

Abstract
Recently, small molecule inhibitors of transforming growth factor‐β (TGF‐β) type I receptor kinase/activin receptor‐like kinase‐5 (ALK5) have been developed to target TGF‐β signalling as a therapeutic strategy for combating cancer. In the present study, the authors examined a novel small molecule inhibitor of ALK5, 3‐((5‐([1,2,4]triazolo[1,5‐a]pyridin‐6‐yl)‐4‐(6‐methylpyridin‐2‐yl)thiazol‐2‐ylamino)methyl)benzonitrile (EW‐7203) in breast cancer cells to determine if it has potential for cancer treatment. The inhibitory effects of EW‐7203 on TGF‐β‐induced Smad signalling and epithelial‐to‐mesenchymal transition (EMT) were investigated in mammary epithelial cells using luciferase reporter assays, immunoblotting, confocal microscopy and wound healing assays. In addition, the suppressive effects of EW‐7203 on mammary cancer metastasis to the lung were examined using a Balb/c xenograft model system. The novel ALK5 inhibitor, EW‐7203, inhibited the TGF‐β1‐stimulated transcriptional activation of p3TP‐Lux and pCAGA12‐Luc. In addition, EW‐7203 decreased phosphorylated Smad2 levels and the nuclear translocation of Smad2 was increased by TGF‐β1. In addition, EW‐7203 inhibited TGF‐β1‐induced EMT and wound healing of NMuMG cells. Furthermore, in xenografted Balb/c mice, EW‐7203 inhibited metastasis to the lung from breast tumors. The novel ALK5 inhibitor, EW‐7203, efficiently inhibited TGF‐β1‐induced Smad signalling, EMT and breast tumor metastasis to the lung in vivo, demonstrating that EW‐7203 has therapeutic potential for breast cancer metastasis to the lung. (Cancer Sci 2011; 102: 1889–1896)
Metastasis is the primary cause of death among cancer patients, and breast cancer patients are no exception.( 1 , 2 ) During breast cancer progression, transforming growth factor‐β (TGF‐β) levels are elevated( 3 , 4 ) and the tumor‐suppressing function of TGF‐β is abrogated.( 5 ) Transforming growth factor‐β has at least two roles during carcinogenesis, for although it was initially found to be a tumor‐suppressing cytokine, it also acts as a mediator of metastasis to the lung.( 6 , 7 ) During the initial stage of metastasis, cancer cells undergo epithelial‐to‐mesenchymal transition (EMT), which involves loss of epithelial cell polarity, acquisition of the mesenchymal phenotype, greater motility and invasiveness, and disruption of cell‐to‐cell adhesion.( 8 )
The EMT is generally induced in epithelial cells by signals released from mesenchymal cells that compose the stroma of normal and neoplastic tissues. Furthermore, members of the TGF‐β cytokine family are the foremost characterized inducers of EMT during embryonic development, wound healing, fibrotic diseases and cancer pathogenesis,( 7 , 9 ) which suggests that TGF‐β might induce EMT via multiple signalling mechanisms. The canonical TGF‐β signalling pathway is activated when TGF‐β binds to TGF‐β type II receptor (TβRII). Initially, this binding facilitates activation of TGF‐β type I receptor (TβRI) kinase (activin receptor‐like kinase 5 [ALK5]),( 10 ) which contains a kinase domain that phosphorylates receptor‐associated Smad2 and Smad3. This enables them to bind to Smad4 to form a heteromeric Smad complex, which in turn translocates to the nucleus and mediates gene transcription by binding to Smad‐binding elements (SBE) in the promoters of its target genes.( 11 )
In addition to the canonical Smad pathway, other pathways have been implicated in the transduction and regulation of TGF‐β signalling.( 12 ) However, the molecular mechanisms that connect Smad‐independent pathways to TGF‐β receptor signalling complex remain elusive and might involve ALK5‐independent mechanisms.( 13 )
Based on the above molecular pathways, diverse strategies have been developed to block TGF‐β signalling including EMT and cancer metastasis.( 14 ) For example, SB‐431542, SD‐208 and SM16 are small molecules that block the catalytic activity of TβRI and have been reported to be potent antitumor or anti‐metastatic agents.( 15 , 16 , 17 ) It has also been reported that neutralizing antibody against TGF‐β ligand has a suppressive effect on lung metastasis,( 18 ) and that a Smad‐binding peptide aptamer inhibits TGF‐β signalling and EMT.( 19 ) To determine whether the targeting of ALK5 offers an effective therapeutic approach to the suppression of breast cancer metastasis, we examined the effects of the novel ALK5 inhibitor EW‐7203 (3‐((5‐([1,2,4]triazolo[1,5‐a]pyridin‐6‐yl)‐4‐(6‐methylpyridin‐2‐yl)thiazol‐2‐ylamino)methyl)benzonitrile) on motility, Smad signalling and EMT in NMuMG and MCF10A cells in vitro and on the development of metastasis in vivo using an orthotopic xenograft model.
Materials and Methods
Reagents and plasmids. EW‐7203 and SB‐505124 were synthesized by D‐K Kim. Human recombinant TGF‐β1 was purchased from R&D Systems (Minneapolis, MN, USA). The reporter constructs, p3TP‐Lux and pCAGA12‐Luc, were kindly provided by Dr Jeong‐Seok Nam, and β‐galactosidase expression plasmid was provided by Dr Seong‐Jin Kim (both of Gachon University of Medicine and Science, Incheon, Korea).
