

Abstract
The present study aimed to investigate the effect of the stimulatory heterotrimeric GTP‐binding (Gs) protein signaling system on cisplatin‐induced apoptosis of lung cancer cells and its underlying mechanism as an attempt to develop a novel strategy to improve the therapeutic efficacy of cisplatin. Overexpression of the constitutively active α subunit of Gs (GαsQL) in A549 human lung cancer cells increased cisplatin‐induced apoptosis, and knockdown of Gαs with small hairpin RNA decreased the percentage of apoptotic cells. GαsQL increased the expression of the proapoptotic proteins B‐cell leukemia/lymphoma‐2 genes (Bcl‐2) homologous antagonist killer protein (Bak) and Bcl‐2 associated X protein (Bax), and decreased the expression of the antiapoptotic proteins Bcl‐2 and Bcl‐Xlong protein. Knockdown of Bak blocked the augmentative effects of GαsQL. GαsQL decreased the degradation rate of the Bak protein, and increased Bak mRNA transcript levels. GαsQL increased Bak‐luciferase activity in a protein kinase A and cyclic AMP response element‐dependent manner. GαsQL also augmented cisplatin‐induced apoptosis of H1299 human lung cancer cells that lack functional p53. From this study, it is concluded that Gαs augments cisplatin‐induced apoptosis of lung cancer cells partially through upregulating Bak expression by increasing transcription and by decreasing the rate of protein degradation. (Cancer Sci 2009; 100: 1069–1074)
Abbreviations:
- Ap‐1, activator protein‐1; Bak
Bcl‐2 homologous antagonist killer protein
- Bax
Bcl‐2 associated X protein
- Bcl‐2
B‐cell leukemia/lymphoma‐2 genes
- Bcl‐XL
Bcl‐2 Xlong protein
- cAMP
cyclic AMP
- cisplatin
cis‐diamminedichloroplatinum
- CRE
cAMP response element
- CREB
cAMP response element binding protein
- Gαs
alpha subunit of stimulatory heterotrimeric GTP‐binding protein Q227L
- G protein
heterotrimeric GTP‐binding protein
- Gs
stimulatory heterotrimeric GTP‐binding protein
- HA
hemaglutinin
- NFAT, nuclear factor of activated T‐cells; NF‐κB, nuclear factor kappa B; PARP
poly (ADP‐ribose) polymerase
- PKA
protein kinase
- PI
propidium iodide
- shRNA
small hairpin RNA
- zVAD‐fmk
enzyloxycarbonyl‐Val‐Ala‐Asp‐(OMe) fluoromethyl ketone
Cisplatin has been used as a chemotherapeutic agent for a wide range of solid tumors, such as testicular, ovarian, bladder, and lung cancer.( 1 , 2 ) The cytotoxic effect of cisplatin is believed to be primarily due to the interaction with DNA: forming interstrand and intrastrand adducts. The resulting DNA abducts hinder both RNA transcription and DNA replication, and activate several signal transduction pathways, including ATR, p53, p73, and mitogen‐activated protein kinase, leading to cell cycle arrest and apoptosis.( 3 ) Cisplatin‐induced apoptosis depends on the generation of reactive oxygen species.( 4 , 5 )
Lung cancer, including non‐small cell lung cancer, is the leading cause of cancer morbidity and mortality worldwide. The most effective systemic chemotherapy for non‐small cell lung cancer is cisplatin‐based combination treatment, which produces a response in most patients. However, relapse is rapid with patients developing resistance to cisplatin. The development of cisplatin resistance causes the outcome of cisplatin therapy to reach a plateau.( 6 , 7 ) Therefore, understanding these mechanisms of cisplatin resistance and developing strategies to circumvent the resistance is important for the continued success of cancer chemotherapy. Numerous cellular mechanisms including inhibition of cisplatin‐induced apoptosis have been proposed to potentially contribute to cisplatin resistance.( 8 , 9 )
G proteins are composed of α, β, and γ subunits and transduce signals from ligand‐activated membrane receptors to intracellular signals by regulating the activity of effectors. G proteins belong to the GTP‐binding protein superfamily that is activated in a GTP‐binding state and inactivated following hydrolysis of GTP by intrinsic GTPase. When G proteins are activated, the GTP‐bound α subunit dissociates from the βγ subunit complex, and both the α subunit and βγ subunit complex independently regulate a variety of effectors including adenylate cyclases, phospholipase Cβ, and ion channels.( 10 , 11 ) Heterotrimeric G proteins are activated by more than 1000 receptors, and regulate various cellular responses including metabolism, proliferation, differentiation, and apoptosis.( 8 , 12 , 13 ) Among the four α subunit families of G proteins, Gαs stimulates adenylate cyclases to increase the cAMP level, which activates cAMP‐dependent PKA. PKA phosphorylates various target molecules to regulate metabolism and expression of various genes. For example, PKA phosphorylates the CREB, which then binds to the CRE and activates the transcription of approximately 4000 target genes, including various genes regulating cellular proliferation and apoptosis.( 14 , 15 ) The cAMP signaling system has been reported to induce apoptosis of various cancer cells, including lung cancer cells.( 16 ) Thus, in an attempt to develop a novel strategy to improve the therapeutic efficacy of cisplatin by activating the cAMP signaling system, the present study was aimed to investigate the effect of the Gαs signaling system on cisplatin‐induced apoptosis of lung cancer cells and its underlying mechanism. From this study, Gαs was found to augment cisplatin‐induced apoptosis by upregulating Bak expression in non‐small cell lung cancer cells in a PKA‐ and CRE‐dependent manner.
