

Abstract
Oxidative stress to DNA is recognized as a mechanism underlying carcinogenic effects of some environmental agents. Here, we hypothesized that dimethylarsinic acid (DMAV), an organic metabolite of inorganic arsenic in humans, might exert carcinogenic potential in a mouse line carrying a mutant Mmh allele of the Mmh/OGG1 gene encoding the enzyme 8‐hydroxyguanine DNA glycosylase 1 (OGG1). Ogg1 mutant and wild type mice were treated with DMAV in their drinking water at a dose of 200 p.p.m. for up to 72 weeks. All DMAV‐treated Ogg1 −/–animals developed tumors, with a tendency for lower total incidences in the Ogg1 +/+ cases. Lung tumors in particular were induced as compared to the lack in non‐carcinogen controls and were significantly more frequent in the homozygotes. At week 4, the levels of DNA 8‐OH‐dG and cell proliferation were significantly elevated in the lungs of non‐treated Ogg1 −/– as compared to Ogg1 +/+ mice and were strongly enhanced by DMAV treatment. Marked induction of Pola1, Cyp7b1, Ndfua3, Mmp13 and other genes specific to cell proliferation, cell signaling and xenobiotic metabolism in the lungs of DMAV‐treated Ogg1 −/– mice was found. Electron microscopic examination revealed the growth of microvilli, with increased numbers of mitochondria only in lungs and lung tumors of DMAV‐exposed Ogg1 −/– mice. Therefore, we strongly suggest that DMAV exerts carcinogenicity in the lungs of Ogg1 −/– mutant mice, with a possible role for persistent accumulation of DNA oxidative adducts. (Cancer Sci 2007; 98: 803–814)
DNAdamage and defective DNA repair are considered to be essential for the initiation of carcinogenesis and age‐related changes in both the efficacy and the rate of DNA repair in mammals to modify susceptibility to exogenous or endogenous carcinogens.( 1 ) Lesions in genomic DNA caused by reactive oxygen species (ROS) are closely associated with aging and various diseases, including cancer, a typical major example being 8‐hydroxy‐2′‐deoxyguanosine (8‐OH‐dG). This is produced by exposure to various carcinogens and is considered to be a critical mutagenic adduct.( 2 ) Mispairing of this oxidized base with adenine results in GC→TA transversion mutations found in mammalian cells.( 3 ) In Saccharomyces cerevisiae and various bacteria, 8‐hydroxyguanine (8‐OH‐G) is excised from DNA by DNA glycosylases (catalyzed by MutM and Ogg1, respectively) with associated lyase activity for chain cleavage. A mammalian homolog of 8‐OH‐G glycosylase/apurinic, apyrimidinic lyase (AP lyase; MutM homolog, MMH) has already been identified and cloned and spontaneously developing tumors in the lungs and ovaries, as well as lymphomas, were recently detected in Myh and Ogg1 knockout mice.( 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 ) Furthermore, chronic ultraviolet (UV) B irradiation of Ogg1 knockout mice have been shown to result in skin cancer development.( 12 ) However, it still remains unclear how deletion of this enzyme affects the carcinogenicity of chemicals in vivo.
The Mmh/OGG1 homozygous mutant mice used in the present study have a physically normal appearance, but lack nicking activity in liver extracts for substrate DNA containing 8‐OH‐dG, exhibiting a 3‐ and 7‐fold increased accumulation of this adduct at 9 and 14 weeks of age, respectively, as compared with wild type or heterozygous mice.( 13 ) Substantial elevation in the spontaneous mutation frequency has been identified in Mmh/OGG1 mutant mice bearing transgenic gpt genes and Arai et al. recently reported that administration of potassium bromate (KBrO3) to Ogg1‐deficient mice resulted in a tremendous increase (∼70‐fold) in 8‐OH‐dG levels in kidney DNA, with consequent GC→TA transversions and deletions.( 13 , 14 ) Furthermore, administration of KBrO3 to Ogg1 mutant mice followed by partial hepatectomy also led to a 3.5‐fold increase in mutation frequency during liver regeneration.( 15 )
Arsenics are of world‐wide concern as environmental carcinogens and epidemiological studies have shown that chronic exposure of humans to inorganic arsenic compounds (trivalent arsenite (As3+) or pentavalent arsenate (As5+)) increases incidences of cancer of the lung, skin, liver, and urinary bladder, also being associated with liver injury and peripheral neuropathy.( 16 , 17 ) Although there is strong evidence for the carcinogenicity of inorganic arsenic compounds in humans, the molecular mechanisms remain incompletely defined. It is reasonable to suspect that more than one mechanism may be involved in arsenicals carcinogenesis because they can be carcinogenic in many diverse tissues in both humans and animals.( 18 ) Defining these mechanisms is critical to determining the nature and extent of the human health hazard presented by environmental arsenic exposure and to the development of intervention and prevention strategies. In this regard, it has become very important to develop the mouse models of arsenic‐induced carcinogenesis. Recent data from biochemical and carcinogenic studies using laboratory animals suggest that methylation of inorganic arsenic to form both dimethylarsinic acid (DMAV) and methylarsonic acid (MMAV), which are the major pathways of biotransformation and detoxification requiring reduced cellular glutathione (GSH), may paradoxically also result in toxification.( 18 ) DMAV, one of the organic methylated arsenics, has been used as a herbicide and is also a major metabolite formed after exposure to As3+ or As5+ inorganic arsenic via ingestion or inhalation in both humans and most rodents.( 19 ) DMAV has been considered to be over 10‐fold less acutely toxic than inorganic arsenic.( 20 )
Initiation‐promotion studies have demonstrated that DMAV acts as a multiorgan tumor promoter in rats and mice and A/J mice treated with 400 p.p.m. DMAV in their drinking water for 50 weeks developed more pulmonary tumors than untreated animals.( 21 , 22 , 23 , 24 , 25 ) A significant dose‐dependent increase in preneoplastic lesions, papillomas and carcinomas in the urinary bladders of rats was also found with DMAV treatment after initiation.( 21 ) Furthermore, in a long‐term carcinogenicity test, DMAV application in the drinking water resulted in induction of bladder carcinomas in rats.( 26 ) It has been suggested that in mice this damage might be due primarily to peroxyl radicals and production of ROS in pulmonary tissues.( 27 ) Tumor promotion might thus be related to oxidative stress.( 28 , 29 ) However, until now, DMAV has not been found to exert initiation activity for mouse tumorigenesis and this is feasible given differences in metabolism from the rat case.( 18 )
In the present study, to cast light on the relationship between human carcinogenic arsenics and oxidative stress, the carcinogenic effects of DMAV were examined in mutant Ogg1 mice. At the end of the treatment period (72 weeks), multi‐organ histopathological, immunohistochemical, biochemical, genomic, proteomic and electron microscopic analyses were conducted with a particular focus on cell proliferation, apoptosis, formation of oxidative DNA modifications and alterations to cell ultrastructure in mouse tissues targeted by DMAV. The findings indicate that lung carcinogenesis in Ogg1 −/– homozygotes is associated with marked elevation of DNA 8‐OH‐dG levels and cellular proliferation.
Materials and Methods
Chemicals. DMAV (purity 99%) was purchased from Sigma (St. Louis, MO, USA). Other chemicals were from Sigma or Wako Pure Chemical Industries (Osaka, Japan).
Maintenance of mice. Animals were housed in an animal facility maintained on a 12 h (07:00–19:00) light/dark cycle, at a constant temperature of 22 ± 1°C, with a relative humidity of 44 ± 5%, and given free access to water and food ad libitum. Throughout the experiment they were administered a common basal pellet diet (CE2; Clea Japan, Tokyo, Japan). In the present study, 14‐week‐old mice at the commencement were used, as our previous data demonstrated that chronic DMAV administration is particularly toxic in young animals.( 19 , 30 )
Experimental protocol. Experiment 1. 14‐week‐old male (20) and female (20) Ogg1 −/–, and male (22) and female (22) Ogg1 +/+mice of a C57Bl/6 J background were divided into two groups each (Ogg1 −/– male and female: five mice per group; Ogg1 −/– male and female: five or six mice per group) and administered DMAV at a concentration of 200 p.p.m. (Ogg1 −/– and Ogg1 +/+ treatment groups) ad libitum in drinking water for 72 weeks or tap water as controls. Their body weight, water intake and food consumption were recorded every week for the first 12 weeks of the study and subsequently once every 4 weeks. Mice were observed daily, killed when becoming moribund during the study or at the end of the experiment at week 72, and autopsied for macroscopic, histopathological (H&E), immunohistochemical, biochemical and electron microscopic examinations.
