

Abstract
Accumulated evidence suggests a major role for the activation of the Sonic Hedgehog (SHH) signaling pathway in the development of neural crest stem cells that give rise to the sympathetic nervous system. We therefore investigated the involvement of SHH signaling in the pathogenesis of neuroblastoma, a common childhood malignant tumor of the sympathetic nervous system. Human neuroblastoma cell lines and a majority of primary neuroblastoma specimens showed high‐level expression of the pathway targets and components, indicating persistent activation of the SHH pathway. All of the neuroblastoma cell lines we examined expressed significant levels of SHH ligand, suggesting an autocrine, ligand‐dependent activation of the SHH pathway in neuroblastoma cells. Inhibition of SHH signaling by cyclopamine induced apoptosis and blocked proliferation in all major types of neuroblastoma cells, and abrogated the tumorigenicity of neuroblastoma cells. Moreover, the knockdown of GLI2 in neuroblastoma BE (2)‐C and SK‐N‐DZ cell lines resulted in the inhibition of colony formation. Our study has revealed a molecular mechanism for the persistent activation of the SHH pathway which promotes the development of neuroblastoma, and suggests a new approach for the treatment of this childhood malignant tumor. (Cancer Sci 2009; 100: 1848–1855)
Sonic Hedgehog (SHH) is a member of the Hedgehog family of signaling proteins that were originally identified by their homology to the Drosophila melanogaster segment polarity gene Hedgehog. SHH induces signaling by binding to its receptor, Patched 1 (PTCH1), which inactivates PTCH1 and prevents it from inhibiting the transmembrane protein Smoothened (SMO). This signaling eventually results in the activation of GLI transcription factors, which, in turn, regulate the expression of many target genes that control cell growth, survival, and differentiation in a wide variety of tissues. Aberrant activation of the SHH signaling pathway has been implicated in the pathogenesis of various types of cancer.( 1 , 2 , 3 )
Neuroblastoma (NB) is a childhood malignant tumor of neural crest origin, arising in the sympathetic nervous system.( 4 ) Several lines of evidence suggest an important role for SHH signaling in regulating the development of neural crest stem cells and the sympathetic nervous system. First, inhibition of SHH signaling in vivo results in neural crest cell death.( 5 ) Second, SHH promotes proliferation but inhibits neuronal differentiation of enteric neural crest stem cells.( 6 ) Third, SHH signaling in neural crest stem cells is essential for the patterning and growth of facial primordia.( 7 ) Finally, SHH signaling promotes the proliferation of postnatal sympathetic cells in culture.( 8 ) As aberrant activation of developmental pathways plays a key role in cancer pathogenesis, we investigated the significance of SHH signaling in NB cell growth and tumorigenesis.
Materials and Methods
Clinical specimens. Forty NB samples were obtained from Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China, with approval from the Office for Scientific Research of the hospitals, and all cases were reviewed by neuropathologists to confirm the diagnosis of NB.
Immunohistochemistry. Tumor sections were deparaffinized, rehydrated, and treated with 10 mM citrate buffer (pH 6.0) at 95°C to retrieve antigens. After quenching endogenous peroxidase activity with H2O2 and blocking with 10% normal horse serum, the sections were incubated sequentially with the primary antibodies goat anti‐SHH (1:50; Santa Cruz Biotechnology, Santa Cruz, CA, USA), goat anti‐PTCH1 (1:100; Santa Cruz Biotechnology), and rabbit anti‐SMO (1:50; Santa Cruz Biotechnology) or rabbit anti‐GLI2 (1:200; Abcam, Cambridge, MA, USA), biotinylated secondary antibodies, and the ABC reagent (Vector Laboratories, Burlingame, CA, USA). The immunostaining was visualized with 3, 3′‐diaminobenzidine (Sigma, St Louis, MO, USA). The sections were then counter‐stained with hematoxylin. Negative controls were performed in each case by replacing the primary antibody with Tris‐buffered saline.
Cell culture. Human NB cell lines SHEP1, SK‐N‐F1, SK‐N‐DZ, and SK‐N‐SH were grown in DMEM supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA). IMR32, CHP212, CHP234, BE(2)‐C, and LAN55n were grown in a 1:1 mixture of DMEM and Ham's nutrient mixture F12 supplemented with 10% FBS. LAN‐6, SMS‐KAN, and SMS‐KCNR were grown in RPMI‐1640 supplemented with 10% FBS. All cells were cultured at 37°C in 5% CO2 humidified incubator and were subcultured at the split ratio of 1:5 every 3 days. The retroviral packaging cell line 293GPG was cultured as previously described.( 9 )
Immunoblotting. Cells were suspended in standard sodium dodecyl sulfate (SDS) sample buffer. Protein concentrations were determined with a Bio‐Rad protein assay kit, using bovine serum albumin (BSA) as the reference. Proteins (80 µg) were separated on SDS‐polyacrylamide gels, transferred to nitrocellulose membranes, and probed with goat anti‐SHH (N‐19, 1:200; Santa Cruz Biotechnology), rabbit anti‐SMO (H‐300, 1:200; Santa Cruz Biotechnology), and rabbit anti‐GLI2 (1:1000; Abcam) or mouse anti‐α‐tubulin (B‐5‐1‐2, 1:5000; Sigma), followed by incubation with horseradish peroxidase–conjugated secondary antibodies (ICN). Proteins were visualized with a SuperSignal West Pico chemiluminescence kit (Pierce, Rockford, IL, USA).