Materials and Methods
Cell culture and reagents. Human non‐small cell lung cancer cells (A549 and H1299) were purchased from American Type Culture Collection (Rockville, MD, USA), and were maintained in RPMI medium containing 10% fetal bovine serum (JBI, Daegu, Korea) and 100 U/mL penicillin and streptomycin in a CO2 incubator at 37°C. Annexin V–fluorescein isothiocyanate, PI, zVAD‐fmk, and cisplatin were obtained from Sigma Chemicals (St Louis, MO, USA). The nucleotide sequences of the decoy oligonucleotide for the CRE and those of the CRE mismatch control were 5′‐TGACGTCATGACGTCATGACGTCA‐3′ and 5′‐CTAGCTAGCTAGCTAGCTAGCTAG‐3′, respectively. The CRE decoy oligonucleotide competes with CRE enhancers for active CREB to block CREB‐dependent gene expression. A shRNA construct targeting Gαs (pLKO.1‐puro‐Gαs) was purchased from Sigma Chemicals, and the target sequence was 5‐CAGAATTTGCTCGCTACACTA‐3. A shRNA for knockdown of Bak (pENTR/H1/TO‐Bak) was cloned in a previous study.( 17 ) Transfection was carried out by electroporation using a Gene Pulser II (Bio‐Rad, Hercules, CA, USA) at 200 V/950 microFaraday. Prostaglandin E2 and H89 were purchased form Sigma Chemicals, and forskolin and isobutylmethylxanthine were from Calbiochem (Nottingham, UK).
Transient and stable expression of constitutively active mutant Ges. The expression plasmid of a HA‐tagged constitutively active mutant of Gαs (GαsQ227L) was purchased from Upstate Biotechnology (Lake Placid, NY, USA). The GαsQ227L mutant has a substitution of Leu227 for Gln227 that results in inactivation of intrinsic GTPase to cause constitutive activation of the protein. For transient expression, subconfluent (80–90%) A549 cells plated in 100 mm dishes were transfected with 15 µg plasmid DNA by electroporation (Bio‐Rad Electroporation Chamber). Control cells were transfected with pcDNA3 vector DNA. Sixteen hours after transfection, the medium was replaced with fresh medium and incubated for an additional 24 h, after which the cells were treated with cisplatin for 24 h and then harvested. For stable expression, the culture medium of GαsQL‐transfected cells was replaced with culture medium containing 500 µg/mL G418, and the cells were incubated for 14–18 days to select the transfected colonies. Expression of GαsQL was confirmed by western blot analysis.
Flow cytometry analysis of annexin V‐ and PI‐stained cells. Flow cytometry analysis of annexin V‐ and PI‐stained cells was used to assess cell death. Briefly, after removal of culture medium, cells were washed twice with phosphate‐buffered saline and incubated in a binding buffer (10 mM HEPES, 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, pH 7.4) containing annexin V–fluorescein isothiocyanate (25 µg/mL) and PI (25 µg/mL) for 30 min. Cells were washed three times in binding buffer and harvested from the plate using a rubber policeman (Nunc, Roskilde, Denmark), and stained cells were quantified using a FacsCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ) using 10 000 cells per measurement.
Western blotting. The expression of proteins was analyzed by western blotting using specific antibodies as described previously.( 18 ) Antibodies against β‐actin and Bcl‐2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), antibodies against PARP, cleaved PARP, cleaved caspase‐3, Bax, Bcl‐XL, and Bak were obtained from Cell Signaling Technology (Beverly, MA, USA). Cytochrome c released into the cytoplasm was analyzed following subcellular fractionation by immunoblotting using an antibody purchased from BD Biosciences. The blot image was developed by incubation with an enhanced chemiluminescence substrate mixture (Pierce, Chester, UK), and captured by a luminescent image analysis system (LAS‐3000; Fuji, Tokyo, Japan). Next, the density of protein bands was quantified using Multi Gauge v2.3 software, and the relative band density was expressed as a percentage or a multiple of corresponding densities of the control.