At death, lungs were removed and the left lobe was fixed in 10% buffered formalin or Bouin solution for histological and immunohistochemical assessment. Part of the right lungs were treated with 2% glutaraldehyde and 2% paraformaldehyde in 0.12 mol/L phosphate buffer (PB) (pH 7.2) for subsequent electron microscopic analysis and the remainder were frozen in liquid nitrogen and stored at −80°C for molecular assessment. Lung nodules were accurately separated from normal lung and divided into separate pieces fixed in 10% buffered formalin, Bouin or 2% glutaraldehyde and 2% paraformaldehyde in 0.12 mol/L PB (pH 7.2).
Experiment 2.
14‐week‐old male and female Ogg1 −/– (40) and Ogg1 +/+ (40) mice on a C57Bl/6 J background (10 animals/group) were treated with DMAV at a concentration of 0 or 200 p.p.m. for 4 weeks. Mice lungs were used for the analysis of 8‐OH‐dG levels, proliferating cell nuclear antigen (PCNA), apoptosis indices and alteration to gene expression using the Affimetrix oligonucleotide microarray system.
Analysis of 8‐OH‐dG formation. In experiment 2, mice lung DNA 8‐OH‐dG levels were determined by a high performance liquid chromatography (HPLC)‐electrochemical detection (ECD) method.( 2 ) Furthermore, in the long term experiment 1, immunohistochemical staining of 8‐OH‐dG was carried out as described previously.( 31 )
Immunohistochemistry for PCNA and apoptosis.
In experiments 1 and 2, immunohistochemical staining for PCNA and apoptosis was carried out using an anti‐PCNA rabbit polyclonal (PC‐10, IgG; Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:500) and single‐stranded DNA ((ssDNA); IgG, 100 µg/mL, DAKO JAPAN, Kyoto, Japan; 1:400) antibodies, respectively, on sections of lung and bladder as described previously.( 31 ) PCNA and apoptosis indices were estimated for epithelial cells of lung alveoli with counts of clearly brown staining positive nuclei per 1000 cells.
Transmission electron microscopy. In long‐term experiment 1, portions of the harvested lungs and urinary bladders were minced into 1‐mm3 pieces, fixed in 0.12 mol/L PB (pH 7.2), containing 2% (v/v) glutaraldehyde and 2% paraformaldehyde (v/v) for 1.5 h, washed in 0.12 mol/L PB (pH 7.2) and 7% sucrose in PB for 7 min twice and postfixed in 2% osmium tetroxide. They were then dehydrated through ascending concentrations of ethanol and propylene oxide, embedded in Durcupan (TAAB Laboratories, Aldermaston, UK), and polymerized at 60°C overnight. Ultrathin sections exhibiting a pale gold interference color from selected blocks were then cut with a diamond knife, mounted on grids, and stained with ethanol uranyl acetate and lead citrate before transmission electron microscopy (TEM) examination (Hitachi H‐7500, Tokyo, Japan).
Affymetrix oligonucleotide microarray analysis. In experiment 2, total RNA was isolated from mouse lung tissue (separate lung lobe without bronchi; pieces <5 mm in diameter) as described previously.( 31 ) Development of lung nodules was not observed at week 4. Therefore, only normal‐appearing lungs were submitted to the microarray analysis. Eight‐microgram pooled aliquots of total RNAs from five mice in each group were treated with DNase 1 and processed for PolyA+ RNA enrichment and generation of cDNA probes using an Affymetrix GeneChip T7‐Oligo(dT) Promoter Primer Kit (Affymetrix) according to the manufacturer's protocol. Biotin‐labeled antisense cRNA was synthesized by in vitro transcription reaction (IVT) using an RNA Transcript Labeling Kit (Affymetrix, P/N 900182), purified and fragmented, and hybridized to Mouse E430 Plus 2.0 arrays (MOE430 Plus 2.0), with 45 101 probe sets, according to the manufacturer's instructions (Affymetrix). Affymetrix GCOS software version 1.0 was used for normalization and for monitoring specific hybridization and microarray data were analyzed using GeneSpring software version 7.2 (Silicon Genetics, Redwood City, CA, USA). Each array was normalized to the 50th percentile and each gene was normalized to the control (non‐treated Ogg1 −/– or Ogg1 +/+ groups). Log2 ratios of the hybridization intensities of mouse lung samples were used to represent the relative gene expression level. To minimize the effects of measurement variation introduced by artificial sources during the experiments, we only included spots with up‐regulation or down‐regulation at least twofold. Microarray analysis was repeated twice to check the reproducibility of the data and mean values for gene expression was calculated. One‐way‐anova was applied to compare replicates mean values of control and experimental groups (Gene Spring). The microarray set for statistical analysis was three. Comparisons of gene expression across treatment groups were carried out using Venn Diagrams and clustering was carried out with the Condition Tree algorithm. In addition, the Gene Ontology grouping was used in conjunction with Gene Spring to identify pathways and functional groups of genes. The Ingenuity program (Ingenuity Systems, Mountain View, CA, USA) was also used to identify networks of interacting genes and other functional groups.
Measurement of mRNA in mouse lung tissues by real‐time quantitative‐polymerase chain reaction. For the confirmation of Affimetrix microarray analysis results, real‐time quantitative‐polymerase chain reaction (Q‐PCR) was carried out using TaqMan probes for 490 C. Primer sequences were designed with Primer Express software (Applied Biosystems, USA): Mm00447106_m1 for polymerase (DNA‐directed), alpha 1 (NM_008892); Mm00484157_m1 for cytochrome P450, 7b1 (NM_007825); Mm00772875_m1 for nicotineamide adenine dinucleotide reduced form (NADH) dehydrogenase (ubiquinone) 1 alpha subcomplex, 3 (NM_025348); Mm00439491_m1 for matrix metalloproteinase 13 (NM_008607); Mm00834384_g1 for heat shock protein 1 (NM_013560); and Mm00607939 for beta‐actin, cytoplasmic (NM_007393). The cDNA product from each sample was used to carry out Q‐PCR according to the manufacturer's instructions. We used beta‐actin mRNA as an internal control for comparison and to normalize the data. Assays were carried out in triplicate using Applied Biosystems Model 7700 instruments.
Statistical analysis. The significance of differences between mean values was analyzed using the Dunnet's two‐tailed test and Fisher's protected least significant difference (PLSD) method using the StatView–J 5.0 program (Berkeley, CA, USA). Kaplan‐Meier analysis was conducted to determine the survival rates for Ogg1 knockout, heterozygous and wild type mice. One‐way‐anova was applied in Affymetrix oligonucleotide microarray analysis to compare replicates mean values of control and experimental groups (Gene Spring). The significance of differences in lesion incidences between groups was assessed by χ2 test or Fisher's exact probability test.
Results
General observations (Experiment 1). During the experiment no toxicity of DMAV in mice was apparent and no significant differences were found among the groups in the quantities of food and water consumed or body weight gained (data not shown). Total DMAV intake was comparable among Ogg1 homozygous and wild type mice.
Significant increase in relative lung, liver and spleen weights was found in the group of Ogg1 −/– mice treated with DMAV (male lung: 0.017 ± 0.003 (P < 0.01); female lung: 0.012 ± 0.005 (P < 0.5); male liver: 0.07 ± 0.02 (P < 0.05); female liver: 0.082 ± 0.025 (P < 0.05); male spleen: 0.006 ± 0.006, female spleen 0.019 ± 0.013 (P < 0.05); as compared to corresponding Ogg1 −/– controls (male lungs: 0.012 ± 0.002; female lungs: 0.007 ± 0.001; male liver: 0.042 ± 0.004; female liver: 0.049 ± 0.003; male spleen: 0.002 ± 0.001; female spleen: 0.003 ± 0.001). Furthermore, lungs weight of control Ogg1 −/– animals was significantly higher (P < 0.05) than that of control Ogg1 +/+ (male and female lungs: 0.005 ± 0.001); relative liver, kidney and spleen weights did not differ between Ogg1 +/+ DMAV‐treated and control groups.
Survival curves for Ogg1 homozygous and wild type age‐matched littermates are shown in Fig. 1a–c, respectively. At week 38, the first Ogg1 +/+ DMAV‐treated male mouse was found dead with a renal adenocarcinoma. The number of surviving Ogg1−/– mice administered DMAV started to decrease at week 58 due to early tumor development as compared to Ogg1 +/+ mice (Fig. 1). The development of various spontaneous tumors in wild type mice was the reason for their mortality, while Ogg1−/– mice (less induction of spontaneous tumors compared to Ogg1 +/+) died predominantly due to the occurrence of malignant lymphomas.
Figure 1.