Drug treatment. Cyclopamine was dissolved in ethanol as 5 mM stock solution. Neuroblastoma cells grown in medium containing 2.5% of FBS were treated with vehicle or cyclopamine (20 µm) for various lengths of time. To enhance the SHH signaling pathway, exogenous SHH (R&D Systems, Minneapolis, MN, USA) dissolved in phosphate‐buffered saline (PBS) (3 µg/mL) was used to treat NB cells. The 5E1 monoclonal antibody to the N‐terminal domain of Sonic Hedgehog (SHH‐N) was obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA, USA), and was used at a concentration of 10 µg/mL. Micrographs showing cell morphology were taken with an Olympus IX70 (Olympus, Tokyo, Japan) before cells were collected. Cell survival was analyzed by Trypan blue exclusion assay.
Soft agar assays. Cells were mixed in 0.3% Noble agar in growth medium containing vehicle or 20 µM cyclopamine and plated onto six‐well plates containing a solidified bottom layer (0.6% Noble agar in growth medium) at 1500 cells per well. Colonies were photographed after 14 to 21 days and scored.
Cell cycle assay. Cells were fixed with 70% ethanol, stained with propidium iodide (PI), and analyzed by flow cytometry (BD FACSCalibur system; BD BioSciences, San Jose, CA, USA). The data were analyzed with CellQuest Pro (BD BioSciences) and ModFit 3.1 (Verity Software, Topsham, ME, USA).
Apoptosis assay. The apoptotic ratios of cells were determined with the Annexin V‐FITC apoptosis detection kit (Biovision, Mountain View, CA, USA). Briefly, after 48 h of drug treatment, the cells were collected and washed twice with cold PBS. They were then resuspended in 100 µL of binding buffer, incubated with 5 µL of Annexin V conjugated to FITC and 10 µL PI for 15 min at room temperature, and then analyzed by flow cytometry (Becton‐Dickinson, San Jose, CA, USA). Untreated cells incubated with 5 µL of Annexin V‐FITC and 10 µL PI were used as the negative control. Annexin V+/PI– cells were recognized as apoptotic, Annexin V+/PI + cells were defined as late apoptotic or secondarily necrotic, and Annexin V–/PI– cells were recognized as normal cells. The experiments were repeated at least three times.
Real‐time PCR. After the cells were harvested, their total RNA was extracted and reverse transcribed into cDNA. The levels of CCND1, p21, and control β‐actin mRNA transcripts were determined by quantitative RT‐PCR using the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA), and the specific primers are listed in Supporting Information Table 1. The individual values were first normalized to that of the β‐actin control, and then the ratio of the relative expression levels over that of the vehicle‐treated cells was calculated.
Retroviral constructs and infection. To generate the GLI2 knockdown, three pairs of 60‐bp oligonucleotides containing the human GLI2 siRNA sequences were synthesized and cloned into the HindIII and BglI sites in pSuper‐retro to generate pSuper‐retro/GLI2si. The siRNA sequence that resulted in the most efficient knockdown of GLI2 expression (5′‐GATGATCC GCACCTCACCC‐3′) was chosen for subsequent experiments. pSuper‐retro‐GFPsi was used as control. Retroviruses were produced using the 293GPG packaging cell line as described.( 9 ) Briefly, 293GPG cells were grown to 70% to 90% confluence in a 6‐cm dish and then transfected with 2 µg pSuper‐retro‐GLI2si or pSuper‐retro‐GFPsi using the LipofectAmine Plus protocol (Life Technologies, Carlsbad, CA, USA). Forty hours after transfection, the retrovirus‐containing medium was filtered through a 0.45‐µm filter (Millipore, Billerica, MA, USA) and supplemented with 4 µg/mL polybrene (Sigma). For retroviral infections, BE(2)‐C cells or SK‐N‐DZ cells were seeded in a 6‐cm dish and incubated overnight. At the time of transduction, BE(2)‐C cells or SK‐N‐DZ cells grown to 30% to 40% confluence were infected by adding 3 mL of media containing retrovirus four times at 12 h intervals. One day after the final round of infection, cells were cultured in the presence of puromycin for 3 days, and drug‐resistant cells were pooled.
Statistical analysis. Quantitative data are expressed as the mean ± SD. The two‐tailed Student's t‐test was performed for paired samples. P < 0.05 was considered statistically significant.