ALK5 in vitro kinase assay. IC50 values of EW‐7203 against ALK5 and p38α protein kinases were determined using FlashPlate (Perkin Elmer, Boston, MA, USA) according to the manual provided by the supplier (ProQinase, Freiburg, Germany). The reaction cocktail used to determine the activity of p38α and ALK5 kinases contained: variable amounts of enzyme and substrate, casein or ATF2; 50 mM HEPES‐NaOH; 10 mM Tris–HCl (pH 7.5); 20 mM NaCl; 3 mM MgCl2; 3 mM MnCl2; 3 μM Na3VO4; 2 mM DTT; 4% glycerol, 1 μM [γ‐33P]‐ATP (∼6 × 105 cpm/well). All assays were performed using the Beckman Coulter Biomek 2000 robotic system (Brea, CA, USA). 32Pi incorporation was determined using a microplate scintillation counter (PerkinElmer).
Cell lines and cultures. Human mammary epithelial cells (MCF10A) and mouse mammary epithelial cells (NMuMG) were grown as previously described.( 6 ) Human keratinocyte cells (HaCaT), mouse mammary tumor cells (4T1) and human hepatoma cells (HepG2) were grown as previously described.( 9 )
SRB assay. 4T1 cells were seeded in 96‐well plates with a density of 5000 cells and various concentrations of EW‐7203 were treated in DMEM containing 0.5% FBS for 72 h. Cells were fixed by treating with cold 10% trichloroacetic acid (TCA) and stained with 0.4% (w/v) sulforhodamine B (SRB) dissolved in 1% acetic acid. Bound dye was solubilized with 10 mM Tris (pH 10.5) and absorbance was measured at 570 nm using an ELISA reader (Bio‐Rad, Hercules, CA, USA).
Establishment of stable HaCaT (3TP‐Lux) cells. To produce 3TP‐Lux stable cell lines, HaCaT cells were transfected with the p3TP‐Lux (neo) expression plasmid using VivaMagic (Vivagen, Seoul, South Korea) according to the manufacturer’s instructions. Transfected cells were cultured for 4 weeks in the presence of 500 μg/mL G418 and several single clones were isolated and their luciferase activities were measured.
Luciferase reporter gene assay. HepG2 cells were transfected with reporter plasmid (p3TP‐Lux or pCAGA12‐Luc) and β‐galactosidase expression plasmid using either VivaMagic (Vivagen) or polyethyleneimine (PEI) (Sigma Aldrich, St Louis, MO, USA). Twenty‐four hours after transfection, cells were treated in 0.2% FBS containing TGF‐β1 (2 ng/mL) in the presence or absence of EW‐7203 for 20 h. Luciferase activities were determined using cell lysates and data were normalized with respect to β‐galactosidase activity. HaCaT (3TP‐Lux) stable cells were treated with 0.2% FBS containing TGF‐β1 (2 ng/mL) with/without EW‐7203 for 20 h and luciferase activities were determined using cell lysates.
Immunoblot analysis. Total cell lysates were prepared using lysis buffer containing 50 mM Tris, 0.5% sodium deoxycholate, 150 mM NaCl, 1% NP‐40, protease inhibitor cocktail (Roche, San Francisco, CA, USA) and phosphatase inhibitor (Na3VO4) for 20 min on ice. Equal amounts of proteins were then subjected to 10% SDS‐PAGE and transferred to membranes. Membranes were then blocked and incubated with one of the following antibodies: anti‐Smad2 (Abfrontier, Seoul, Korea), anti‐E‐cadherin, anti‐N‐cadherin, anti‐Vimentin (BD Transduction Laboratories, San Jose, CA, USA), anti‐phospho‐Smad2 (Upstate, Boston, MA, USA) or anti‐β‐actin (Sigma, St Louis, MO, USA). Proteins were detected using an ECL kit (Amersham Biosciences, Piscataway, NJ, USA).
Wound healing assay. NMuMG cells were serum‐starved in medium containing 0.2% FBS and wounded using plastic micropipette tips. Wounded cells were then treated with TGF‐β1 (2 ng/mL) in the presence or absence of EW‐7203 (1 μM) for several hours. Cell motilities were assessed by phase‐contrast microscopy at ×100 magnification.
Confocal microscopy. For Smad2/3 staining, MCF10A cells were treated in 0.5% horse serum containing TGF‐β1 (2 ng/mL) in the presence or absence of EW‐7203 for 1.5 h. For E‐cadherin and ZO‐1 staining, NMuMG cells were treated with 0.2% FBS containing TGF‐β1 (2 ng/mL) in the presence or absence of EW‐7203 for 48 h. Anti‐Smad2/3, anti‐E‐cadherin (BD Transduction Laboratories, San Jose, CA, USA) and anti‐ZO‐1 (Invitrogen, Carlsbad, CA, USA) antibodies were used as primary antibodies, and Cy3‐conjugated goat anti‐mouse IgG, FITC‐conjugated goat anti‐mouse IgG and Rhodamine (TRITC)‐conjugated goat anti‐rabbit IgG antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) were used as secondary antibodies. Cells were analyzed using the LSM 510 META laser confocal microscopy system (Carl Zeiss Microimaging Inc., Thornwood, NY, USA). Cells were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) to label nuclei.