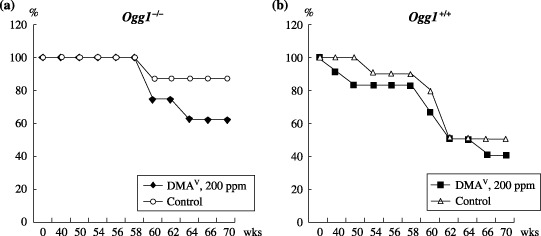
Survival curves for Ogg1 −/– (a) and Ogg1 +/+ (b) mice. Non‐treated Ogg1 −/– mice were found to be more healthy and long‐lived as compared to the Ogg1 +/+ control mice. The number of surviving Ogg1 −/– mice administered dimethylarsinic acid (DMAV) started to decrease at week 58 due to tumor development.
Lesion incidence, multiplicity and histopathology. Data for tumor incidences, multiplicities and organ distributions of tumors in DMAV‐treated and non‐treated mice with reference to the Ogg1 −/– and Ogg1 +/+ mice genotypes are presented in Table 1. Because of highly similar tumor induction in male and female mice, combined data are shown. Ogg1 −/– mice treated with DMAV were more susceptible to the induction of tumors as compared to wild type littermates, although no tumors were found in untreated Ogg1 −/– control mice, this being the reason for the high survival rate. Significant increases in total tumor incidence (P < 0.0001) and multiplicity (P < 0.001) were observed in the DMAV‐applied Ogg1 −/– group as compared to the control Ogg1 −/– or DMAV‐treated or non‐treated wild type animals.
Table 1.
Tumor incidence, multiplicity and histopathology
| Site and type of tumor† | Incidence (%) | (Multiplicity (no. tumors/mouse) | ||
|---|---|---|---|---|
| DMAV, 200 p.p.m. | Control | DMAV, 200 p.p.m. | Control | |
| Ogg1 −/– | ||||
| No. mice | 10 | 10 | 10 | 10 |
| No. tumor bearing mice (%) | 10 (100%)**, *** | 0 | ||
| M. lymphoma/leukemia | 4 (40) | 0 | 0.4 ± 0.5 | 0 |
| Liver | ||||
| HCC | 1 (10) | 0 | 0.1 ± 0.31 | 0 |
| Lung | ||||
| Hyperplasia | 10 (100) | 10 (100) | 5.0 ± 1.0*, **** | 6.8 ± 1.09 |
| Adenoma | 2 (20) | 0 | 0.2 ± 0.42 | 0 |
| Adenocarcinoma | 3 (30) | 0 | 0.3 ± 0.48 | 0 |
| Total tumors | 5 (50)*, *** | 0 | 0.5 ± 0.52*, *** | 0 |
| Mammary gland | 1 (10) | 0 | 0.1 ± 0.3 | 0 |
| Adenocarcinoma | ||||
| Ogg1 +/+ | ||||
| No. mice | 12 | 10 | 12 | 10 |
| No. tumor bearing mice (%) | 6 (50%) | 5 (50%) | ||
| M. lymphoma/leukemia | 2 (16.7) | 2 (20) | 0.17 ± 0.38 | 0.2 ± 0.3 |
| Liver | ||||
| HCC | 1 (8.3) | 0 | 0.08 ± 0.28 | 0 |
| Lung | ||||
| Hyperplasia | 10 (100) | 2 (20) | 3.00 ± 2.76* | 0.33 ± 0.10 |
| Adenoma | 0 | 1 (10) | 0 | 0.10 ± 0.31 |
| Adenocarcinoma | 0 | 0 | 0 | 0 |
| Total tumors | 0 | 1 (10) | 0 | 0.10 ± 0.31 |
| Mammary gland | ||||
| Adenocarcinoma | 1 (8.3) | 0 | 0.08 ± 0.28 | 0 |
| Renal | ||||
| Adenocarcinoma | 1 (8.3) | 0 | 0.08 ± 0.28 | 0 |
| Subcutis | ||||
| Fibrosarcoma | 0 | 1 (10) | 0 | 0.1 ± 0.31 |
| Skin | ||||
| Sarcoma | 1 (8.3) | 0 | 0.08 ± 0.28 | 0 |
Only organs with neoplastic lesions are listed.
P < 0.05,
P < 0.0001 versus corresponding non‐treated controls;
P < 0.05,
P < 0.0001 versus Ogg1 +/+ DMAV‐treated or control groups. DMAV, dimethylarsinic acid; HCC; hepatocellular carcinoma; M. lymphoma, malignant lymphoma.
The tumors induced in the DMAV‐treated group of Ogg1 −/– mice were mainly in the lungs (50%) and malignant lymphomas/leukemias (40%) (Table 1, Fig. 2a,b). Significant elevation of lung tumor multiplicity was apparent in DMAV‐treated Ogg1 homozygous mice when compared to their control Ogg1 –/– and DMAV‐treated Ogg1 +/+ counterparts (P < 0.05). Total incidence of lung adenoma and adenocarcinoma in male Ogg1 –/– mice was 60%, a little higher than in females (40%). Both adenomas and carcinomas were induced at the same rate (1:1). On the contrary, no lung tumors were observed in DMAV‐treated male Ogg1 + /+ animals, and only one female Ogg1 +/+ mouse in the control group developed spontaneous adenocarcinoma (20%). Furthermore, a significant increase of hyperplastic epithelial lesion multiplicity was observed in the lungs of non‐treated Ogg1 –/– mice as compared to non‐treated wild type mice (Table 1). The trend for increase of lung hyperplasia was found in the lungs of Ogg1 + /+ animals treated with DMAV. A slight decrease in the number of hyperplastic lesions but an increase of tumor development was observed in the lungs of DMAV‐treated Ogg1 –/– animals as compared to non‐treated Ogg1 –/– mice, indicating that DMAV exerted a promoting effect on lung carcinogenesis in Ogg1 –/– mice (Table 1). Malignant lymphomas/leukemias were mainly observed in Ogg1 −/– mice (40%) treated with DMAV, although intergroup differences in incidence and multiplicity were not significant. Interestingly, no tumors were found in non‐treated Ogg1 −/– animals (Table 1). In contrast, 50% of DMAV‐treated or control wild type littermates developed tumors, which included malignant lymphomas/leukemias, mammary tumors, renal adenocarcinomas, subcutis fibrosarcomas and skin sarcomas (Table 1). Spontaneous tumors of non‐treated Ogg1 +/+ mice were malignant lymphomas, lung adenomas and subcutis fibrosarcomas.
Figure 2.
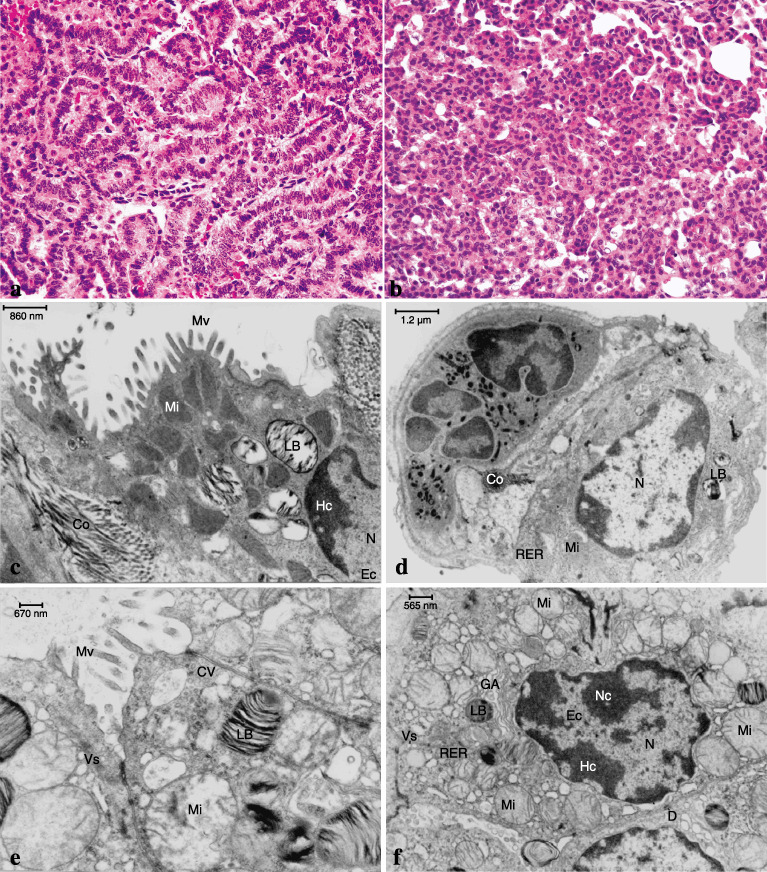
Results of histopathological (H&E) staining and transmission electron microscopy (TEM) in the lungs of Ogg1 −/– mice maintained on 200 p.p.m. dimethylarsinic acid (DMAV). (a) (× 400) and (b) (× 400) Adenocarcinoma and adenoma, respectively, in the lung of the DMAV‐treated Ogg1 −/– mouse (H&E). (c) TEM of alveoli from a non‐tumorous area in a Ogg1 −/– animal treated with DMAV and (d) alveoli of a non‐treated Ogg1 −/– animal. (e, f) TEM of a lung adenocarcinoma from an Ogg1 −/– animal treated with DMAV. Note the numerous mitochondria, microvilli and abundant vesicles. Those changes were absent in Ogg1 +/+ mice (data not shown). Co, collagen fibres; CV, clatrin vesicles; D, desmosomes; Ec, euchromatin; GA, Golgy apparatus; Hc, heterochromatin; LB, lamella bodies; Mi, mitochondria; Mv, microvilli; N, nucleus; Nc, nucleolus; RER, rough endoplasmic reticulum; Vs, vesicles. Original magnifications: × 400 (a); × 400 (b); × 25 000 (c–f), Scale bar: 860 nm (c); 1.2 µm (d); 670 nm (e); 565 nm (f).