Results
Sonic Hedgehog (SHH) signaling pathway components are highly expressed in primary NBs and NB cell lines. To investigate the possible involvement of the SHH signaling pathway in NB pathogenesis, we analyzed 40 human NB specimens for the expression of SHH signaling pathway components by immunohistochemistry. Fifty‐eight to 70% of the tumor samples expressed significant levels of SHH, PTCH1, SMO, and GLI2 (Fig. 1a). The SHH, PTCH1, and SMO staining patterns were consistent with their membrane localization (Fig. 1a), while GLI2 was predominantly localized in the nucleus (Fig. 1a). These observations were also confirmed in human NB cell lines. Immunoblotting revealed high‐level expression of SMO and GLI2 in all NB cell lines we examined (Fig. 1b,c). Using an antibody against the N‐terminus of SHH, we found that all of the cell lines expressed significant levels of SHH‐N (Fig. 1c, SHH‐N), an active 19‐kDa ligand generated by autocatalytic processing of the 45‐kDa SHH precursor (Fig. 1c, SHH). This finding suggests autocrine and/or juxtacrine activation of the SHH signaling pathway in NB cell lines.
Figure 1.
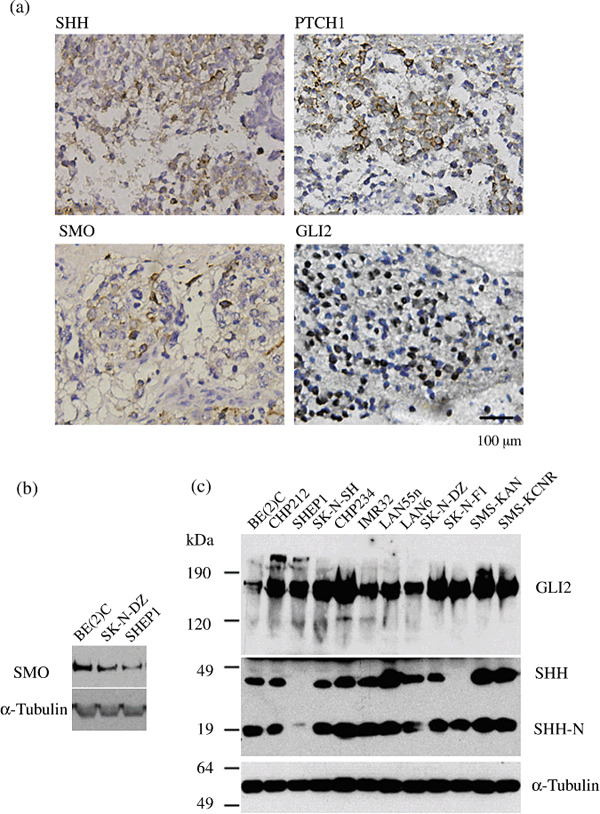
Sonic Hedgehog (SHH) pathway components were highly expressed in human primary neuroblastomas (NBs) and NB cell lines. The expression of SHH, Patched 1 (PTCH1), Smoothened (SMO), and GLI2 in 40 human NB specimens was characterized by immunohistochemistry. Protein expression of SMO, GLI2, and SHH in human NB cells was characterized by immunoblotting. Data are representative images from groups of tissues or immunoblots. The expression of α‐tubulin was used as the control. (a) Immunohistochemical (IHC) staining for SHH, PTCH1, SMO, and GLI2 in NB specimens. Positive cells stained brown. Scale bars, 100 µm. (b) Immunoblot analysis of SMO in human NB cell lines. (c) Immunoblot analysis of SHH and GLI2 expression in NB cell lines. SHH undergoes autocatalytic processing to generate SHH‐N, a 19‐kDa protein with SHH signaling activity.
Sonic Hedgehog (SHH) pathway activation is essential for the survival and proliferation of NB cells. We next examined the biological significance of the activation of SHH signaling in NB cell lines. Three morphologically distinct NB cell lines, which represent the three major human NB cell types, the N‐(neuroblastic, SK‐N‐DZ), S‐(substrate‐adherent and non‐neuronal, SHEP1), and I‐type (intermediate, BE(2)‐C) NB cells, were chosen for the investigation.( 10 ) These cells were treated with cyclopamine, a steroidal alkaloid that specifically antagonizes the SHH signaling pathway through direct interaction with SMO.( 11 , 12 , 13 ) Treatment with 20 µM cyclopamine for 48 h resulted in a significant reduction in the number of viable cells compared to vehicle‐treated cells (Fig. 2a–f). Most of the cyclopamine‐treated cells detached from culture plates, and a significant number of remaining cells exhibited morphological changes, such as cell shrinkage, that are characteristic of apoptosis (Fig. 2).
Figure 2.