RNA isolation and RT‐PCR. MCF10A cells were treated in 0.5% horse serum containing TGF‐β1 (1 ng/mL) in the presence or absence of EW‐7203 and total RNA were isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. cDNA were synthesized using AMV RTase (Promega, Madison, WI, USA) and oligo dT primer over 1 h at 42°C. The synthesized cDNA was subjected to PCR amplification using Taq polymerase (Promega) and the following gene‐specific primers (for human gene): GAPDH, (forward) 5‐ACATCGCTCAGACACCATGG‐3 and (reverse) 5‐GTAGTTGAGGTCAATGAAGGG‐3; E‐cadherin, (forward) 5‐ATTCTGGGGATTCTTGGAGG‐3 and (reverse) 5‐GGTCAGTATCAGCCGCTTTC‐3; and N‐cadherin, (forward) 5‐GTGCCATTAGCCAAGGGAATTCAGC‐3 and (reverse) 5‐GCGTTCCTGTTCCACTCATAGGAGG‐3. Amplified DNA was analyzed by agarose gel electrophoresis.
Matrigel invasion assay. 4T1 cells were seeded at 4 × 104 cells/well on the upper chamber of a transwell in serum‐free medium with or without TGF‐β1 (2 ng/mL) in the presence or absence of ALK5 inhibitors. After incubation for 20 h at 37°C in 5% CO2, the cells remaining on the upper surface of the membrane were removed with a cotton swab, and DAPI‐stained cells remaining on the bottom surface were observed using fluorescence microscopy. Average cell number per view field was obtained from five random fields.
Animal study. Eight‐week‐old female Balb/c mice were purchased from Orient Bio Inc. (Seoul, Korea). Animals were maintained according to the Guidelines for the welfare and use of animals in cancer research( 20 ) in a temperature‐controlled room (at 22°C) and supplied food and water ad libitum. 4T1 cells (1 × 104) were transplanted into the left thoracic mammary fat pads and tumor‐bearing mice were treated with either saline (vehicle) or EW‐7203 (40 mg/kg, in 100 μL saline) intraperitoneally. Metastatic nodules on left lobe lung surfaces were counted by Indian ink injection into tracheas, as previously described.( 21 )
Results
Specific inhibition of ALK5 by EW‐7203. Radioisotopic kinase assay was used in order to quantify ALK5 inhibition by EW‐7203. The IC50 values of EW‐7203 against ALK5 and p38α protein kinases were determined to be 0.00926 μM and 3.78 μM, respectively (Table 1). EW‐7203 strongly inhibited ALK5 kinase, and its effect on ALK5 was 400‐fold than that on p38α protein kinase.
Table 1.
IC50 values for EW‐7203 against activin receptor‐like kinase 5 (ALK5) and p38α†
| Protein kinase | IC50 (μM) |
|---|---|
| ALK5‡ | 0.00926 |
| p38α | 3.78 |
†ALK5 was expressed as human recombinant glutathione‐S‐transferase‐fusion proteins in Sf9 insect cells by using a baculovirus expression system. p38α was expressed as human recombinant proteins in an Escherichia coli expression system. ‡Casein and myelein basic protein were used as substrates against ALK5 and p38α, respectively.
EW‐7203 inhibits TGF‐β1‐stimulated luciferase activity. To examine the effect of EW‐7203 on Smad signaling, we performed luciferase reporter assays using HepG2 cells transiently transfected with either TGF‐β‐responsive pCAGA12‐Luc plasmid or 3TP‐Lux plasmid. TGF‐β1 treatment resulted in 8‐ and 17‐fold increases in luciferase activity in HepG2 cells containing pCAGA12‐Luc plasmid or 3TP‐Lux plasmid, respectively. However, these TGF‐β1‐induced luciferase activities were completely blocked by co‐treatment with 1 μM EW‐7203 and TGF‐β1 (Fig. 1A). In addition, EW‐7203 completely blocked TGF‐β1‐induced luciferase activity in HaCaT (3TP‐Lux) stable cells (Fig. 1B) co‐treated with 1 μM EW‐7203 and TGF‐β1. As shown in Figure 1(B), EW‐7203 inhibited luciferase activity below the basal level in HaCaT (3TP‐Lux) stable cells (the basal activity might have been due to residual TGF‐β in culture medium) (Fig. 1B). These results indicate that EW‐7203 strongly blocks TGF‐β1‐induced transcriptional activation.
Figure 1.
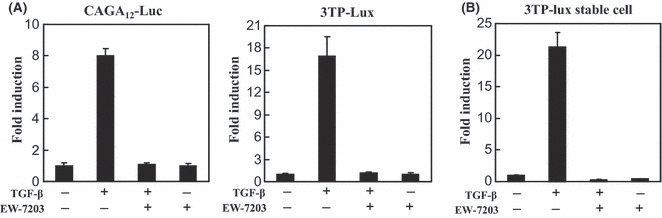
Inhibition of transforming growth factor‐β type I (TGF‐β1)‐induced transcriptional activation by EW‐7203. (A) HepG2 cells were transfected with β‐galactosidase expression plasmid, and pCAGA12‐Luc (left panel) or p3TP‐Lux (right panel) reporter plasmids, and treated with TGF‐β1 (2 ng/mL) ± EW‐7203 (1 μM). Luciferase activities were measured as described in the Methods. (B) HaCaT (3TP‐Lux) cells were treated with TGF‐β1 (2 ng/mL) ± EW‐7203 (1 μM) and luciferase activities in lysates were normalized versus β‐galactosidase activity. Data represent means ± SD. All experiments were repeated three times. +, presence; −, absence.