Extensive examination of the urinary bladders of all treated and non‐treated mice revealed simple hyperplasia only in DMAV‐administered homozygous animals (data not shown). No urinary calculi were found in Ogg1 −/– and Ogg1 +/+ mice after DMAV application.
Changes of cell ultrastructure detected in the lungs and lung tumors (Experiment 1). Increased numbers of mitochondria, amounts of endoplasmic reticulum, and collagen fibers, with elongated microvilli only in the lungs of Ogg1 −/– mice were detected by TEM after DMAV treatment for 72 weeks. Ten sections from each animal were analyzed to ensure that there real quantitative differences existed. Those changes were mostly observed in Type II pneumocytes of Ogg1 −/– mice but not wild type animals treated with DMAV (Fig. 2c). TEM examination of lung tumors developing in Ogg1 −/– animals after DMAV treatment, revealed an increased number of mitochondria and lamella bodies and markedly developed rough endoplasmic reticulum (Fig. 2e,f) and large numbers of pleomorphic microvilli (data not shown), in line with changes in Type II pneumocytes of non‐tumorous areas (Fig. 2c).
Lung 8‐OH‐dG, cellular proliferation and apoptosis indices (Experiments 1 and 2). Elevated levels of DNA 8‐OH‐dG, detected by HPLC‐ECD analysis, were found in the lungs of non‐treated Ogg1 −/– animals as compared to their wild‐type counterparts (Fig. 3a). Furthermore, treatment with DMAV caused a significant increase of 8‐OH‐dG levels in Ogg1 −/– mouse lungs. Immunohistochemical determination of 8‐OH‐dG also showed a strong overexpression in the lungs of Ogg1 −/– mice due to the DMAV treatment (Fig. 3b).
Figure 3.
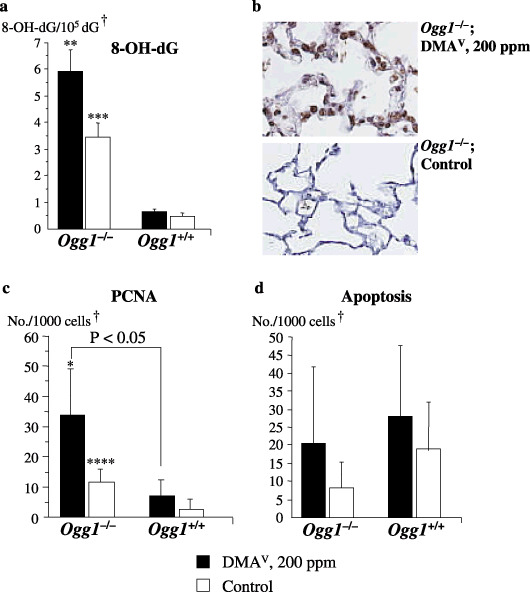
Formation of 8‐hydroxy‐2′‐deoxyguanosine (8‐OH‐dG) and cellular proliferation and apoptosis indices in the lungs of Ogg1 −/– and Ogg1 +/+ mice treated with dimethylarsinic acid (DMAV) at 200 p.p.m. for 72 weeks. (a) 8‐OH‐dG (c) proliferating cell nuclear antigen (PCNA) and (d) apoptosis indices. (b) 8‐OH‐dG staining pattern in DMAV‐treated and control Ogg1 −/– mouse. †Normal‐appearing area; *Significant at P < 0.05 versus Ogg1 −/– control group; **Significant at P < 0.001 versus Ogg1 −/– control group; ***Significant at P < 0.0001 and ****Significant at P < 0.05 versus Ogg1 +/+ control group.
Proliferating cell nuclear antigen positive indices for the lungs of non‐treated Ogg1 −/– mice were found to be significantly higher than in control Ogg1 +/+ animals. These data were confirmed in a short experiment (Experiment 2), in which Ogg1 −/– and wild type mice were administered DMAV for 4 weeks. DMAV treatment resulted in further significant elevation of PCNA positive indices in the normal‐appearing areas of lungs and urinary bladder mucosa (data not shown) of homozygous mice as compared to control Ogg1 −/– or their wild‐type age‐matched littermates (Fig. 3c).
Apoptosis indices tended to be elevated only in the lungs of Ogg1 −/– animals administered DMAV but without significance (Fig. 3d). Tumors were found to be negative for apoptosis. ssDNA staining patterns in serial sections were similar to those for 8‐OH‐dG and increase of the apoptotic indices paralleled elevation in 8‐OH‐dG and the PCNA indices (data not shown).
Changes in gene expression triggered by DMAV (Experiment 2). The results of the Affymetrix oligonucleotide microarray analysis of differentially expressed genes in the lungs of mice, obtained in the short‐term experiment after 4 weeks of DMAV administration are presented in Table 2. The results were compared with Gene Spring software and a strong concordance was found. Furthermore, the data obtained by microarrays on the expression of Pola1, Cyp7b1, Ndfua3, Mmp13, and Hspb1 were checked by real‐time Q‐PCR analysis, which showed the same patterns (Fig. 4a–e).
Table 2.