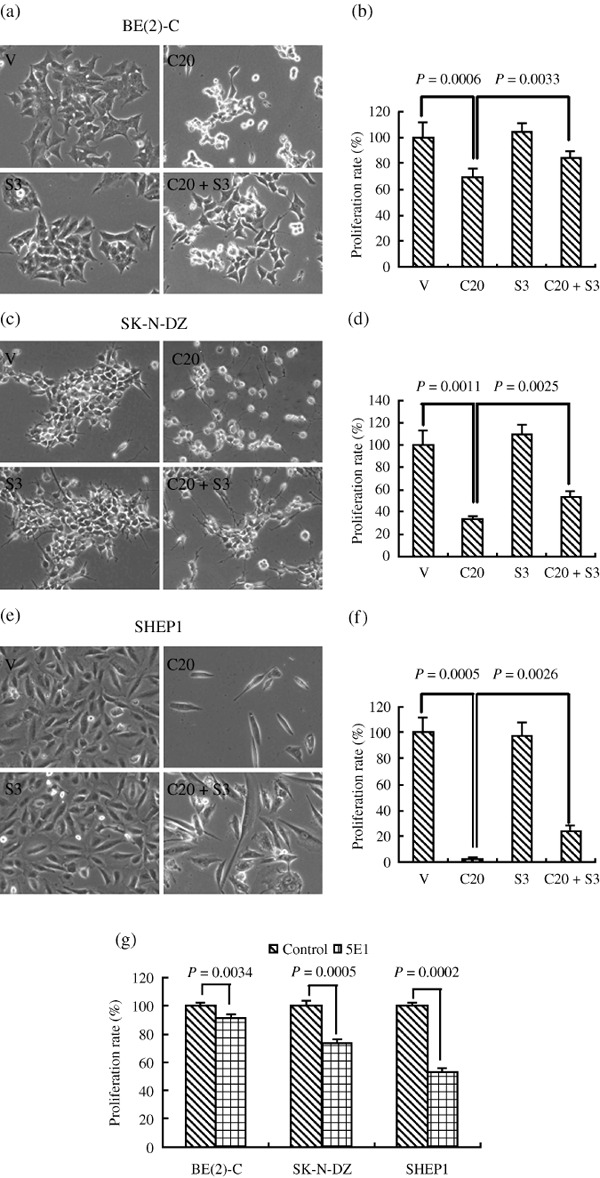
Sonic Hedgehog (SHH) pathway activation is essential for the survival and growth of neuroblastoma (NB) cells. Three representative NB cell lines were treated with vehicle, 20 µM cyclopamine, 3 µg/mL SHH, or 5E1 for 2 days. Cell viability was measured by a Trypan blue exclusion assay and is expressed as a percentage of vehicle control ± SD. The proliferation of control cells at each time‐point was designated as 100%. (a) Representative images of BE(2)‐C cells. (b) The proliferation rate of BE(2)‐C cells (%). (c) Representative images of SK‐N‐DZ cells. (d) The proliferation rate of SK‐N‐DZ cells (%). (e) Representative images of SHEP1 cells. (f) The proliferation rate of SHEP1 cells (%). (g) The proliferation rate of three NB cell lines with 5E1 treatment. Data were obtained from at least three independent experiments. Statistical analysis was performed using the two‐tailed Student's t‐test. V, treated with ethanol alone; C20, 20 µmol/L Cyclopamine; S3, 3 µg/mL SHH.
We also tested the impact of exogenous SHH on NB cell proliferation and the inhibitory effect of cyclopamine. NB cells were treated with 20 µM cyclopamine and/or 3 µg/mL SHH or vehicle for 48 h and their cell growth status was examined under a microscope and quantified as described in the ‘Materials and Methods’ (Fig. 2). Treatment with exogenous SHH alone did not significantly enhance the proliferation of NB cell lines. However, treatment with SHH together with cyclopamine significantly attenuated cyclopamine‐mediated inhibition of proliferation in vitro. In addition, treatment with the 5E1 antibody inhibited the proliferation of NB cell lines (Fig. 2g).
Sonic Hedgehog (SHH) pathway activation is required for the tumorigenicity of NB cells. Growing evidence suggests that many tumors arise from a discrete population of cancer stem cells capable of self‐renewal and differentiation along multiple lineages.( 14 , 15 ) The SHH signaling pathway has been shown to play an important role in the maintenance of cancer stem cells.( 16 , 17 , 18 ) Based on these findings, we investigated the role of SHH signaling in NB cell tumorigenesis. Compared with vehicle control, BE(2)‐C NB cells treated with cyclopamine gave rise to significantly fewer and smaller colonies in a soft agar assay, an in vitro assay of tumorigenicity (Fig. 3). Strikingly, cyclopamine treatment completely abrogated the growth of SK‐N‐DZ cells in soft agar (Fig. 3). SHH alone had no significant effect, but in cells pre‐treated with SHH, the inhibition by cyclopamine of NB cells was only partially effective, as evidenced by colony growth (Fig. 3). We also examined several other NB cell lines, including IMR32, SMS‐SAN, and SMS‐KCNR (data not shown). All cell lines showed reduced ability to grow in soft agar following cyclopamine treatment. These data suggest that SHH signaling is essential for the tumorigenicity of NB cells.
Figure 3.