Luciferase activity assays. A549 cells were transfected with plasmids containing a luciferase reporter gene under the control of the Bak promoter by electroporation using a Gene Pulser II at 200 V/950 microfarads. The Bak promoter luciferase‐expressing plasmid containing 3500 bp of 5′ untranslated region with a CRE site at –1703 bp was constructed previously.( 17 ) Luciferase activities were assayed using the Bioluminescent Reporter Gene Assay System (Tropix, Bedford, MA, USA) according to the manufacturer's instructions. At least three independent experiments were carried out in duplicate and promoter activities were normalized against β‐galactosidase activity.
Data analysis. All experiments were independently repeated at least three times, and data are presented as means ± SE. The non‐parametric Mann–Whitney U‐test was used to analyze mean values, and P‐values <0.05 were considered to be statistically significant.
Results
Ges augments cisplatin‐induced apoptosis of A549 human lung cancer cells. The effect of Gαs on cisplatin‐induced apoptosis was examined in A549 human lung cancer cells. Gαs was found to augment cisplatin‐induced apoptosis of A549 cells after transient transfection of constitutively active GαsQL (data not shown). Thus, to investigate the mechanism in detail by which Gαs augments apoptosis, GαsQL was expressed stably in A549 cells. The expression of HA‐tagged GαsQL was confirmed by western blot analysis using an antibody specific to Gαs or the HA tag (Fig. 1a). GαsQL‐expressing cells had a large increase in CREB phosphorylation, indicating that the cAMP signaling pathway was activated following expression of GαsQL.
Figure 1.
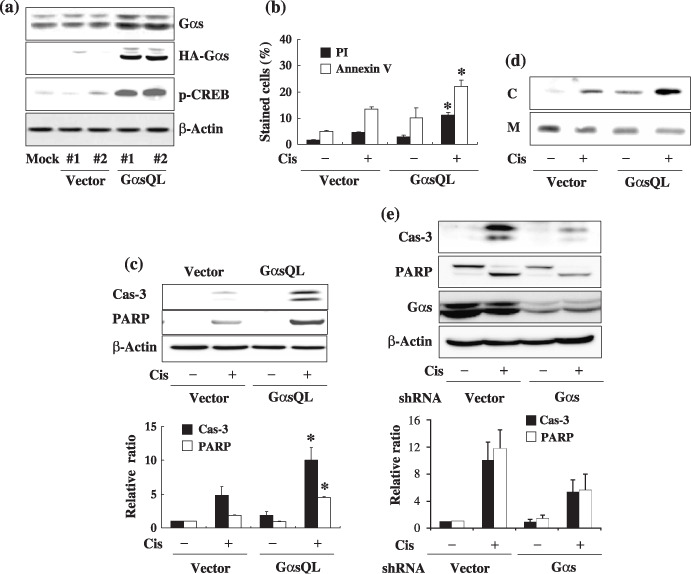
Alpha subunit of stimulatory heterotrimeric GTP‐binding protein (Gαs) augments cisplatin‐induced apoptosis of A549 human lung cancer cells. (a) Stable expression of alpha subunit of stimulatory heterotrimeric GTP‐binding protein Q227L (GαsQL) in A549 cells. Expression of GαsQL and phosphorylation of cyclic AMP response element binding protein (CREB) in hemaglutinin (HA)‐tagged GαsQL‐transfected cells were confirmed by western blot analysis using a specific antibody against Gαs, HA, or phosphorylated CREB (Ser‐133). β‐Actin was analyzed as a loading control; #1 and #2 are the respective clone numbers of vector‐ or GαsQL‐transfected cells. (b–d) Assessment of cisplatin‐induced apoptosis of A549 cells by (b) flow cytometry of annexin V‐ and propidium iodide (PI)‐stained cells, (c) western blot analysis of caspase‐3 and poly (ADP‐ribose) polymerase (PARP) cleavage, and (d) cytosolic release of cytochrome c (C, cytosolic fraction; M, mitochondrial fraction). A549 cells were then treated with 40 µM cisplatin for 24 h prior to assessment of apoptosis. (e) Knockdown of Gαs expression decreased cisplatin‐induced apoptosis. Subconfluent A549 cells were transfected with Gαs small hairpin RNA (shRNA), and 48 h after transfection cells were treated with 40 µM cisplatin for 24 h, after which apoptosis was assessed by western blot analysis of caspase‐3 and PARP cleavage. Histograms represent mean ± SE, and the asterisks indicate a significant difference from vector‐transfected controls (P < 0.05, Mann–Whitney U‐test).
Using A549 cells stably expressing GαsQL, the effect of Gαs on cisplatin‐induced apoptosis was examined. Treatment with cisplatin caused more cell death in GαsQL‐expressing cells than in vector‐transfected cells, as assessed by microscopic examination (data not shown), by FACS analysis of annexin V‐stained cells (Fig. 1b), and by western blot analysis of the cleavage of caspase‐3 and PARP (Fig. 1c), and cytoplasmic translocation of cytochrome c (Fig. 1d). The effect of Gαs on cisplatin‐induced apoptosis was further confirmed by analyzing the effect of loss of Gαs on cisplatin‐induced cell death. Transfection of Gαs shRNA reduced Gαs expression, and decreased the cleavage of caspase‐3 and PARP compared to vector‐transfected cells (Fig. 1e).