Differentially expressed genes in the lungs of Ogg1 −/– and Ogg1 +/+ mice treated with dimethylarsinic acid (DMAV) for 4 weeks
| Animals | Function Gene | Accession No. | Ratio | P‐value |
|---|---|---|---|---|
| Male Ogg1 −/– | Cell proliferation | DMA, 200 p.p.m. vs vehicle | ||
| Polymerase (DNA directed), alpha 1 | NM_008892 | 2.268 | 0.011 | |
| Mitogen activated protein kinase kinase 6 | BB540608 | 4.46 | 0.001 | |
| Topoisomerase (DNA) II alpha | BB749838 | 2.922 | 0.006 | |
| Heat shock protein 1A | AW763765 | 2.547 | 0.009 | |
| Heat shock protein 1B | M12573 | 2.344 | 0.009 | |
| Vascular endothelial growth factor C | AW228853 | 2.687 | 0.006 | |
| Sulfotransferase, hydroxysteroid preferring 2 | NM_009286 | 5.381 | 0.001 | |
| Phospholipase A2, group VII | AK005158 | 3.321 | 0.003 | |
| Extracellular matrix | ||||
| Catenin delta 2 | BB431091 | 3.561 | 0.002 | |
| Fibroblast growth factor 23 | BB840359 | 0.292 | 0.015 | |
| Stress response | ||||
| Cytochrome P450, family 7, subfamily b, polypeptide 1 | NM_007825 | 2.318 | 0.012 | |
| NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 3 | AA060162 | 3.345 | 0.002 | |
| Female Ogg1 −/– | Cell proliferation | |||
| Polymerase (DNA directed), alpha 1 | NM_008892 | 2.021 | 0.018 | |
| Polymerase (DNA directed), gamma | AV351762 | 2.023 | 0.019 | |
| Polymerase (DNA directed), iota | NM_011972 | 2.193 | 0.011 | |
| Polymerase (DNA directed), epsilon 2 (p59 subunit) | AF036898 | 2.255 | 0.022 | |
| Topoisomerase (DNA) I | BB127876 | 2.015 | 0.029 | |
| Mitogen‐activated protein kinase kinase kinase kinase 5 | AW555664 | 2.356 | 0.027 | |
| Mitogen‐activated protein kinase kinase kinase kinase 14 | BG072756 | 2.867 | 0.02 | |
| Ornithine decarboxylase antizyme inhibitor | BI155585 | 0.301 | 0.043 | |
| Stress response | ||||
| Heat shock protein 4 | NM_015765 | 2.281 | 0.028 | |
| Heat shock 70 kDa protein 12A | BQ17716 | 2.245 | 0.023 | |
| Cytochrome P450, family 7, subfamily b, polypeptide 1 | NM_007825 | 2.318 | 0.03 | |
| NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 3 | AA060162 | 4.282 | 0.004 | |
| Superoxide dismutase 1, soluble | BB530180 | 0.308 | 0.042 | |
| Matrix metalloproteinase 13 | NM_008607 | 2.564 | 0.029 | |
| Carbonic anhydrase 13 | AK010166 | 2.431 | 0.033 | |
| Guanine nucleotide binding protein, gamma 5 subunit | BC010725 | 2.062 | 0.042 | |
| Apoptosis | ||||
| Transforming growth factor, beta receptor I | NM_009370 | 0.197 | 0.027 | |
| Ubiquitin specific protease 7 | C77542 | 0.0581 | 0.001 | |
| Male Ogg1 +/+ | Stress response | |||
| Metallothionein 1 | BC027262 | 5.491 | 0.005 | |
| Cytochrome b‐245, beta polypeptide | AV373944 | 2.105 | 0.045 | |
| NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 3 | AA060162 | 3.282 | 0.009 | |
| Matrix metalloproteinase 9 | D12712 | 2.604 | 0.011 | |
| Adipsin | NM_013459 | 2.464 | 0.021 | |
| Cell signaling | ||||
| Mitogen activated protein kinase kinase 6 | BB540608 | 0.444 | 0.034 | |
| Mitogen activated protein kinase kinase kinase 12 | AW556977 | 0.422 | 0.018 | |
| Extracellular matrix | ||||
| Procollagen, type XVII, alpha 1 | NM_007732 | 0.182 | 0.039 | |
| Female Ogg1 +/+ | Stress response | |||
| Matrix metalloproteinase 17 | NM_011846 | 2.478 | 0.043 | |
| Cytochrome P450, family 7, subfamily b, polypeptide 1 | NM_007825 | 2.214 | 0.002 | |
| Superoxide dismutase 1, soluble | BB530180 | 0.428 | 0.043 | |
| Carbonic anhydrase 3 | AK003671 | 0.266 | 0.017 | |
| Extracellular matrix | ||||
| Titin | BC025840 | 2.783 | 0.042 | |
| Male Ogg1 −/– vs Ogg1 +/+ | Vehicle vs vehicle | |||
| Xenobiotic metabolism | ||||
| Cytochrome P450, family 7, subfamily b, polypeptide 1 | NM_007825 | 2.032 | 0.001 | |
| Cell proliferation | ||||
| Polymerase (DNA‐directed), delta 4 | BB825816 | 2.065 | 0.002 | |
| Metabolism | ||||
| Phosphoenolpyruvate carboxykinase 1, cytosolic | AW106963 | 10.75 | 0.033 | |
| Amine oxidase, copper containing 3 | NM_009675 | 2.179 | 0.048 | |
| Extracellular matrix, intracellular transport | ||||
| Karyopherin (importin) beta 1 | BC004096 | 2.483 | 0.037 | |
| Cadherin 4 | NM_009867 | 2.784 | 0.028 | |
| Stress response | ||||
| Osmotic stress protein | NM_011020 | 0.371 | 0.041 | |
| Heat shock protein 1 | NM_013560 | 0.364 | 0.046 | |
| Heat shock protein 1A | BC006722 | 0.228 | 0.022 | |
| Heat shock protein 1B | M12573 | 0.256 | 0.033 | |
| Heat shock protein 105 | D67017 | 0.251 | 0.02 | |
| Heat shock protein 8 | AW763765 | 0.211 | 0.011 | |
| Adipsin | NM_013459 | 5.094 | 0.001 | |
| Female Ogg1 ‐/‐ vs Ogg1 +/+ | Xenobiotic metabolism | |||
| Cytochrome P450, family 7, subfamily b, polypeptide 1 | NM_007825 | 2.032 | 0.046 | |
| Cell proliferation and angiogenesis | ||||
| Cyclin C | AK014079 | 2.049 | 0.049 | |
| Mitogen activated protein kinase 8 | BB184171 | 3.009 | 0.013 | |
| Matrix metalloproteinase 13 | NM_008607 | 11.08 | 0.001 | |
| Matrix metalloproteinase 14 (membrane‐inserted) | AI325305 | 2.121 | 0.045 | |
| Matrix metalloproteinase 17 | NM_0118 | 2.585 | 0.035 | |
| Stress response | ||||
| Heat shock protein 1 | AF047377 | 0.469 | 0.041 | |
| Heat shock protein 1A | BC006722 | 0.301 | 0.021 | |
| Heat shock protein 1B | M12573 | 0.409 | 0.04 | |
| Extracellular matrix | ||||
| Protocadherin beta 16 | BB027682 | 4.741 | 0.004 | |
Genes, that were up‐regulated or down‐regulated versus vehicle control more than two‐fold in Ogg1 −/– and Ogg1 +/+ animals. The significance of differences in gene expression was analyzed by statistical analysis anova, one‐way test (GeneSpring, n = 3). NADH, nicotineamide adenine dinucleotide reduced form.
Figure 4.
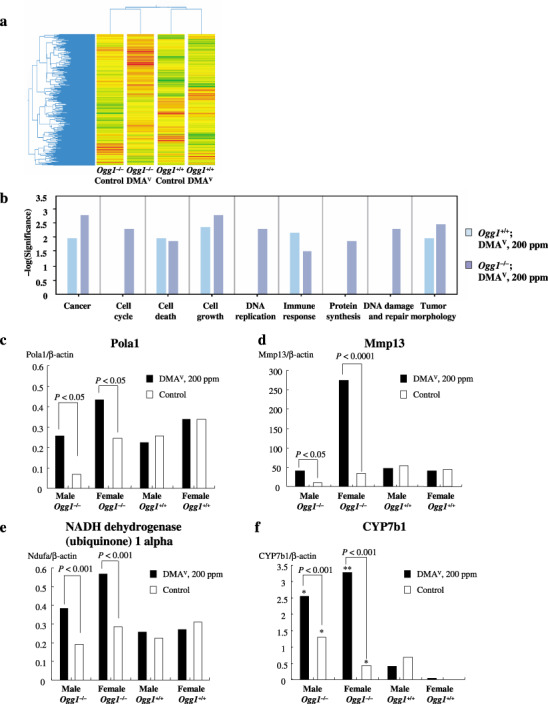
Gene Spring clustering analysis of the Affymetrix oligonucleotide microarray data (a) Ingenuity pathway analysis (b) and conformation of microarray results by real‐time quantitative polymerase chain reaction (Q‐PCR) (c–f). (c) Polymerase (DNA‐directed) alpha 1 (d) Mmp13 (e) nicotineamide adenine dinucleotide reduced form (NADH) dehydrogenase (ubiquinone) 1 alpha, and (f) CYP7b1 mRNA expression. *P < 0.05 and **P < 0.0001 versus Ogg1 +/+ control and dimethylarsinic acid (DMAV)‐treated groups.
Expression of 165 genes in Ogg1 −/– males and 182 genes in Ogg1 −/– females was changed in lungs following treatment with the 200 p.p.m. carcinogenic dose of DMAV. Among them we observed up‐regulation of metabolism associated genes, including cytochrome P450 family 7, subfamily b, polypeptide 1 (CYP7b1), NADH dehydrogenase (ubiquinone) 1 alpha, cell signaling and cell proliferation related polymerase (DNA‐directed) alpha 1, gamma, iota, epsilon 2 (p59 subunit), MAPKKK6, MAPKKK5 and MAPKKK14, topoisomerase (DNA) II alpha, stress proteins like heat shock protein 1A and 1B, vascular endothelial growth factor C, sulfotransferase and phospholipase A2, group VII (Table 2 and Fig. 4).
In the lungs of animals in the Ogg1 −/– control group, when compared with wild type mice, overexpression of steroid and lipid metabolizing mono‐oxygenase CYP7b1, cell proliferation regulating polymerase (DNA‐directed), delta 4 (Pold4), and cytosolic phosphoenolpyruvate carboxykinase 1 (Pck1) was found in males and cyclin C, mitogen activated protein kinase 8 (MAPK8), and involved in angiogenesis matrix metalloproteinases 13, 14, 17 (Mmp13, Mmp14 and Mmp17) in females (Table 2). Furthermore, in control male and female Ogg1 −/– mutant mice, genes including stress proteins like heat shock proteins (HSP1, 1 A, 1B, 8 and 105) were down‐regulated.