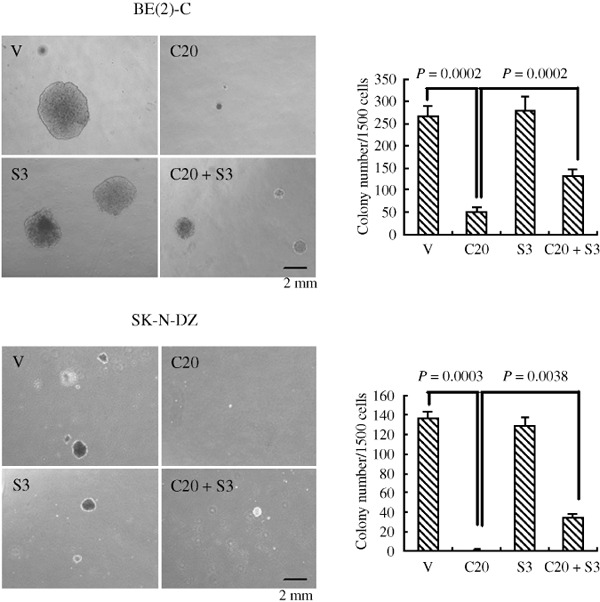
Sonic Hedgehog (SHH) pathway activation is required for the tumorigenicity of neuroblastoma (NB) cells. BE(2)‐C and SK‐N‐DZ cells were treated with 20 µM cyclopamine and/or 3 µg/mL SHH or the vehicle in a soft agar culture system for 2–3 weeks, and the formed colonies were stained with MTT and imaged in order to count colonies. Data represent the average ± SD of at least three independent experiments. Statistical analysis was performed using the two‐tailed Student's t‐test. V, treated with ethanol alone; C20, 20 µmol/L Cyclopamine; S3, 3 µg/mL SHH. Scale bar, 2 mm.
Inhibition of the SHH signaling pathway induces cell G0/G1 arrest and apoptosis in NB cell lines. To elucidate the mechanisms involved in SHH‐mediated proliferation and survival of NB cells, we examined the effects of the SHH signaling pathway on cell cycle and apoptosis in NB cell lines. Flow cytometric analysis was carried out on the three major types of NB cell lines, as shown in Figure 4. Cell cycle analysis of cyclopamine‐treated BE(2)‐C cells revealed a significant increase in the proportion of cells in the G0/G1 phase (79.36 ± 1.39%), along with a significant reduction in both S (8.78 ± 0.16%) and G2/M phases (11.86 ± 1.30%), compared with vehicle‐treated cells (G0/G1, 60.76 ± 1.30%; S, 20.46 ± 0.73%; G2/M, 18.79 ± 0.80%). Similar results were obtained in the SK‐N‐DZ and SHEP1 cell lines (Fig. 4). These data show that cyclopamine treatment blocks the G0/G1‐S transition and induces G0/G1 arrest in NB cell lines. It also demonstrated a significant inhibition of NB proliferation by cyclopamine. To investigate the cell death induced by cyclopamine, we assessed the apoptosis of NB cells by flow cytometry. As shown in Figure 5, cyclopamine treatment increased the apoptosis rate of BE(2)‐C cells significantly, compared with vehicle‐treated cells. Similar results were obtained in the SK‐N‐DZ and SHEP1 cells (Fig. 5). Real‐time PCR revealed that cyclopamine decreases the mRNA level of CCND1, while increasing that of p21. SHH partially reversed these effects (Fig. 6).
Figure 4.
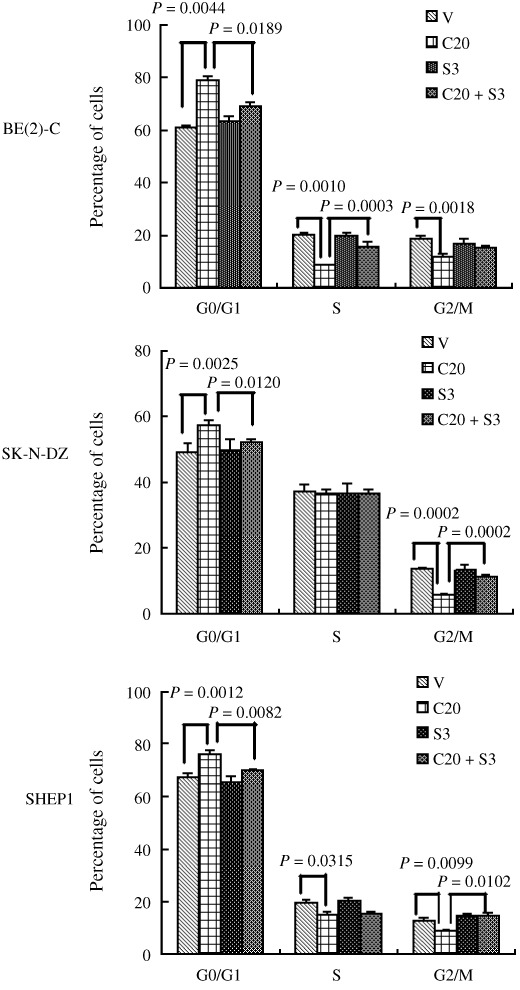
Sonic Hedgehog (SHH) pathway activation affects the cell cycle of neuroblastoma (NB) cells. Three types of NB cell lines were treated with 20 µM cyclopamine and/or 3 µg/mL SHH or the vehicle alone for 48 h and the status of cell cycling was analyzed by FACS analysis. Data represent the average ± SD of at least three independent experiments. Statistical analysis was performed using the two‐tailed Student's t‐test. V, treated with ethanol alone; C20, 20 µmol/L Cyclopamine; S3, 3 µg/mL SHH.