Ges augments cisplatin‐induced apoptosis by upregulation of Bak expression in lung cancer cells. Next, to elucidate the underlying mechanism by which Gαs augments cisplatin‐induced apoptosis in A549 cells, the effect of Gαs on expression of Bcl‐2 family proteins that mediate both the pro‐apoptotic and the anti‐apoptotic responses was examined. Expression of GαsQL increased the cisplatin‐induced expression of pro‐apoptotic molecules such as Bak and Bax, but slightly decreased the cisplatin‐induced expression of anti‐apoptotic molecules such as Bcl‐XL and Bcl‐2 (Fig. 2a,b). The most prominent change in expression was that of Bak, which increased 2.1‐fold in cisplatin‐treated GαsQL‐expressing cells in comparison to cisplatin‐treated vector‐transfected control cells. Because Gαs activates adenylate cyclase to increase cAMP production, which results in phosphorylation of CREB and the expression of various genes, the effect of forskolin, an adenylate cyclase activator, on cisplatin‐induced apoptosis was examined. Treatment with forskolin together with isobutylmethylxanthine, a cAMP phosphodiesterase inhibitor, also augmented the cisplatin‐induced cleavage of PARP and caspase‐3, and it caused an increase in the cisplatin‐induced expression of Bak and Bax and decrease in the expression of Bcl‐2 and Bcl‐XL, which is very similar to results following GαsQL overexpression (Fig. 2c).
Figure 2.
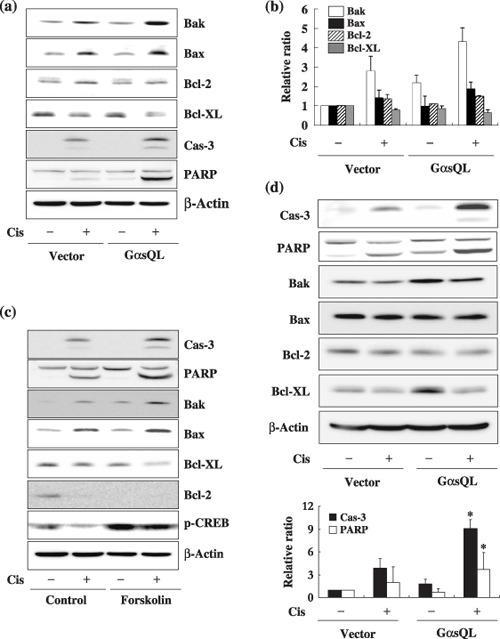
Alpha subunit of stimulatory heterotrimeric GTP‐binding protein (Gαs) augments cisplatin‐induced apoptosis by regulating the expression of B‐cell leukemia/lymphoma‐2 genes (Bcl‐2) family proteins in A549 cells. (a,b) Effects of Gαs on the expression level of Bcl‐2 family proteins in cisplatin‐treated A549 cells. (a) A549 cells were treated with 40 µM cisplatin for 24 h, and expression of Bcl‐2 family proteins was analyzed by western blotting. (b) The densitometric results are presented as histograms. (c) Effects of forskolin on the expression of Bcl‐2 family proteins and cisplatin‐induced apoptosis. A549 cell were pretreated with 7.5 µM forskolin and 100 µM isobutylmethylxanthine in dimethyl sulfoxide for 1 h and then with 40 µM cisplatin for 24 h before analysis of the expression of Bcl‐2 family proteins by western blotting. (d) Augmentation of cisplatin‐induced apoptosis of H1299 lung cancer cell by Gαs. GαsQL was also stably expressed in H1299 human lung cancer cells, which were treated with 40 µM cisplatin for 24 h, and the Bcl‐2 family proteins, and cleavage of caspase‐3 and poly (ADP‐ribose) polymerase (PARP) were analyzed by western blotting. Bak, Bcl‐2 homologous antagonist killer protein; Bax, Bcl‐2 associated X protein; CREB, cyclic AMP response element binding protein.
Then, to examine whether augmentation of cisplatin‐induced apoptosis by Gαs is common to other lung cancer cells, H1299 human lung cancer cells, where the p53 gene is deleted, were used for additional analysis. Similar to A549 cells, stable expression of GαsQL in H1299 cells increased the cisplatin‐induced cleavage of caspase‐3 and PARP (Fig. 2d). The expression of Bak was increased 1.9‐fold in cisplatin‐treated GαsQL‐expressing cells in comparison to cisplatin‐treated vector‐transfected control cells, but the expression of Bax and Bcl‐2 were not changed by GαsQL and cisplatin. The protein level of Bcl‐XL was increased 2.8‐fold in the GαsQL stable cell line in comparison to vector‐transfected control cells (Fig. 2d). This result indicates that Gαs may augment cisplatin‐induced apoptosis by regulating the expression of Bcl‐2 family members via cAMP, and that this effect does not require the p53 protein.