DMAV administration to Ogg1 +/+ male mice resulted in up‐regulation of stress response‐related genes including metallothionein, genes encoding xenobiotic metabolizing enzymes and matrix metalloproteinase nine. In females Mmp17, CYP7b1, SOD1 and titin were found to be induced (Table 2, Fig. 4).
A total of more than two‐fold of up‐ or down‐regulated genes were used for further comparative analyses (e.g. Gene Spring and Ingenuity Pathway analyses). When the microarray data from the treatment groups of combined replicates of male and female mice were clustered into a condition tree by Gene Spring analysis in a standard correlation using the average replicates for signature genes, the relationship of one treatment group to another was as expected. The Ogg1 −/– and Ogg1 +/+ male and female DMAV‐treated groups were clustered together (Fig. 4). The data suggest that there are patterns of early gene expression changes that clearly distinguish DMAV‐treated Ogg1 −/– with large numbers of overexpressed genes from all other groups, as well as the non‐treated Ogg1 −/– and Ogg1 +/+ groups.
Analysis of the microarray averaged data by uploading to Ingenuity Pathway Analysis revealed suppression of drug, lipid metabolism and endocrine system development and function in the non‐treated Ogg1 −/– mice lung comparing to the Ogg1 +/+ case. However, genes related to cancer, cellular growth, proliferation and the cell cycle were found to be up‐regulated (data not shown). Expression of genes interfering with cancer, cell cycle, cellular proliferation, DNA replication, recombination and repair, lipid metabolism, cell–cell signaling and interaction, molecular transport and tumor morphology pathways was found to be induced, but the immune response pathway was suppressed when Ogg1 −/– was administered by DMAV. When DMAV‐treated wild type and Ogg1 mutant mice were compared, it was found that genes involved in cancer, cellular growth and proliferation, lipid metabolism and cell–cell signaling and interaction were overexpressed in Ogg1 −/– animals. Furthermore, free radical scavenging was not found to be activated in Ogg1 −/– mice.
Discussion
The present investigation revealed that long‐term administration of DMAV in drinking water exerts carcinogenicity in the lung of homozygous Ogg1 mutant mice that are deficient in the capacity to repair oxidative DNA base modifications. To our knowledge, this is the first evidence that DMAV, which is negative in the Ames test for mutagenicity, exerts carcinogenic activity in a mouse lung associated with high levels of 8‐OH‐dG. The mechanism of DMAV carcinogenicity in the lungs of Ogg1 −/– mice might thus be an accumulation of non‐repaired 8‐OH‐dG with high levels of cell proliferation, possibly leading to mutations and further elevation in cell proliferation, thus, resulting in initiation and promotion of lung carcinogenesis in those animals.( 15 ) It was earlier proposed that promotion by DMAV might be due mostly to the induction of enzymes involved in xenobiotic metabolism, peroxyl ((CH3)2AsOO) radicals, and additional damage to DNA and cell proliferation.( 32 ) Constitutively high levels of cell proliferation in the lung tissue of Mmh/Ogg1‐deficient mice might cause accumulation of DNA damage as the new cell cycle begins with already damaged and mutated DNA.
Interestingly, no spontaneously developed tumors were found in non‐treated Ogg1 homozygous mice, which might be due to the activation of backup DNA repair or other adaptation mechanisms to high levels of oxidative stress. Our data differ from those recently obtained by Xie et al. and Sakumi et al. reporting spontaneously developed lymphomas, lung and ovary tumors in Ogg1 knockout mice.( 10 , 11 ) This is possibly due to the differences in the method of Ogg1 knockout mice generation. Variation in the levels of cell proliferation in the lungs of Ogg1‐deficient and wild type mice are in line with previously reported data demonstrating differential expression of DNA repair enzymes in animal tissues and by the high production of reactive oxygen species in the lung due to respiration.( 33 )
The results of the present study are in concordance with our recent data obtained on investigation of the effects of potassium bromate in Ogg1 −/– mouse liver after partial hepatectomy, causing an increase in the mutation frequency.( 15 ) Recently, carcinogenicity of DMAV has been accessed in Muta and p53 heterozygous knockout mice.( 34 , 35 ) When DMAV was intraperitoneally injected into Muta mice at a dose of 10.6 mg/kg for five consecutive days it caused only a weak 1.3‐fold increase in the mutation frequency of the lacZ gene in the lung, and the authors suggested that the mutagenicity of DMAV is too low to be detected in Muta mice.( 34 ) However, administration of DMAV in drinking water to p53 heterozygous knockout mice at a dose of 200 p.p.m. led to significantly early induction of more numerous tumors in p53+/– as compared to wild type mice.( 35 ) However, no evidence for organ‐tumor specificity was observed. Similar to the study with p53 heterozygous knockout mice, we observed a tendency for increase in the development of malignant lymphomas in female Ogg1 −/– mice, but did not find significant differences in the numbers of developing tumors between DMAV‐treated and control wild type mice. Furthermore, DNA damage was not observed in the lungs of wild type and heterozygous animals, possibly because they were more resistant to DMAV treatment than their Ogg1 −/– counterparts.
Recently, oxidative stress has been postulated as a novel mode of arsenic carcinogenicity and it has been shown that DMAV‐induced lung‐specific DNA damage in mice can be attributed to free radicals, particularly peroxyl, superoxide or hydroxyl radicals, arising from the reaction of DMAV with molecular oxygen in vivo.( 18 , 26 ) Depletion in cellular glutathione may be correlated with oxidative stress mediated by reactive oxygen species and induction of nitric oxide. The reaction and interaction of these reactive species with target molecules lead to oxidative stress, lipid peroxidation, DNA damage, and activation of signaling cascades associated with tumor promotion and/or progression.( 36 ) Antioxidants that can inhibit, reduce, or scavenge the production of ROS and reactive nitrogen species (RNS) induced by arsenic can not only decrease direct cellular damage such as lipid peroxidation, enzyme inactivation and DNA oxidation caused by arsenic, but also ameliorate cell injuries or death by redox signaling pathways activated by arsenic exposure.( 36 ) The interaction of arsenic with essential metals is not clear. It has been suggested that peroxyl radicals may play an important role in DNA damage and in tumor promotion.( 23 , 37 ) Apurinic/apyriminidic sites may form where initial damage occurs, and altered DNA may undergo beta‐elimination to form DNA single‐strand scissions or Schiff‐base reactions to form DNA‐protein cross‐links.( 38 ) Those forms of DNA damage are also specifically produced in mouse and rat lungs after acute oral administration of DMAV, but are not observed in other tissues.( 39 , 40 ) Mice treated with DMAV in drinking water excrete DMAV primarily in the urine, though it may be in the form of an oxidizable complex,( 41 ) and approximately 3–4% of an oral dose of DMAV (40 mg As/kg) administered to mice is excreted as trimethylarsine oxide (TMAO) over 48 h.( 42 ) No demethylated products of DMA are observed in urine or tissues of mice administered DMA.( 42 ) For rodents the putative pathway of arsenic metabolism has been proposed (Fig. 5). When B6C3F1 mice were administered 1.11 or 111 mg/kg of DMAV, kidney had the highest concentration of DMAV of all tissues up to 60 min postadministration. It has been also reported that gavage and i.v. administration of DMAV in mice results in dose‐dependent prolonged retention in the lung as compared with liver and kidney and the dose‐dependent distribution of DMAV in the lung was suggested to have a role in the toxic effects DMAV elicits in this organ.( 41 , 42 ) DMA could be binding to a critical macromolecule and this binding is related to its toxic effects. The present observation that the administration of DMAV to Ogg1 −/– mice caused significant elevation of 8‐OH‐dG in lung DNA is in line with data obtained by other authors with regard to the promotion of lung carcinogenesis.( 23 ) Furthermore, recent results implicated 8‐OH‐dG repair defects in certain lung cancers.( 43 )
Figure 5.
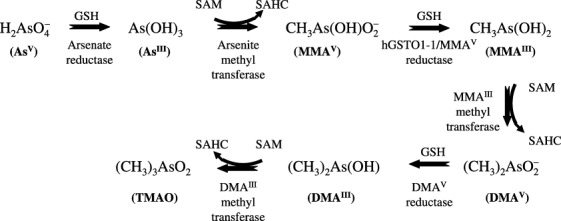
The putative pathway of arsenic metabolism in rodents. AsIII, arsenite; AsV, arsenate; DMAIII, monomethylarsinous acid; DMAV, dimethylarsinic acid; hGSTO1‐1, human glutathione‐S‐transferase omega 1‐1; MMAIII, monomethylarsonous acid; MMAV, monomethylarsonic acid; SAHC, S‐adenosyl‐L‐homocysteine; SAM, S‐adenosyl‐L‐methionine; TMAO, trimethylarsine oxide.