Figure 5.
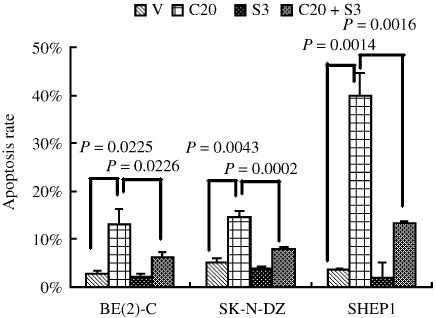
Sonic Hedgehog (SHH) pathway activation affects apoptosis of neuroblastoma (NB) cells. Three types of NB cell lines were treated with 20 µM cyclopamine and/or 3 µg/mL SHH or the vehicle for 48 h and apoptosis was analyzed by FACS. The Annexin V+/PI– cells were considered to be in the early stage of apoptosis, the Annexin V+/PI+ cells were in the late stage of apoptosis, Annexin V–/PI+ cells were necrotic, and Annexin V–/PI– cells were healthy cells. Data are expressed as the mean percent ± SD of each group of cells from three independent experiments. V, treated with ethanol alone; C20, 20 µmol/L Cyclopamine; S3, 3 µg/mL SHH.
Figure 6.
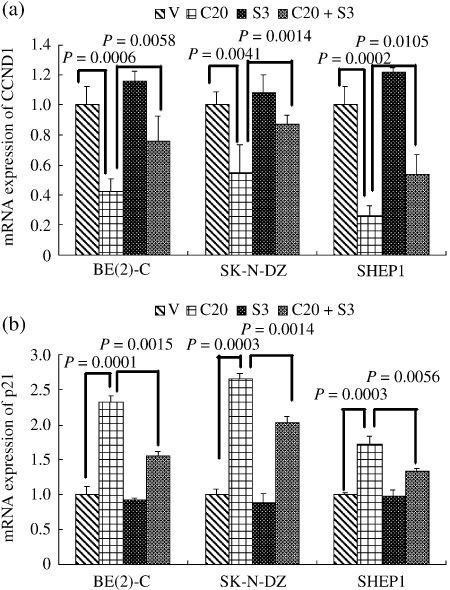
Relative mRNA levels of CCND1 and p21 determined by RT‐PCR in three representative neuroblastoma (NB) cell lines treated with vehicle or 20 µM cyclopamine and/or SHH 3 µg/mL for 2 days. Data represent the average ± SD of at least three independent experiments. Statistical analysis was performed using the two‐tailed Student's t‐test. Cyclopamine. V, treated with ethanol alone; C20, 20 µmol/L Cyclopamine; S3, 3 µg/mL SHH.
GLI2 is essential for the tumorigenicity of human NB cells. The observation that GLI2 is highly expressed in almost all of the NB cell lines and in most human NBs (Fig. 1) implicates a more general role for GLI2 in the development of NB. We speculated that GLI2 might be important for the tumorigenicity of NB cells. We generated three GLI2 siRNA‐expressing retroviral constructs that targeted different regions of the GLI2‐coding sequence, and chose the most efficient sequence (Fig. 7a). Retroviruses produced from the constructs were able to knock down the level of GLI2 by about 40–50% in the NB cell lines BE(2)‐C and SK‐N‐DZ (Fig. 7a). Both BE(2)‐C cells and SK‐N‐DZ cells with reduced levels of Gli2 gave rise to significantly fewer and smaller colonies in soft agar than the control cells (Fig. 7b–c). Thus, the downregulation of GLI2 inhibited the clonogenic activity of NB cells. Downregulation of GLI2 also decreased the mRNA level of CCND1, while increasing that of p21, in NB cell lines (Fig. 7d–e).
Figure 7.