Inhibition of the phosphorylation and nuclear translocation of Smad2 by EW‐7203. To examine the effect of EW‐7203 on the phosphorylation of Smad2, we performed western blot analysis for phosphorylated‐Smad2 protein in NMuMG cells treated with either SB‐505124 or EW‐7203 in the presence of TGF‐β1. As shown in Figure 2(A), levels of phosphorylated Smad2 were rapidly increased by TGF‐β1 in NMuMG cells (Fig. 2A, lane 2) and 4T1 cells (Fig. 2B, lane 2), whereas EW‐7203 inhibited this increase (Fig. 2A, lanes 5 and 6; Fig. 2B, lane 4). Comparison of the inhibitory potencies of EW‐7203 and SB‐505124 showed that EW‐7203 more potently inhibited these phosphorylated Smad2 increases than SB‐505124 (Fig. 2A, lanes 3 and 5, and lanes 4 and 6, respectively). In addition, we examined the effect of EW‐7203 on the nuclear translocation of Smad2/3 in MCF10A cells. After treating MCF10A cells with EW‐7203 in the presence or absence of TGF‐β1 for 1.5 h, Smad2/3 protein was visualized using immunofluorescence assay. As expected, treatment with TGF‐β1 increased in the nuclear translocation of Smad2/3, and co‐treatment with EW‐7203 inhibited this nuclear translocation (Fig. 2C). These results indicate that EW‐7203 blocks TGF‐β1‐induced phosphorylation of Smad2 and the subsequent translocation of Smad2/3 to the nucleus.
Figure 2.
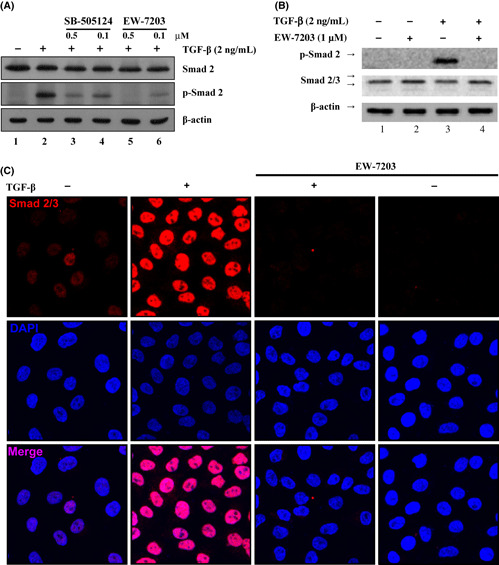
Inhibition of transforming growth factor‐β type I (TGF‐β1)‐induced Smad2 phosphorylation and nuclear translocation by EW‐7203. (A) NMuMG cells were treated with 0.2% FBS containing TGF‐β1 (2 ng/mL) ± SB‐505124 or EW‐7203 for 1.5 h. Total protein extracts were western blotted with anti‐Smad2 monoclonal antibody, anti‐phospho‐Smad2 polyclonal antibody and anti‐β‐actin monoclonal antibody. (B) 4T1 cells were treated with 0.2% FBS containing TGF‐β1 (2 ng/mL) ± EW‐7203 for 1.5 h. Total protein extracts were western blotted with anti‐Smad2 monoclonal antibody, anti‐phospho‐Smad2 polyclonal antibody and anti‐β‐actin monoclonal antibody. (C) MCF10A cells grown on cover glasses were treated with TGF‐β1 (2 ng/mL) in ±EW‐7203 (1 μM) for 1.5 h. Cells were fixed and incubated with anti‐Smad2/3 monoclonal antibody. Fluorescence was visualized using Cy3‐conjugated goat anti‐mouse IgG or DAPI and analyzed using a confocal microscopy. All experiments were repeated three times. +, presence; −, absence.
Effects of EW‐7203 on TGF‐β1‐induced morphological changes. During metastasis, the overexpression of TGF‐β contributes to cellular morphological changes and promotes EMT by cancer cells. 22 To investigate whether EW‐7203 blocks TGF‐β1‐induced morphological changes in MCF10A and NMuMG cells, we treated these cells with EW‐7203 with or without TGF‐β1. Treatment of MCF10A and NMuMG cells with TGF‐β1 was found to change cell morphology from a cuboid to a spindle shape, possibly because it reduced cell‐to‐cell contact. However, co‐treatment with EW‐7203 inhibited this TGF‐β1‐induced morphological change (Fig. 3). Interestingly, EW‐7203 (100 nM) appeared to be better at maintaining a cuboidal shape than SB‐505124 (100 nM) in MCF10A cells (Fig. 3A). These results indicate that EW‐7203 blocks the induction of a mesenchymal morphology by TGF‐β1.
Figure 3.
