 1119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Erp NP, Eechoute K, Van Der Veldt AA et al. Pharmacogenetic pathway analysis for determination of sunitinib‐induced toxicity. J Clin Oncol 2009; 27: 4406–12. [DOI] [PubMed] [Google Scholar]
- 32. Noguchi K, Kawahara H, Kaji A, Katayama K, Mitsuhashi J, Sugimoto Y. Substrate‐dependent bidirectional modulation of P‐glycoprotein‐mediated drug resistance by erlotinib. Cancer Sci 2009; 100: 1701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tamura A, Watanabe M, Saito H et al. Functional validation of the genetic polymorphisms of human ATP‐binding cassette (ABC) transporter ABCG2: identification of alleles that are defective in porphyrin transport. Mol Pharmacol 2006; 70: 287–96. [DOI] [PubMed] [Google Scholar]
- 34. Miwa M, Tsukahara S, Ishikawa E, Asada S, Imai Y, Sugimoto Y. Single amino acid substitutions in the transmembrane domains of breast cancer resistance protein (BCRP) alter cross resistance patterns in transfectants. Int J Cancer 2003; 107: 757–63. [DOI] [PubMed] [Google Scholar]
- 35. Honjo Y, Hrycyna CA, Yan QW et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP‐overexpressing cells. Cancer Res 2001; 61: 6635–9. [PubMed] [Google Scholar]
- 36. Ozvegy‐Laczka C, Koblos G, Sarkadi B, Varadi A. Single amino acid (482) variants of the ABCG2 multidrug transporter: major differences in transport capacity and substrate recognition. Biochim Biophys Acta 2005; 1668: 53–63. [DOI] [PubMed] [Google Scholar]
- 37. Morisaki K, Robey RW, Ozvegy‐Laczka C et al. Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother Pharmacol 2005; 56: 161–72. [DOI] [PubMed] [Google Scholar]
- 38. Hegedus T, Orfi L, Seprodi A, Varadi A, Sarkadi B, Keri G. Interaction of tyrosine kinase inhibitors with the human multidrug transporter proteins, MDR1 and MRP1. Biochim Biophys Acta 2002; 1587: 318–25. [DOI] [PubMed] [Google Scholar]
- 39. Ozvegy‐Laczka C, Cserepes J, Elkind NB, Sarkadi B. Tyrosine kinase inhibitor resistance in cancer: role of ABC multidrug transporters. Drug Resist Updat 2005; 8: 15–26. [DOI] [PubMed] [Google Scholar]
- 40. Noguchi K, Katayama K, Mitsuhashi J, Sugimoto Y. Functions of the breast cancer resistance protein (BCRP/ABCG2) in chemotherapy. Adv Drug Deliv Rev 2009; 61: 26–33. [DOI] [PubMed] [Google Scholar]
- 41. Katayama K, Shibata K, Mitsuhashi J, Noguchi K, Sugimoto Y. Pharmacological interplay between breast cancer resistance protein and gefitinib in epidermal growth factor receptor signaling. Anticancer Res 2009; 29: 1059–65. [PubMed] [Google Scholar]
- 42. Loo TW, Clarke DM. Mutational analysis of ABC proteins. Arch Biochem Biophys 2008; 476: 51–64. [DOI] [PubMed] [Google Scholar]
- 43. Saito H, Hirano H, Nakagawa H et al. A new strategy of high‐speed screening and quantitative structure‐activity relationship analysis to evaluate human ATP‐binding cassette transporter ABCG2‐drug interactions. J Pharmacol Exp Ther 2006; 317: 1114–24. [DOI] [PubMed] [Google Scholar]
- 44. Li YF, Polgar O, Okada M, Esser L, Bates SE, Xia D. Towards understanding the mechanism of action of the multidrug resistance‐linked half‐ABC transporter ABCG2: a molecular modeling study. J Mol Graph Model 2007; 25: 837–51. [DOI] [PubMed] [Google Scholar]
- 45. Hazai E, Bikadi Z. Homology modeling of breast cancer resistance protein (ABCG2). J Struct Biol 2008; 162: 63–74. [DOI] [PubMed] [Google Scholar]
- 46. Ozvegy‐Laczka C, Varady G, Koblos G et al. Function‐dependent conformational changes of the ABCG2 multidrug transporter modify its interaction with a monoclonal antibody on the cell surface. J Biol Chem 2005; 280: 4219–27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Reversal of drug resistance by sunitinib. (a) Breast cancer resistance protein (BCRP)‐mediated resistance to SN‐38 (open circles) or mitoxantrone (MXR) (filled circles) in the presence of sunitinib was determined as IC50 values, and the relative resistance was calculated as the ratio of the IC50 value in the presence of sunitinib divided by the IC50 value in the absence of sunitinib. Similar analyses were performed for P‐gp‐mediated resistance to MXR (filled circles), doxorubicin (DOX) (filled triangles), vincristine (VCR) (open circles), and paclitaxel (PTX) (open diamonds) to investigate the effects of sunitinib (b) and cyclosporine A (CsA) (c).
Fig. S2. Positions of the substituted amino acids in the breast cancer resistance protein (BCRP) protein (a). Variant BCRP protein expression levels were analyzed by western blotting (b) and flow cytometry (c) in selected BCRP‐transduced PA317 cells. For western blotting, cell lysates (10 μg/lane) were resolved by SDS‐PAGE and the expression of BCRP and GAPDH was detected using anti‐BCRP (3488) or anti‐GAPDH antibodies, respectively. For FACS analysis, cells were incubated with or without a biotinylated anti‐BCRP 5D3 antibody, and labeled with R‐phycoerythrin‐conjugated streptavidin.
Fig. S3. Binding of the 5D3 antibody to F431L‐breast cancer resistance protein (BCRP). Interactions between sunitinib and BCRP were monitored with the 5D3 reactivity change, as described in the Materials and Methods. The cells were incubated in the presence or absence of 1, 3, and 10 μmol/L FTC for 5 min at 37°C followed by incubation with the 5D3 antibody (2 μg/mL) for 1 h at 37°C. After incubation with the secondary antibody, the cells were washed and fluorescence intensity was measured using a BDTM LSR II system.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item