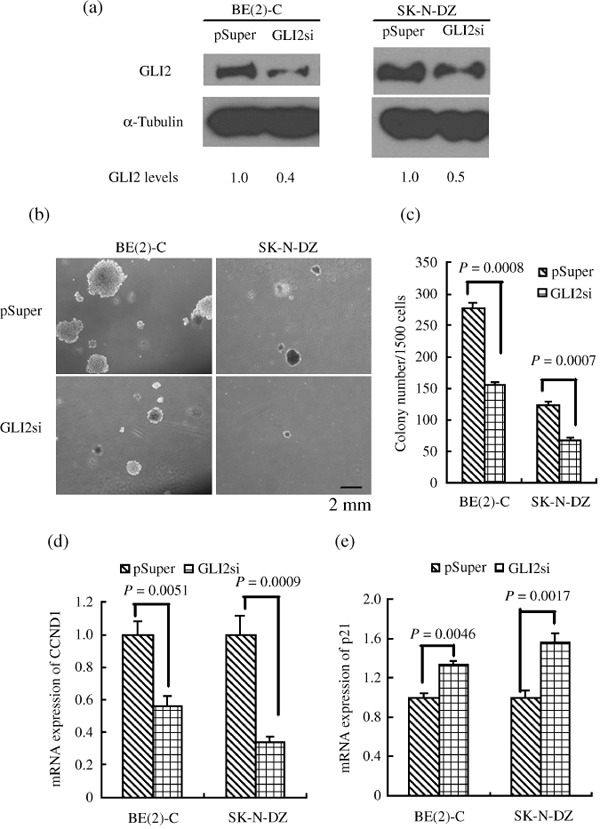
Knockdown of GLI2 inhibited colony growth of neuroblastoma (NB) cell lines in soft agar. (a) Immunoblot analysis of GLI2 expression levels in the indicated NB cell lines. Forty to 50% reduction of GLI2 levels was obtained. α‐Tubulin levels are shown as the loading control. (b) Phase contrast images of the colony growth of the indicated NB cell lines in soft agar after culture for 14 to 21 days. (c) Numbers of colonies per 1500 plating cells after 14 to 21 day culture. Colonies that contained more than 50 cells or were larger than 0.5 mm were scored. (d) Relative mRNA levels of CCND1 as determined by RT‐PCR in NB GLI2 knockdown cells. (e) Relative mRNA levels of p21 as determined by RT‐PCR in NB GLI2 knockdown cells. Each bar represents the average ± SD of three independent experiments. Statistical analysis was performed using the two‐tailed Student's t‐test. Scale bar, 2 mm.
Discussion
The results presented in this paper establish that the SHH signaling pathway is widely activated in human NB cell lines and primary tumors, manifesting as high‐level expression of the pathway targets and components SHH, PTCH1, SMO, and GLI2. We further demonstrated that the activation of this pathway is critical for the growth and survival of NB cells. Cyclopamine treatment, which blocks SHH signaling, led to decreased proliferation and increased apoptosis in NB cells, and abolished the ability of NB cells to grow in soft agar. Interestingly, SHEP1 cells were much more sensitive to the inhibition of cyclopamine than BE(2) C and SK‐N‐DZ cells. The different susceptibility of SHEP1 cells may stem from their differential expression of SHH signaling components. SHEP1 cells with lower level expression of SHH had a higher susceptibility to cyclopamine. SHH alone had no significant effect, but for the cells pre‐treated with SHH, the inhibition of cyclopamine on NB cells was partially reversed in cell and colony growth. More importantly, knocking down the expression of GLI2 in NB cell lines resulted in inhibition of colony formation. These findings suggest an essential role for SHH pathway activation in the maintenance of the tumorigenicity of NB cells.
This study reveals a mechanism for the activation of the SHH signaling pathway in NB cells. There were significant levels of SHH expression in all of the NB cell lines we examined. The tumor cells expressed both the SHH precursor and the mature signaling form, SHH‐N, which can activate the SHH pathway in an autocrine and/or juxtacrine manner. Moreover, 5E1, a monoclonal antibody to the N‐terminal domain of Sonic Hedgehog (SHH‐N), could inhibit the proliferation of NB cell lines. Since SHH pathway activation is essential for NB cell survival and proliferation, the finding that NB cells are capable of producing their own SHH ligand suggests that ligand‐dependent activation of the SHH pathway( 19 ) may play a key role in NB pathogenesis.
Blockage of the SHH signaling pathway by cyclopamine treatment induced apoptosis and G1 arrest of NB cell lines. These results indicated that the SHH pathway affects the end‐point for NB cell lines through cell apoptosis and cell cycle arrest. Previous studies have shown that the activity of the SHH pathway can increase levels of the anti‐apoptotic protein BCL2 and promote survival of medulloblastoma cells via upregulation of BCL2.( 20 ) On the other hand, SHH pathway activation also has mitogenic effects. SHH signaling can promote continued cell cycle progression in proliferating neural precursor cells by maintaining expression of G1‐phase cyclins.( 21 ) D‐cyclin activity is necessary for cell cycle progression through G1 phase to S phase, and p21/CIP1 can induce G1 arrest and block S‐phase entry by inactivating Cyclin‐dependent kinases (CDK)s.( 22 ) We therefore examined the expression of CCND1 and p21 in NB cells. Our results show that the blockage of SHH signaling induced G0/G1 arrest with decreased CCND1 and increased p21 expression, which are consistent with a previous study.( 23 ) These data suggest that SHH signaling may regulate the cell cycle of NB cells by influencing the expression of CCND1 and p21 proteins.