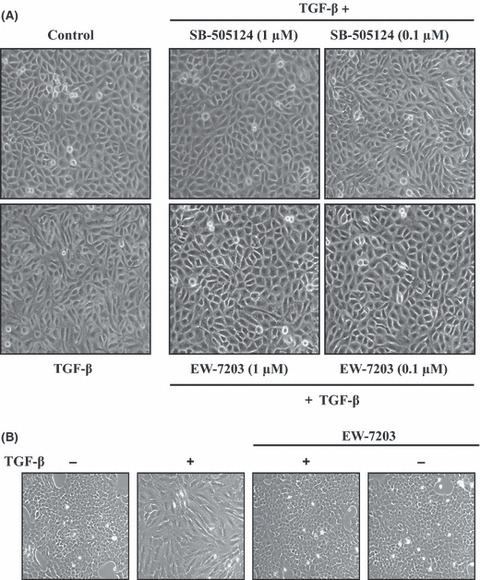
Effect of EW‐7203 on transforming growth factor‐β type I (TGF‐β1)‐induced morphological changes. (A) MCF10A cells were treated with TGF‐β1 (3 ng/mL) ± SB‐505124 or EW‐7203 for 72 h. (B) NMuMG cells were treated with TGF‐β1 (2 ng/mL) ± EW‐7203 (1 μM) for 72 h. Cell morphologies were assessed under a phase‐contrast microscope at ×100 magnification. +, presence; −, absence.
EW‐7203 inhibition of TGF‐β1‐induced EMT. To examine the effect of EW‐7203 on EMT, we measured the levels of epithelial markers, E‐cadherin and ZO‐1 and levels of mesenchymal markers, N‐cadherin and vimentin, in mammary epithelial cells treated with EW‐7203 in the presence or absence of TGF‐β1. As shown in Figure 4(A), TGF‐β1 caused the disappearance of membrane E‐cadherin and ZO‐1 protein. However, this was blocked when TGF‐β1 and EW‐7203 were co‐treated (Fig. 4A). We then examined the effect of EW‐7203 on EMT markers by western blotting. As expected, TGF‐β1 downregulated E‐cadherin levels and upregulated N‐cadherin and vimentin protein levels (Fig. 4B, lane 2), and EW‐7203 co‐treatment inhibited these changes (Fig. 4B, lanes 2 and 5). Furthermore, EW‐7203 had more potent effects on EMT marker proteins than SB‐505124 (Fig. 4B, lanes 4 and 6). We also examined the effect of EW‐7203 on the mRNA levels of EMT markers by RT‐PCR, and similarly, we found that EW‐7203 dose‐dependently inhibited the effects of TGF‐β1 on the mRNA levels of the abovementioned EMT markers (Fig. 4C,D).
Figure 4.
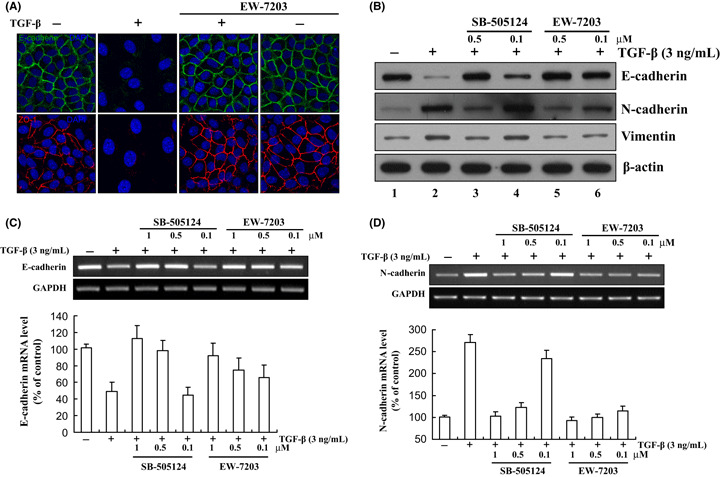
Inhibition of transforming growth factor‐β type I (TGF‐β1)‐induced epithelial‐to‐mesenchymal transition (EMT) by EW‐7203. (A) NMuMG cells were treated with TGF‐β1 (2 ng/mL) ± EW‐7203 (1 μM) for 48 h. Cells were fixed and incubated with anti‐E‐cadherin monoclonal antibody or anti‐ZO‐1 polyclonal antibody. Fluorescence was visualized using FITC‐conjugated goat anti‐mouse IgG or Rhodamine (TRITC)‐conjugated goat anti‐rabbit IgG or DAPI. Samples were analyzed using a confocal microscope as described in the Methods. All experiments were repeated three times. (B) MCF10A cells were treated with TGF‐β1 (3 ng/mL) ± SB‐505124 or EW‐7203 for 72 h. Total protein extracts from treated cells were western blotted with anti‐E‐cadherin, anti‐N‐cadherin, anti‐Vimentin or anti‐β‐actin monoclonal antibody. β‐actin was used as a loading control. (C,D) MCF10A cells were treated with TGF‐β1 (3 ng/mL) ± EW‐7203 (1 μM) for 72 h. Total RNA were isolated and the expressions of E‐cadherin and N‐cadherin were determined by RT‐PCR. GAPDH was used as a loading control. Data represent the means ± SD of three independent experiments. +, presence; −, absence.