Because the change in Bak expression was most prominent among Bcl‐2 family members, we examined whether Bak mediates the augmentation of cisplatin‐induced apoptosis by Gαs. Knockdown of Bak by transfection with Bak shRNA reduced the expression of Bak, and the cleavage of caspase‐3 and PARP following cisplatin treatment compared to GαsQL‐expressing control cells (Fig. 3a). Treatment of A549 cells with prostaglandin E2, an activator of Gαs‐coupled receptors, also increased expression of Bak and augmented cisplatin‐induced cleavage of caspase‐3 and PARP (Fig. 3b). This result suggests that Bak mediates the augmentation of cisplatin‐induced apoptosis by the Gαs signaling system in A549 cells.
Figure 3.
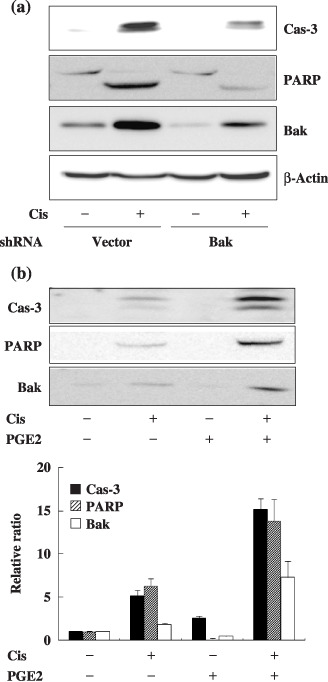
Alpha subunit of stimulatory heterotrimeric GTP‐binding protein (Gαs) augments cisplatin‐induced apoptosis by upregulating the Bak expression level in A549 cells. (a) Knockdown of B‐cell leukemia/lymphoma‐2 genes homologous antagonist killer protein (Bak) expression by small hairpin RNA (shRNA) decreased cisplatin‐induced apoptosis. A549 cells stably expressing Gαs were transfected with Bak shRNA for 48 h, after which cells were treated with 40 µM cisplatin for 24 h prior to apoptosis analysis. (b) Prostaglandin E2 (PGE2), a Gs‐coupled receptor agonist, increased Bak expression and cisplatin‐induced apoptosis. A549 cell were pretreated with 10 µM PGE2 for 1 h and then with 40 µM cisplatin for 24 h prior to apoptosis analysis. PARP, poly (ADP‐ribose) polymerase.
Ges delays degradation of Bak protein and induces Bak mRNA level in A549 lung cancer cells. Next, to investigate how Gαs increases Bak expression, the effect of Gαs on the degradation rate of Bak protein was examined in cycloheximide‐treated A549 cells. Interestingly, the degradation rate of Bak was slower in GαsQL‐expressing cells (half life 21.5 h) than in vector‐transfected control cells (half life 7.3 h) (Fig. 4a). The effect of Gαs on Bak mRNA expression was also examined by reverse transcription–polymerase chain reaction in A549 cells. The expression level of Bak mRNA increased to 1.7‐fold of the control in GαsQL‐expressing cells (Fig. 4b). To examine whether Gαs increases transcription of the Bak gene, Bak luciferase activity was analyzed. Bak luciferase activity increased in GαsQL‐expressing cells to 2.8‐fold of the control, and it was inhibited by treatment with a PKA inhibitor (H89) and a CRE decoy (Fig. 4c). The effect of H89 and decoy CRE was confirmed by showing that H89 inhibited forskolin‐induced phosphorylation of CREB, and that CRE decoy inhibited forskolin‐induced CRE luciferase expression. This result suggests that Gαs increases Bak expression by decreasing the degradation rate of the Bak protein as well as increasing transcription of the Bak gene, which is dependent on PKA and CRE.
Figure 4.
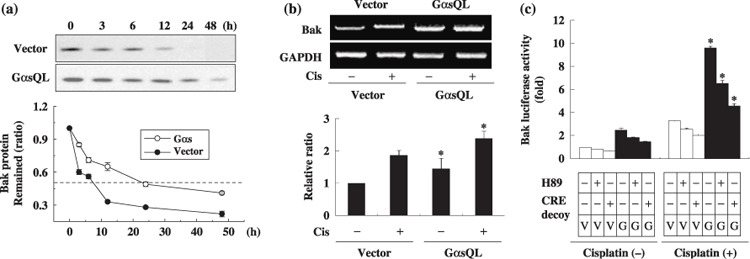
Alpha subunit of stimulatory heterotrimeric GTP‐binding protein (Gαs) upregulates B‐cell leukemia/lymphoma‐2 gene homologous antagonist killer protein (Bak) expression by delaying degradation of Bak protein and by increasing transcription of the Bak gene. (a) Gαs reduced the degradation rate of the Bak protein. Subconfluent A549 cells were treated with a protein synthesis inhibitor, cycloheximide (30 µg/mL), and the remaining Bak protein was monitored at the indicated times by western blotting analysis. (b) Gαs increased Bak mRNA levels. Subconfluent A549 cells were treated with cisplatin for 24 h and Bak mRNA level was measured by reverse transcription–polymerase chain reaction. The expression level of Bak mRNA was normalized to that of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). (c) Gαs increased the activity of Bak promoter. For luciferase assays, A549 cells were transfected with 2.5 µg of (–3500 bp) Bak‐pLuc and 2.5 µg of a β‐galactosidase construct. After 24 h, cells were pretreated with H89 (15 mM) for 30 min or transfected with cyclic AMP response element (CRE) decoy (200 nM), and then with 40 µM cisplatin for 24 h. Bak luciferase activities were measured and normalized against β‐galactosidase activity and are presented as ratio against vector‐transfected controls. Three independent experiments were carried out in duplicate, and asterisks indicate significant difference from vector‐transfected controls (P < 0.05, Mann–Whitney U‐test). G, GαsQL; V, vector.