We also found that the GLI2 transcription factor plays an important role in the tumorigenicity of NB cells. The GLI2 transcription factor is indispensable for mouse development, as GLI2 knockout mice die prenatally and exhibit neural tube defects that include a complete loss of the floor plate and a reduction of V3 interneurons.( 24 , 25 ) On the other hand, mice lacking GLI1 seem to be normal and fertile, suggesting that the GLI1 protein is not required for SHH signaling in mice.( 26 ) As found in many other cancer cell types,( 27 , 28 , 29 ) our data show that most NB tumor samples and all of the 12 NB cell lines examined express a high level of GLI2 but not GLI1. In addition, knocking down GLI2 inhibited colony formation. Thus, GLI2 may become an attractive target for therapeutic intervention for the treatment of NB.
Supporting information
Table S1. Primer sequences
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (no. 30600189). We thank Dr Han‐Fei Ding (Cancer Center, The Medical College of Georgia) for providing NB cell lines and technical assistance.
Correction added after publication 20 July 2009. Hongjuan Cui was added in the authors list.
References
- 1. Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol 2005; 6: 306–17. [DOI] [PubMed] [Google Scholar]
- 2. Riobo NA, Manning DR. Pathways of signal transduction employed by vertebrate Hedgehogs. Biochem J 2007; 403: 369–79. [DOI] [PubMed] [Google Scholar]
- 3. Rubin LL, De Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov 2006; 5: 1026–33. [DOI] [PubMed] [Google Scholar]
- 4. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 2003; 3: 203–16. [DOI] [PubMed] [Google Scholar]
- 5. Ahlgren SC, Bronner‐Fraser M. Inhibition of sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Curr Biol 1999; 9: 1304–14. [DOI] [PubMed] [Google Scholar]
- 6. Fu M, Lui VC, Sham MH, Pachnis V, Tam PK. Sonic hedgehog regulates the proliferation, differentiation, and migration of enteric neural crest cells in gut. J Cell Biol 2004; 166: 673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev 2004; 18: 937–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Williams Z, Tse V, Hou L, Xu L, Silverberg GD. Sonic hedgehog promotes proliferation and tyrosine hydroxylase induction of postnatal sympathetic cells in vitro . Neuroreport 2000; 11: 3315–9. [DOI] [PubMed] [Google Scholar]
- 9. Ory D, Neugeboren B, Mulligan R. A stable human‐derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci USA 1996; 93: 11400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ross RA, Biedler JL, Spengler BA. A role for distinct cell types in determining malignancy in human neuroblastoma cell lines and tumors. Cancer Lett 2003; 197: 35–9. [DOI] [PubMed] [Google Scholar]
- 11. Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev 2002; 16: 2743–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen‐mediated inhibition of target tissue response to Shh signaling. Science 1998; 280: 1603–7. [DOI] [PubMed] [Google Scholar]
- 13. Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998; 125: 3553–62. [DOI] [PubMed] [Google Scholar]
- 14. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414: 105–11. [DOI] [PubMed] [Google Scholar]
- 15. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med 2006; 355: 1253–61. [DOI] [PubMed] [Google Scholar]
- 16. Bar EE, Chaudhry A, Lin A et al . Cyclopamine‐mediated hedgehog pathway inhibition depletes stem‐like cancer cells in glioblastoma. Stem Cells 2007; 25: 2524–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu S, Dontu G, Mantle ID et al . Hedgehog signaling and Bmi‐1 regulate self‐renewal of normal and malignant human mammary stem cells. Cancer Res 2006; 66: 6063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small‐cell lung cancer. Nature 2003; 422: 313–7. [DOI] [PubMed] [Google Scholar]
- 19. Jacob L, Lum L. Deconstructing the hedgehog pathway in development and disease. Science 2007; 318: 66–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bar EE, Chaudhry A, Farah MH et al . Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am J Pathol 2007; 170: 347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kenney AM, Rowitch DH. Sonic hedgehog promotes G (1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol 2000; 20: 9055–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gartel AL, Serfas MS, Tyner AL. p21‐negative regulator of the cell cycle. Proc Soc Exp Biol Med 1996; 213: 138–49. [DOI] [PubMed] [Google Scholar]
- 23. Morton JP, Mongeau ME, Klimstra DS et al . Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. PNAS 2007; 104: 5103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ding Q, Motoyama J, Gasca S et al . Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 1998; 125: 2533–43. [DOI] [PubMed] [Google Scholar]
- 25. Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 1998; 125: 2759–70. [DOI] [PubMed] [Google Scholar]
- 26. Park HL, Bai C, Platt KA et al . Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 2000; 127: 1593–605. [DOI] [PubMed] [Google Scholar]
- 27. Kim Y, Yoon JW, Xiao X et al . Selective down‐regulation of glioma‐associated oncogene 2 inhibits the proliferation of hepatocellular carcinoma cells. Cancer Res 2007; 67: 3583–93. [DOI] [PubMed] [Google Scholar]
- 28. Fan L, Pepicelli CV, Dibble CC et al . Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004; 145: 3961–70. [DOI] [PubMed] [Google Scholar]
- 29. Grachtchouk M, Mo R, Yu S et al . Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat Genet 2000; 24: 216–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item