Inhibition of TGF‐β1‐induced cell motility by EW‐7203. A wound healing assay was conducted with NMuMG cells to examine the effect of EW‐7203 on TGF‐β1‐induced cell motility. Wounded cells were treated with EW‐7203 in the presence or absence of TGF‐β1 for 0, 15 or 36 h. As shown in Figure 5(A), TGF‐β1 accelerated cell motility and wound closure; however, EW‐7203 completely inhibited TGF‐β1‐increased cell motility (Fig. 5A). Because we confirmed that EW‐7203 strongly inhibited cell motility by wound healing assay, we further studied the effect of EW‐7203 on cell invasion using 4T1 cells. As shown in Figure 5(B), EW‐7203 strongly inhibited TGF‐β1‐induced cell invasion.
Figure 5.
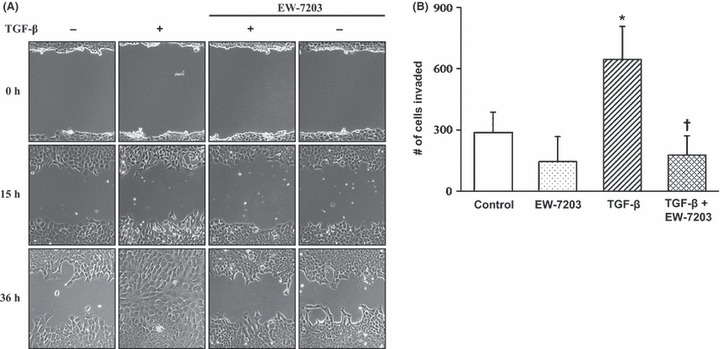
Inhibition of transforming growth factor‐β type I (TGF‐β1)‐induced cell motility by EW‐7203. (A) NMuMG cells grown on six‐well plates were serum starved in medium containing 0.2% FBS for 12 h prior to wounding. Wounded cells were treated with TGF‐β1 (2 ng/mL) ± EW‐7203 (1 μM) for the indicated times. Cellular motilities were assessed using phase‐contrast microscopy at ×100 magnification. (B) 4T1 cells in a transwell plate were treated in serum‐free medium with or without TGF‐β1 (2 ng/mL) ± EW‐7203 (1 μM). DAPI‐stained cells remaining on the bottom surface were observed using fluorescence microscopy as described in the Methods. Data represent the mean number of invaded cells per view field ±SD (n = 3). †P < 0.05. *P < 0.05. +, presence; −, absence.
Suppression of breast cancer metastasis to lung in vivo by EW‐7203. 4T1 cells were transplanted into the mammary fat pad of Balb/c mice and orthotopically grafted mice were treated with EW‐7203 (40 mg/kg) daily for 18 days. The number of metastatic nodules on the surfaces of lung left lobes were then counted. The number of metastatic nodules in EW‐7203‐treated mice were lower than those in saline‐treated mice (Fig. 6A, lower panel). To examine the effect of EW‐7203 on the phosphorylation of Smad2 in mammary tumors, we performed western blot analysis for phosphorylated‐Smad2 protein in tumors from mice treated with EW‐7203 in the presence or absence of TGF‐β1 challenge. As shown in Figure 6(B), levels of phosphorylated Smad2 in tumors were elevated by TGF (Fig. 6B, lane 2), whereas EW‐7203 inhibited this increase (Fig. 6B, lanes 3 and 4). The primary tumor sizes of tumor‐bearing Balb/c mice were monitored until the mice were killed, and no differences in primary tumor growth were observed between saline‐treated and EW‐7203‐treated mice (Fig. 6C). To examine the effect of EW‐7203 on the proliferation of 4T1 cells, we carried out a SRB assay. As shown in Figure 6(D), EW‐7203 treatment did not inhibit cell proliferation of 4T1 cells with a concentration of 100 nM. This might explain why EW‐7203 did not change the primary tumor growth (Fig. 6C). The bodyweights of tumor‐bearing Balb/c mice were monitored until they were killed, and no differences in bodyweight were observed between saline‐treated and EW‐7203‐treated mice (data not shown). These results show that EW‐7203 inhibits the metastasis of breast cancer cells to the lung in vivo.
Figure 6.
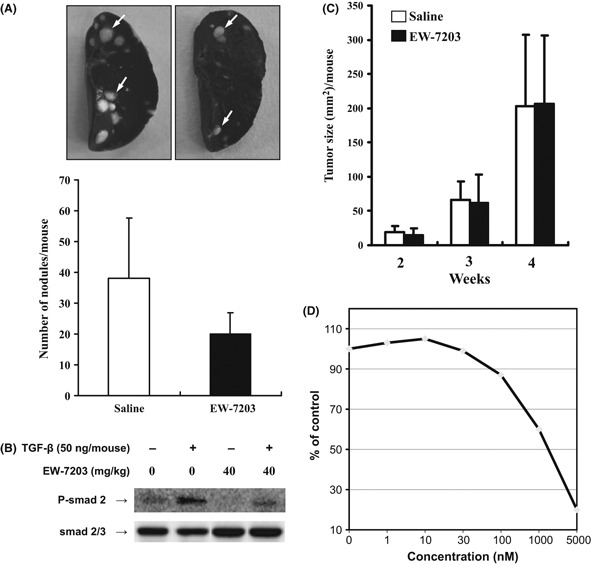
Suppression of breast cancer metastasis to lung in vivo by EW‐7203. Two weeks after transplantation of 4T1 cells on the mammary fat pad, Balb/c mice were intraperitoneally administered saline or EW‐7203 (40 mg/kg per day) for 18 days. (A) Indian ink‐stained metastatic nodules on lung surfaces were counted (white arrows, upper panel). Average numbers of metastatic nodules on lobes are shown in the lower panel (means ± SD, n = 3). (B) Mammary tumors from mice treated with 40 mg/kg per day for 18 days were removed and homogenized in radioimmunoprecipitation assay (RIPA) buffer. Total protein extracts were western blotted with anti‐Smad2 monoclonal antibody, anti‐phospho‐Smad2 polyclonal antibody and anti‐β‐actin monoclonal antibody. (C) Primary tumor sizes were monitored and data represent means ± SD (mm2) (n = 3). (D) 4T1 cells were treated with vehicle (0.1% DMSO) or indicated concentrations of EW‐7203 for 72 h. Numbers of cells were determined by sulforhodamine B (SRB) assay. All experiments were repeated at least three times. +, presence; −, absence.