Discussion
The present study examined the effect of Gαs on cisplatin‐induced apoptosis of human lung cancer cells. Gαs augmented cisplatin‐induced apoptosis of lung cancer cells by upregulating Bak expression. This conclusion is based on several lines of evidence. First, stable expression of a constitutively active mutant of Gαs augmented cisplatin‐induced apoptosis of both A549 and H1299 lung cancer cells. Knockdown of Gαs expression using snRNA decreased cisplatin‐induced apoptosis. Second, Gαs upregulated Bak expression, and knockdown of Bak expression by snRNA abolished the augmenting effect of Gαs in A549 cells. Finally, Gαs upregulated Bak expression by decreasing the degradation rate of Bak protein and by increasing the transcriptional activity of the Bak gene in a PKA‐ and CRE‐dependent manner.
Gαs, which is activated by a variety of membrane receptors, stimulates adenylate cyclase and subsequently increases the cAMP level, resulting in activation of the cAMP signaling system to regulate various cellular responses, including proliferation, differentiation, and apoptosis. The cAMP signaling system has been reported to stimulate apoptosis of thymocytes,( 19 ) malignant glioma cells,( 20 ) and leukemia cells.( 21 ) Treatment of human lung cancer cells with cAMP phosphodiesterase inhibitors or cholera toxin, which activates Gαs, triggered apoptosis and reduced cell viability.( 16 , 22 ) These reports are in agreement with our results that Gαs augments cisplatin‐induced apoptosis of lung cancer cells in a p53‐independent manner. Nonetheless, the mechanism of how the cAMP signaling system stimulates apoptosis of lung cancer cells is not clearly understood. Cholera toxin triggers apoptosis of lung cancer cells without changing the expression of Bcl‐2 and Bax, and theophylline, a cAMP phosphodiesterase inhibitor, synergized with cisplatin to induce programmed cell death of non‐small cell lung cancer cells involving reduction of intracellular levels of Bcl‐2.( 23 ) However, results from our study show that the Gαs–cAMP signaling system augments cisplatin‐induced apoptosis by upregulating the expression level of pro‐apoptotic molecules such as Bak and Bax and downregulating the expression level of anti‐apoptotic Bcl‐2 and Bcl‐XL in A549 lung cancer cells. The Gαs–cAMP signaling system also augmented cisplatin‐induced apoptosis of H1299 cells with increased expression of Bak, but the expression of antiapoptotic Bcl‐XL was also increased in GαsQL‐expressing cells as reported in SH‐SY5Y cells.( 18 ) It is speculated that the effect of increased expression of Bcl‐XL on the apoptosis of H1299 cells was repressed by the effect of increased expression of pro‐apoptotic Bak. The discrepancy of the observed effects of the Gαs–cAMP signaling system on the Bcl‐2 family of proteins may be due to the difference in cell lines and in the apoptosis‐triggering stimuli. We also showed that the Gαs–cAMP signaling system upregulated Bak expression by decreasing the degradation rate of Bak proteins, which is the first report to the best of our knowledge, and by increasing Bak mRNA in a PKA‐ and CRE‐dependent manner in lung cancer cells. Bak protein is a multidomain pro‐apoptotic member of the Bcl‐2 family, and plays an essential role in intrinsic cell death pathways by stimulating the release of cytochrome c from mitochondria.( 24 , 25 ) We previously reported that Gαs increases the binding of CREB to a variant CRE in the promoter region of the Bak gene to increase Bak expression in SH‐SY5Y neuroblastoma cells. Thus, Gαs is suggested to induce Bak gene transcription by increasing CREB binding to the variant CRE in A549 cells as in SH‐SY5Y neuroblastoma cells. However, Gαs augmented apoptosis in A549 cells whereas it protected against apoptosis in the neuroblastoma cells.( 17 ) Induction of Bak by the cAMP signaling pathway was also observed when prostate and colon cancer cells were treated with antisense oligodeoxynucleotides targeted against the RIα subunit of PKA.( 26 ) Cisplatin increased the expression of Bak even though it inhibited phosphorylation of CREB in lung cancer cells as hydrogen peroxide did in SH‐SY5Y cells.( 17 ) Hydrogen peroxide induced Bak expression through activation of activator protein‐1 (Ap‐1), nuclear factor kappa B (NF‐κB) and nuclear factor of activated T‐cells (NFAT) in SH‐SY cells, which suggests that cisplatin might induce Bak expression by activation of the same transcription factors because hydrogen peroxide was reported to play an essential role in cisplatin‐induced apoptotic cell death of lung cancer cells.( 27 )