The results of the oligonucleotide microarray analysis on uploading to Ingenuity Pathway Analysis indicated that DMAV treatment induced the expression of genes involved with cancer, the cell cycle, cellular proliferation, DNA replication, recombination and repair, lipid metabolism, cell–cell signaling and interaction, molecular transport and immunological disease pathways in Ogg1 −/– mice. Those alterations, including up‐regulation of Pola1, Cyp7b1, Ndfua3, Mmp13, and Hspb1 are most likely to contribute to the lung carcinogenicity of DMAV. To our knowledge these are the first data for alterations in such gene expression in mouse lung by DMAV. Recently, in utero treatment with arsenic of pregnant mice was shown to result in CYP7B1 overexpression in their adult offspring.( 44 )
In line with our results, it has recently been shown that short‐term transplacental inorganic arsenic exposure in C3H mice increases lung adenocarcinoma incidence from 0% in controls to over 20%.( 45 ) Furthermore, gestational inorganic arsenic exposure in C3H mice have been shown to initiate pulmonary carcinogenesis that was promoted by the phorbol ester, 12‐O‐tetradecanoyl phorbol‐13‐acetate (TPA) in male or female offspring.( 44 ) Arsenic was accumulated dose‐dependently in the lung tissues of iAsV‐exposed mice. Recent results demonstrated an increase in lung tumor number and lung tumor size and the rate of poorly differentiated lung adenocarcinoma in iAsV‐exposed mice compared to the control. Methylation rates of p16INK4a and RASSF1A appeared to be higher in a dose‐related tendency in lung tumors from iAsV‐exposed mice compared to the control.( 46 ) Altogether these results are in accord with human studies that show the lung is a target of arsenic carcinogenesis.( 47 , 48 ) Characterization of new mouse models, will stimulate further research aimed at a better understanding of proliferative lesions of the lung.( 49 )
In non‐treated control Ogg1 −/– mice lungs, analysis of all the microarrays averaged data revealed lower expression levels than normal for genes whose products contribute to drug, lipid metabolism and endocrine system development and function. In a large number of heat shock protein cases, down‐regulation was evident. This might be a possible explanation for why the spontaneous tumors were less evident in control Ogg1 −/– comparing to Ogg1 +/+ animals. On the other hand, genes related to the cancer, cellular growth and proliferation and cell cycle were shown to be up‐regulated in the lung, which might explain the increased levels of cellular proliferation, as assessed in terms of PCNA positive indices, in Ogg1 −/–. When Ogg1 −/– mice were administered DMAV, Ingenuity pathway analysis revealed overexpression of genes involved with cancer, cell cycle, cellular proliferation, DNA replication damage and repair, lipid metabolism, cell–cell signaling and interaction, tumor morphology, molecular transport protein synthesis but suppression of immune response pathways. Alterations to cell proliferation included up‐regulation of CYP7b1, NADH dehydrogenase (ubiquinone 1 alpha), HSP1A, 1B, 4 and 70 kDa protein 12 A, polymerases (DNA‐directed) alpha 1, gamma, iota, epsilon 2, MAPKKK 5, 6 and 14. Suppression of the immune response pathway detected in Ogg1 −/– mice by DMAV might explain the increased malignant lymphoma development in female Ogg1 −/– mice. When DMAV‐treated wild type and Ogg1 mutant mice were compared, it was found that genes involved in cancer, cellular growth and proliferation, lipid metabolism and cell–cell signaling and interaction were overexpressed in Ogg1 −/– animals. The fact that the free radical scavenging pathway was not activated in Ogg1 −/– mice might be the reason for massive 8‐OH‐dG formation in the lung.
Interestingly, immunohistochemical examination of normal‐appearing tissue of Ogg1 −/– mice lungs induced by DMAV treatment, tumors showed high levels of concordance between induction of 8‐OH‐dG and apoptosis (ssDNA). The observation that oxidative DNA damage in Ogg1 −/– mouse lung is mostly observed in apoptotic cells could mean that strong oxidative degradation of DNA is the direct cause.
Electron microscopic and immunohistochemical examinations revealed significant elevation of cellular proliferation in the lungs of Ogg1 −/– mice as compared to control Ogg1 −/– and Ogg1 +/+ animals. Both in the non‐neoplastic lungs and lung tumors of type II origin of Ogg1 −/– mice treated with DMAV microvilli, increased and enlarged mitochondria were observed. These results indicated that changes of ultrastructure pointing to the induction of cell proliferation in pneumocyte type II cells induced by DMAV are related to the development of lung tumors.
In conclusion, DMAV, an organic methylated arsenical, here exerted carcinogenicity in the lungs of homozygous Mmh/Ogg1‐deficient mice defective in repair of oxidative DNA base modifications. Our data indicate that significant persistent elevation of 8‐OH‐dG, with induction of Pola1, Cyp7b1, Ndfua3, Mmp13, and Hspb1 genes expression induced by DMAV with high background levels of cell proliferation might be responsible for DMAV carcinogenicity in the Ogg1 −/– mutant mouse lung.
Acknowledgments
This work was supported by a Grant‐in‐Aid for Scientific Research from the Ministry of Health, Labor and Welfare of Japan. We thank Masayo Imanaka, Shoko Araki, Kaori Touma, and Hideki Nakagawa for their technical assistance, and Mari Dokoh, Yuko Onishi and Yoko Shimada for their help during preparation of this manuscript.
References
- 1. Anisimov VN, Alimova IN. The use of mutagenic and transgenic mice for the study of aging mechanisms and age pathology. Adv Gerontol 2001; 7: 72–94. [PubMed] [Google Scholar]
- 2. Kasai H. A new automated method to analyze urinary 8‐hydroxydeoxyguanosine by a high‐performance liquid chromatography‐electrochemical detector system. J Radiat Res (Tokyo) 2003; 44: 185–9. [DOI] [PubMed] [Google Scholar]
- 3. Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation‐damaged base 8‐oxodG. Nature 1991; 349: 431–4. [DOI] [PubMed] [Google Scholar]
- 4. Roldan‐Arjona T, Wei YF, Carter KC et al. Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8‐hydroxyguanine‐DNA glycosylase. Proc Natl Acad Sci USA 1997; 94: 8016–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Radicella JP, Dherin C, Desmaze C, Fox MS, Boiteux S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 1997; 94: 8010–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arai K, Morishita K, Shinmura K et al. Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene 1997; 14: 2857–61. [DOI] [PubMed] [Google Scholar]
- 7. Lu R, Nash HM, Verdine GL. A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr Biol 1997; 7: 397–407. [DOI] [PubMed] [Google Scholar]
- 8. Bjoras M, Luna L, Johnsen B et al. Opposite base‐dependent reactions of a human base excision repair enzyme on DNA containing 7,8‐dihydro‐8‐oxoguanine and abasic sites. Embo J 1997; 16: 6314–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aburatani H, Hippo Y, Ishida T et al. Cloning and characterization of mammalian 8‐hydroxyguanine‐specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res 1997; 57: 2151–6. [PubMed] [Google Scholar]
- 10. Xie Y, Yang H, Cunanan C et al. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K‐ras oncogene in lung tumors. Cancer Res 2004; 64: 3096–102. [DOI] [PubMed] [Google Scholar]
- 11. Sakumi K, Tominaga Y, Furuichi M et al. Ogg1 knockout‐associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res 2003; 63: 902–5. [PubMed] [Google Scholar]
- 12. Kunisada M, Sakumi K, Tominaga Y et al. 8‐oxoguanine formation induced by chronic UVB exposure makes Ogg1 knockout mice susceptible to skin carcinogenesis. Cancer Res 2005; 65: 6006–10. [DOI] [PubMed] [Google Scholar]