Discussion
In the present study, we examined the anti‐metastatic effect of EW‐7203, a selective ALK5 inhibitor that was originally synthesized using a target‐based approach. Small molecule inhibitors devised to directly block the catalytic activity of TβRI, including SB‐431542( 23 ) and SB‐505124 (GlaxoSmithKline, Brentford, Middlesex, UK)( 24 ), SD‐093( 25 ) and SD‐208 (Scios, New Brunswick, NJ, USA)( 26 ), and LY‐580276 (Lilly Research Laboratories, Indianapolis, IN, USA)( 27 ), which all act as competitive inhibitors by attacking the ATP‐binding site of TβRI kinase.( 28 ) The present study shows that TGF‐β1 induces EMT in mammary epithelial cells and that this process can be effectively blocked by EW‐7203 (Fig. 4). Recently, Ge and colleagues published similar results for a different TβRI kinase inhibitor, SD‐093.( 29 ) TGF‐β1‐induced EMT plays important roles in the progression of both cancer and chronic renal disease. In cancer, EMT generally predicts a more aggressive tumor cell behavior.( 30 ) During carcinogenesis, epithelial cells can undergo mesenchymal transformation to a fibroblast‐like phenotype, and thus gain the ability to migrate, invade and metastasize. Furthermore, TGF‐β has been shown to play an important role in EMT( 31 ) and in the malignant progression of fibroblast‐like squamous carcinoma cells to highly invasive spindle cell carcinomas during in vivo skin carcinogenesis,( 32 ) and the tumor‐promoting role of TGF‐β is known to be associated with its ability to induce EMT in late‐stage cancers. The EMT is characterized by a reduction in cell‐to‐cell adhesion, increased cell motility and by the activation of proteolysis, properties that are associated with tumor cell invasion and metastasis.( 22 , 33 ) Furthermore, Smads act as regulators of EMT and directly or indirectly activate the transcriptions of mesenchymal markers.( 34 )
The present study demonstrates that EW‐7203 inhibits TGF‐β1‐induced Smad2 phosphorylation within mammary epithelial cells more potently than SB‐505124 (Fig. 2A). Furthermore, functional analysis revealed that EW‐7203 is capable of blocking TGF‐β1‐induced EMT in mammary epithelial cells, and its kinase selectivity against p38α showed that EW‐7203 selectively targets the ALK5 pathway (Table 1).
Collectively, the potency shown by EW‐7203 for inhibiting TβRI kinase activity, its favorable kinetic properties and its ability to inhibit TGF‐β1‐induced Smad2 phosphorylation and EMT within mammary epithelial cells in conjunction with its remarkable specificity for p38α make it an interesting candidate for further drug development. Inhibition of autocrine TGF‐β1 signaling in carcinoma cells is known to reduce cell invasiveness and tumor metastasis, and these effects of TGF‐β1 are associated with its ability to induce EMT.( 31 ) Therefore, TGF‐β1‐induced EMT is likely to be an important requirement for the pathogenesis of cancer and fibrotic diseases. Our finding that EW‐7203 effectively inhibits TGF‐β1‐induced EMT of breast cancer cells and their metastatic activity in vivo suggests that EW‐7203 is capable of blocking TGF‐β1‐mediated functions in vivo, and thus it has substantial potential as a treatment for cancer, fibrosis and other diseases.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgment
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (No. 20090093972).