This study showed that treatment with prostaglandin E2 augmented cisplatin‐induced apoptosis of A549 cells, suggesting the potential use of ligands for G protein‐coupled receptors in increasing the therapeutic efficiency of cisplatin. Phosphodiesterase inhibitors were used to induce cell death alone or in combination with cisplatin, as high intracellular levels of cAMP have been shown to effectively kill cancer cells in vitro, but substances elevating cAMP, such as forskolin, cause high cytotoxicity.( 23 ) Although phosphodiesterase inhibitors might be less toxic, they do not act in a cancer cell‐specific manner. However, the expression of G protein‐coupled receptors is differentially regulated depending on cell type as evidenced from the fact that only limited cell types that express a specific receptor can respond to a hormone or neurotransmitters. Thus, if cancer cells have derived from a cell type that expressed a specific G protein‐coupled receptor, the cancer cells might continue to express the G protein‐coupled receptor. Therefore, if the apoptosis of cancer cells can be modulated selectively by stimulation of a specific G protein‐coupled receptor that is expressed in cancer cells, it might be possible to improve the therapeutic efficiency of radiotherapy and chemotherapy of the cancer. The present study suggests that cisplatin‐induced apoptosis of lung cancer cells can be sensitized with more selectivity by stimulating the Gs‐coupled receptor specific to cancer cells. However, the effect of the Gαs–cAMP signaling system on apoptosis was different among lung cancer cells. The Gαs–cAMP signaling system protects lung cancer cells from ultraviolet radiation‐induced apoptosis( 28 ) and plays an important role in cell growth and survival of H1734 non‐small cell lung cancer cells.( 29 ) Therefore, it is essential to investigate the mechanisms that determine the effects of the cAMP signaling system on the cell death of lung cancer cells for the development of a new strategy to increase the therapeutic effect of cytotoxic drugs or radiation by modifying the cAMP signaling system.
We conclude that Gαs augments cisplatin‐induced apoptosis of lung cancer cells in part by upregulating Bak expression through an increase in transcription and a decrease in protein degradation in non‐small cell lung cancer cells. This suggests that the Gs‐coupled receptor might be a useful molecular target for improving the therapeutic efficiency of anticancer drugs and ionizing radiation.
Acknowledgments
This study was supported by a grant from the Korea Health 21 R. & D. Project, Ministry of Health and Welfare (A050335), by a Korea Science and Engineering Foundation grant funded by the Korea government (MOST) (no. R01‐2005‐000‐10230‐0), and by a grant (no. 2007‐01258) from the Nuclear R. & D. Program of the Ministry of Science and Technology, Republic of Korea.
References
- 1. Wong E, Giandomenico CM. Current status of platinum‐based antitumor drugs. Chem Rev 1999; 99: 2451–66. [DOI] [PubMed] [Google Scholar]
- 2. Cohen SM, Lippard SJ. Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucl Acid Res Mol Biol 2001; 67: 93–130. [DOI] [PubMed] [Google Scholar]
- 3. Jamieson ER, Lippard SJ. Structure. Recognition, Processing Cisplatin-DNA Adducts Chem Rev 1999; 99: 2467–98. [DOI] [PubMed] [Google Scholar]
- 4. Miyajima A, Nakashima J, Yoshioka K, Tachibana M, Tazaki H, Murai M. Role of reactive oxygen species in cis‐dichlorodiammineplatinum‐induced cytotoxicity on bladder cancer cells. Br J Cancer 1997; 76: 206–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schweyer S, Soruri A, Heintze A, Radzun HJ, Fayyazi A. The role of reactive oxygen species in cisplatin‐induced apoptosis in human malignant testicular germ cell lines. Int J Oncol 2004; 25: 1671–6. [DOI] [PubMed] [Google Scholar]
- 6. Group N‐sCLCC . Chemotherapy in non‐small cell lung cancer: a meta‐analysis using updated data on individual patients from 52 randomised clinical trials. BMJ 1995; 311: 899–909. [PMC free article] [PubMed] [Google Scholar]