- 13. Minowa O, Arai T, Hirano M et al. Mmh/Ogg1 gene inactivation results in accumulation of 8‐hydroxyguanine in mice. Proc Natl Acad Sci USA 2000; 97: 4156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arai T, Kelly VP, Minowa O, Noda T, Nishimura S. High accumulation of oxidative DNA damage, 8‐hydroxyguanine, in Mmh/Ogg1 deficient mice by chronic oxidative stress. Carcinogenesis 2002; 23: 2005–10. [DOI] [PubMed] [Google Scholar]
- 15. Arai T, Kelly VP, Komoro K, Minowa O, Noda T, Nishimura S. Cell proliferation in liver of Mmh/Ogg1‐deficient mice enhances mutation frequency because of the presence of 8‐hydroxyguanine in DNA. Cancer Res 2003; 63: 4287–92. [PubMed] [Google Scholar]
- 16. Taylor PR, Qiao YL, Schatzkin A et al. Relation of arsenic exposure to lung cancer among tin miners in Yunnan Province, China. Br J Ind Med 1989; 46: 881–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leonard A, Lauwerys RR. Carcinogenicity, teratogenicity and mutagenicity of arsenic. Mutat Res 1980; 75: 49–62. [DOI] [PubMed] [Google Scholar]
- 18. Kitchin KT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol Appl Pharmacol 2001; 172: 249–61. [DOI] [PubMed] [Google Scholar]
- 19. Kenyon EM, Hughes MF. A concise review of the toxicity and carcinogenicity of dimethylarsinic acid. Toxicology 2001; 160: 227–36. [DOI] [PubMed] [Google Scholar]
- 20. Kaiser J. Environmental health. Second look at arsenic finds higher risk. Science 2001; 293: 2189. [DOI] [PubMed] [Google Scholar]
- 21. Yamamoto S, Konishi Y, Matsuda T et al. Cancer induction by an organic arsenic compound, dimethylarsinic acid (cacodylic acid), in F344/DuCrj rats after pretreatment with five carcinogens. Cancer Res 1995; 55: 1271–6. [PubMed] [Google Scholar]
- 22. Wanibuchi H, Yamamoto S, Chen H et al. Promoting effects of dimethylarsinic acid on N‐butyl‐N‐(4‐hydroxybutyl) nitrosamine‐induced urinary bladder carcinogenesis in rats. Carcinogenesis 1996; 17: 2435–9. [DOI] [PubMed] [Google Scholar]
- 23. Yamanaka K, Ohtsubo K, Hasegawa A et al. Exposure to dimethylarsinic acid, a main metabolite of inorganic arsenics, strongly promotes tumorigenesis initiated by 4‐nitroquinoline 1‐oxide in the lungs of mice. Carcinogenesis 1996; 17: 767–70. [DOI] [PubMed] [Google Scholar]
- 24. Yamanaka K, Katsumata K, Ikuma K, Hasegawa A, Nakano M, Okada S. The role of orally administered dimethylarsinic acid, a main metabolite of inorganic arsenics, in the promotion and progression of UVB‐induced skin tumorigenesis in hairless mice. Cancer Lett 2000; 152: 79–85. [DOI] [PubMed] [Google Scholar]
- 25. Hayashi H, Kanisawa M, Yamanaka K et al. Dimethylarsinic acid, a main metabolite of inorganic arsenics, has tumorigenicity and progression effects in the pulmonary tumors of A/J mice. Cancer Lett 1998; 125: 83–8. [DOI] [PubMed] [Google Scholar]
- 26. Wei M, Wanibuchi H, Yamamoto S, Li W, Fukushima S. Urinary bladder carcinogenicity of dimethylarsinic acid in male F344 rats. Carcinogenesis 1999; 20: 1873–6. [DOI] [PubMed] [Google Scholar]
- 27. Yamanaka K, Hoshino M, Okamoto M, Sawamura R, Hasegawa A, Okada S. Induction of DNA damage by dimethylarsine, a metabolite of inorganic arsenics, is for the major part likely due to its peroxyl radical. Biochem Biophys Res Commun 1990; 168: 58–64. [DOI] [PubMed] [Google Scholar]
- 28. Kitchin KT, Ahmad S. Oxidative stress as a possible mode of action for arsenic carcinogenesis. Toxicol Lett 2003; 137: 3–13. [DOI] [PubMed] [Google Scholar]
- 29. Wanibuchi H, Hori T, Meenakshi V et al. Promotion of rat hepatocarcinogenesis by dimethylarsinic acid. association with elevated ornithine decarboxylase activity and formation of 8‐hydroxydeoxyguanosine in the liver. Jpn J Cancer Res 1997; 88: 1149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. U.S. Interagency Staff Group on Carcinogens. Chemical carcinogens: a review of the science and its associated principles. Environ Health Perspect 1986; 67: 201–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kinoshita A, Wanibuchi H, Imaoka S et al. Formation of 8‐hydroxydeoxyguanosine and cell‐cycle arrest in the rat liver via generation of oxidative stress by phenobarbital: association with expression profiles of p21 (WAF1/Cip1), cyclin D1 and Ogg1. Carcinogenesis 2002; 23: 341–9. [DOI] [PubMed] [Google Scholar]
- 32. Nishikawa T, Wanibuchi H, Ogawa M et al. Promoting effects of monomethylarsonic acid, dimethylarsinic acid and trimethylarsine oxide on induction of rat liver preneoplastic glutathione S‐transferase placental form positive foci: a possible reactive oxygen species mechanism. Int J Cancer 2002; 100: 136–9. [DOI] [PubMed] [Google Scholar]
- 33. Russo MT, De Luca G, Degan P et al. Accumulation of the oxidative base lesion 8‐hydroxyguanine in DNA of tumor‐prone mice defective in both the Myh and Ogg1 DNA glycosylases. Cancer Res 2004; 64: 4411–14. [DOI] [PubMed] [Google Scholar]
- 34. Noda Y, Suzuki T, Kohara A et al. In vivo genotoxicity evaluation of dimethylarsinic acid in MutaMouse. Mutat Res 2002; 513: 205–12. [DOI] [PubMed] [Google Scholar]
- 35. Salim EI, Wanibuchi H, Morimura K et al. Carcinogenicity of dimethylarsinic acid in p53 heterozygous knockout and wild‐type C57BL/6J mice. Carcinogenesis 2003; 24: 335–42. [DOI] [PubMed] [Google Scholar]
- 36. Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and Carcinogenesis. Mol Cellular Biochem 2004; 255: 67–78. [DOI] [PubMed] [Google Scholar]
- 37. Yamanaka K, Okada S. Induction of lung‐specific DNA damage by metabolically methylated arsenics via the production of free radicals. Environ Health Perspect 1994; 102 (Suppl 3): 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamanaka K, Hayashi H, Kato K, Hasegawa A, Okada S. Involvement of preferential formation of apurinic/apyrimidinic sites in dimethylarsenic‐induced DNA strand breaks and DNA‐protein crosslinks in cultured alveolar epithelial cells. Biochem Biophys Res Commun 1995; 207: 244–9. [DOI] [PubMed] [Google Scholar]
- 39. Yamanaka K, Hasegawa A, Sawamura R, Okada S. Dimethylated arsenics induce DNA strand breaks in lung via the production of active oxygen in mice. Biochem Biophys Res Commun 1989; 165: 43–50. [DOI] [PubMed] [Google Scholar]
- 40. Brown JL, Kitchin KT, George M. Dimethylarsinic acid treatment alters six different rat biochemical parameters: relevance to arsenic carcinogenesis. Teratog Carcinog Mutagen 1997; 17: 71–84. [PubMed] [Google Scholar]
- 41. Vahter M, Marafante E, Dencker L. Tissue distribution and retention of; 74As‐dimethylarsinic acid in mice and rats. Arch Environ Contam Toxicol 1984; 13: 259–64. [DOI] [PubMed] [Google Scholar]
- 42. Marafante E, Vahter M, Norin H et al. Biotransformation of dimethylarsinic acid in mouse, hamster and man. J Appl Toxicol 1987; 7: 111–7. [DOI] [PubMed] [Google Scholar]
- 43. Mambo E, Chatterjee A, Souza‐Pinto NC et al. Oxidized guanine lesions and hOgg1 activity in lung cancer. Oncogene 2005; 24: 4496–508. [DOI] [PubMed] [Google Scholar]
- 44. Waalkes MP, Liu J, Chen H et al. Estrogen signaling in livers of male mice with hepatocellular carcinoma induced by exposure to arsenic in utero. J Natl Cancer Inst 2004; 96: 466–74. [DOI] [PubMed] [Google Scholar]
- 45. Waalkes MP, Ward JM, Liu J, Diwan BA. Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol Appl Pharmacol 2003; 186: 7–17. [DOI] [PubMed] [Google Scholar]
- 46. Cui X, Wakai T, Shirai Y, Hatakeyama K, Hirano S. Chronic oral exposure to inorganic arsenate interferes with methylation status of p16INK4a and RASSF1A and induces lung cancer in A/J mice. Toxicol Sci 2006; 91: 372–81. [DOI] [PubMed] [Google Scholar]
- 47. NRC. Arsenic in Drinking Water. Washington, DC: National Research Council, 1999: 1–310. [Google Scholar]
- 48. IARC (Intermational Agency for Research on Cancer) 2004. IARC monographs on the evaluation of carcinogenic risks to humans, Vol. 84: Arsenic in drinking water. In: Some Drinking Water Disinfectants and Contaminants, Including Arsenic. Lyon, France: IARC, 2004: 269–477. [Google Scholar]
- 49. Nikitin AY, Alcaraz A, Anver MR et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res 2004; 64: 2307–16. [DOI] [PubMed] [Google Scholar]