References
- 1. Weigelt B, Peterse JL, van‘t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer 2005; 5: 591–602. [DOI] [PubMed] [Google Scholar]
- 2. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell 2006; 127: 679–95. [DOI] [PubMed] [Google Scholar]
- 3. Gorsch SM, Memoli VA, Stukel TA, Gold LI, Arrick BA. Immunohistochemical staining for transforming growth factor beta 1 associates with disease progression in human breast cancer. Cancer Res 1992; 52: 6949–52. [PubMed] [Google Scholar]
- 4. Ivanovic V, Todorovic‐Rakovic N, Demajo M et al. Elevated plasma levels of transforming growth factor‐beta 1 (TGF‐beta 1) in patients with advanced breast cancer: association with disease progression. Eur J Cancer 2003; 39: 454–61. [DOI] [PubMed] [Google Scholar]
- 5. Buck MB, Knabbe C. TGF‐beta signaling in breast cancer. Ann N Y Acad Sci 2006; 1089: 119–26. [DOI] [PubMed] [Google Scholar]
- 6. Muraoka‐Cook RS, Shin I, Yi JY et al. Activated type I TGFbeta receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene 2006; 25: 3408–23. [DOI] [PubMed] [Google Scholar]
- 7. Massague J. TGFbeta in cancer. Cell 2008; 134: 215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tomaskovic‐Crook E, Thompson EW, Thiery JP. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res 2009; 11: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang J, Weinberg RA. Epithelial‐mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008; 14: 818–29. [DOI] [PubMed] [Google Scholar]
- 10. Brown KA, Pietenpol JA, Moses HL. A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF‐beta signaling. J Cell Biochem 2007; 101: 9–33. [DOI] [PubMed] [Google Scholar]
- 11. Shi Y, Massague J. Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell 2003; 113: 685–700. [DOI] [PubMed] [Google Scholar]
- 12. Derynck R, Zhang YE. Smad‐dependent and Smad‐independent pathways in TGF‐beta family signalling. Nature 2003; 425: 577–84. [DOI] [PubMed] [Google Scholar]
- 13. Yu L, Hebert MC, Zhang YE. TGF‐beta receptor‐activated p38 MAP kinase mediates Smad‐independent TGF‐beta responses. EMBO J 2002; 21: 3749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pennison M, Pasche B. Targeting transforming growth factor‐beta signaling. Curr Opin Oncol 2007; 19: 579–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Halder SK, Beauchamp RD, Datta PK. A specific inhibitor of TGF‐beta receptor kinase, SB‐431542, as a potent antitumor agent for human cancers. Neoplasia 2005; 7: 509–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ge R, Rajeev V, Ray P et al. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor‐beta type I receptor kinase in vivo . Clin Cancer Res 2006; 12: 4315–30. [DOI] [PubMed] [Google Scholar]
- 17. Rausch MP, Hahn T, Ramanathapuram L et al. An orally active small molecule TGF‐beta receptor I antagonist inhibits the growth of metastatic murine breast cancer. Anticancer Res 2009; 29: 2099–109. [PMC free article] [PubMed] [Google Scholar]
- 18. Nam JS, Terabe M, Mamura M et al. An anti‐transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res 2008; 68: 3835–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao BM, Hoffmann FM. Inhibition of transforming growth factor‐beta1‐induced signaling and epithelial‐to‐mesenchymal transition by the Smad‐binding peptide aptamer Trx‐SARA. Mol Biol Cell 2006; 17: 3819–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Workman P, Aboagye EO, Balkwill F et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer 2010; 102: 1555–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kataoka M, Schumacher G, Cristiano RJ et al. An agent that increases tumor suppressor transgene product coupled with systemic transgene delivery inhibits growth of metastatic lung cancer in vivo . Cancer Res 1998; 58: 4761–5. [PubMed] [Google Scholar]
- 22. Guarino M, Rubino B, Ballabio G. The role of epithelial‐mesenchymal transition in cancer pathology. Pathology 2007; 39: 305–18. [DOI] [PubMed] [Google Scholar]
- 23. Saunier EF, Akhurst RJ. TGF beta inhibition for cancer therapy. Curr Cancer Drug Targets 2006; 6: 565–78. [DOI] [PubMed] [Google Scholar]
- 24. Hjelmeland MD, Hjelmeland AB, Sathornsumetee S et al. SB‐431542, a small molecule transforming growth factor‐beta‐receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol Cancer Ther 2004; 3: 737–45. [PubMed] [Google Scholar]
- 25. Subramanian G, Schwarz RE, Higgins L et al. Targeting endogenous transforming growth factor beta receptor signaling in SMAD4‐deficient human pancreatic carcinoma cells inhibits their invasive phenotype1. Cancer Res 2004; 64: 5200–11. [DOI] [PubMed] [Google Scholar]
- 26. Uhl M, Aulwurm S, Wischhusen J et al. SD‐208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo . Cancer Res 2004; 64: 7954–61. [DOI] [PubMed] [Google Scholar]
- 27. Yi JY, Shin I, Arteaga CL. Type I transforming growth factor beta receptor binds to and activates phosphatidylinositol 3‐kinase. J Biol Chem 2005; 280: 10870–6. [DOI] [PubMed] [Google Scholar]
- 28. Yingling JM, Blanchard KL, Sawyer JS. Development of TGF‐beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov 2004; 3: 1011–22. [DOI] [PubMed] [Google Scholar]
- 29. Ge R, Rajeev V, Subramanian G et al. Selective inhibitors of type I receptor kinase block cellular transforming growth factor‐beta signaling. Biochem Pharmacol 2004; 68: 41–50. [DOI] [PubMed] [Google Scholar]
- 30. Thiery JP. Epithelial‐mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2: 442–54. [DOI] [PubMed] [Google Scholar]
- 31. Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol 1998; 8: 1243–52. [DOI] [PubMed] [Google Scholar]
- 32. Cui W, Fowlis DJ, Bryson S et al. TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 1996; 86: 531–42. [DOI] [PubMed] [Google Scholar]
- 33. Kang Y, Massague J. Epithelial‐mesenchymal transitions: twist in development and metastasis. Cell 2004; 118: 277–9. [DOI] [PubMed] [Google Scholar]
- 34. Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF‐beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial–mesenchymal cell transition. Mol Biol Cell 2005;16:1987–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]