- 7. Gebbia V, Oniga F, Agueli R, Paccagnella A. Treatment of advanced non‐small cell lung cancer: chemotherapy with or without cisplatin? Ann Oncol 2006; 17 (Suppl. 2): ii83–7. [DOI] [PubMed] [Google Scholar]
- 8. McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. G‐protein signaling: back to the future. Cell Mol Life Sci 2005; 62: 551–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev 2007; 33: 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clapham DE, Neer EJ. New roles for G‐protein beta gamma‐dimers in transmembrane signalling. Nature 1993; 365: 403–6. [DOI] [PubMed] [Google Scholar]
- 11. Conklin BR, Bourne HR. Structural elements of G alpha subunits that interact with G beta gamma, receptors, and effectors. Cell 1993; 73: 631–41. [DOI] [PubMed] [Google Scholar]
- 12. Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature 1990; 348: 125–32. [DOI] [PubMed] [Google Scholar]
- 13. Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science 1991; 252: 802–8. [DOI] [PubMed] [Google Scholar]
- 14. Zhang X, Odom DT, Koo SH et al . Genome‐wide analysis of cAMP‐response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA 2005; 102: 4459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Siu YT, Jin DY. CREB – a real culprit in oncogenesis. FEBS J 2007; 274: 3224–32. [DOI] [PubMed] [Google Scholar]
- 16. Allam M, Bertrand R, Zhang‐Sun G, Pappas J, Viallet J. Cholera toxin triggers apoptosis in human lung cancer cell lines. Cancer Res 1997; 57: 2615–18. [PubMed] [Google Scholar]
- 17. Kim SY, Seo M, Kim Y et al . Stimulatory heterotrimeric GTP‐binding protein inhibits hydrogen peroxide‐induced apoptosis by repressing BAK induction in SH‐SY5Y human neuroblastoma cells. J Biol Chem 2008; 283: 1350–61. [DOI] [PubMed] [Google Scholar]
- 18. Kim SY, Seo M, Oh JM, Cho EA, Juhnn YS. Inhibition of gamma ray‐induced apoptosis by stimulatory heterotrimeric GTP binding protein involves Bcl‐xL down‐regulation in SH‐SY5Y human neuroblastoma cells. Exp Mol Med 2007; 39: 583–93. [DOI] [PubMed] [Google Scholar]
- 19. Gu C, Ma YC, Benjamin J, Littman D, Chao MV, Huang XY. Apoptotic signaling through the beta‐adrenergic receptor. A new Gs effector pathway. J Biol Chem 2000; 275: 20 726–33. [DOI] [PubMed] [Google Scholar]
- 20. Chen TC, Hinton DR, Zidovetzki R, Hofman FM. Up‐regulation of the cAMP/PKA pathway inhibits proliferation, induces differentiation, and leads to apoptosis in malignant gliomas. Laboratory Invest 1998; 78: 165–74. [PubMed] [Google Scholar]
- 21. Ruchaud S, Seite P, Foulkes NS, Sassone‐Corsi P, Lanotte M. The transcriptional repressor ICER and cAMP‐induced programmed cell death. Oncogene 1997; 15: 827–36. [DOI] [PubMed] [Google Scholar]
- 22. Shafer SH, Phelps SH, Williams CL. Reduced DNA synthesis and cell viability in small cell lung carcinoma by treatment with cyclic AMP phosphodiesterase inhibitors. Biochem Pharmacol 1998; 56: 1229–36. [DOI] [PubMed] [Google Scholar]
- 23. Hirsh L, Dantes A, Suh BS et al . Phosphodiesterase inhibitors as anti‐cancer drugs. Biochem Pharmacol 2004; 68: 981–8. [DOI] [PubMed] [Google Scholar]
- 24. Chittenden T, Harrington EA, O’Connor R et al . Induction of apoptosis by the Bcl‐2 homologue Bak. Nature 1995; 374: 733–6. [DOI] [PubMed] [Google Scholar]
- 25. Wei MC, Zong WX, Cheng EH et al . Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cho YS, Kim MK, Tan L, Srivastava R, Agrawal S, Cho‐Chung YS. Protein kinase A RIalpha antisense inhibition of PC3M prostate cancer cell growth: Bcl‐2 hyperphosphorylation, Bax up‐regulation, and Bad‐hypophosphorylation. Clin Cancer Res 2002; 8: 607–14. [PubMed] [Google Scholar]
- 27. Wang L, Chanvorachote P, Toledo D et al . Peroxide is a key mediator of Bcl‐2 down‐regulation and apoptosis induction by cisplatin in human lung cancer cells. Mol Pharmacol 2008; 73: 119–27. [DOI] [PubMed] [Google Scholar]
- 28. Hastings RH, Araiza F, Burton DW, Bedley M, Deftos LJ. Parathyroid hormone‐related protein regulates apoptosis in lung cancer cells through protein kinase A. Am J Physiol Cell Physiol 2004; 287: C1616–22. [DOI] [PubMed] [Google Scholar]
- 29. Aggarwal S, Kim SW, Ryu SH, Chung WC, Koo JS. Growth suppression of lung cancer cells by targeting cyclic AMP response element‐binding protein. Cancer Res 2008; 68: 981–8. [DOI] [PMC free article] [PubMed] [Google Scholar]