

Abstract
The relationship between apoptosis and the cell cycle remains unclear. In the present study we have investigated the relationship between cell cycle progression and the activation of caspases (caspase‐3 and caspase‐8) in Fas (CD95)‐mediated apoptosis in asynchronously growing leukemia cells. We found that cells expressing the active form of caspase‐3 were cyclin A/B1 and Ki‐67 negative but cyclin E positive, whereas expression of the active form of caspase‐8 was detected in cyclin A/B1/E‐negative and Ki‐67‐negative cells. In addition, both the activation of caspases and Fas‐mediated apoptosis were completely abolished when leukemia cells were arrested in early G1 phase. Using post‐sorting western blot analysis, we demonstrated that caspase‐3 and caspase‐8 were activated in p27‐negative cells. These results suggest that caspase‐3 would be activated in cells entering into late G1 or early S phase, and caspase‐8 would be activated in middle or late G1 phase. The speed of cell cycle progression from G1 to S phase might be influential in the speed of caspase activation and induction of Fas‐mediated apoptosis. (Cancer Sci 2007; 98: 1174–1183)
Apoptosis is a mechanism of cell death that is fundamental in the control of cellular homeostasis in most multicellular organisms. A major mechanism controlling cell apoptosis relies on the interaction of the Fas receptor (CD95) with its ligand, FasL. Fas receptor activation first requires its trimerization by FasL. The trimerized receptor binds the receptor‐specific adaptor protein Fas‐associated protein with death domain (FADD) through interaction of the death domain (DD), which is present in the cytoplasmic region of the Fas receptor.( 1 ) FADD, in turn, recruits pro‐caspase‐8 to form the death‐inducing signaling complex (DISC).( 2 ) In DISC, pro‐caspase‐8 is cleaved and then propagates the apoptotic signal via a mitochondria‐independent or ‐dependent (type I or type II, respectively) pathway to cleave and activate downstream caspase‐3.( 3 ) In the mitochondria‐dependent pathway, Bid is cleaved by activated caspase‐8, and cleaved Bid conveys apoptotic signals to the mitochondria.( 4 ) The truncated Bid then translocates to the mitochondria and induces the release of apoptosis factors, such as cytochrome c, which form an essential part of the apoptosis complex to trigger activation of downstream caspases, including caspase‐3 and caspase‐9.( 5 ) Activated caspase‐3 cleaves the substrate, such as DNA fragmentation factor,( 6 ) and apoptosis then occurs.
Cyclins and cyclin‐dependent kinases (cdk) have been shown to be subunits of cell cycle‐dependent protein kinases that regulate key events during progression of the cell cycle. Some cyclins are expressed discontinuously during the cell cycle, their synthesis and degradation being strictly scheduled. Cyclin E is expressed and forms complexes with cdk2 in middle‐to‐late G1, several hours before the onset of S phase, and promotes cell entry into S phase.( 7 , 8 , 9 ) The cyclin A–cdc2 complex is formed in S phase.( 10 ) Cyclin B1 is necessary for mitosis to occur; its expression is normally low in G0/G1, increases in S, is maximal during G2/M, and it is rapidly degraded at the end of mitosis.( 11 , 12 ) Immunocytochemical detection of cyclins by multiparameter flow cytometry has provided an approach to cell cycle studies.( 13 , 14 )
The relationship between apoptosis and the cell cycle remains unclear, although more and more evidence has accumulated in the past decade. Apoptosis is almost exclusively found in proliferating cells,( 15 ) such as activation‐induced cell death (AICD) in T cells.( 16 , 17 ) Our group found that activation‐induced T cell death occurs at the G1A phase of the cell cycle.( 18 ) In our previous work, we showed that Fas‐mediated apoptotic signals can be transduced into cells in G1B phase, and G1A to G1B transition might support the induction of Fas‐mediated apoptosis.( 19 , 20 )
In the present study we have investigated the relationship between cell cycle progression and the activation of caspases in Fas‐mediated apoptosis in asynchronously growing leukemia cells. We demonstrated that cells expressing the active form of caspase‐3 were cyclin A/B1 negative but were positive for cyclin E, whereas expression of the active form of caspase‐8 was detected in cyclin A/B1/E‐negative cells. We also demonstrated that the expression of both caspase‐3 and caspase‐8 was detected in Ki‐67‐negative cells. In addition, activation of caspase‐8 or caspase‐3 was abolished when cells were arrested in early G1 phase of the cell cycle. These results suggest that caspase‐8 was cleaved in middle or late G1 phase, whereas caspase‐3 was activated in late G1 or early S phase. Using post‐sorting western blot analysis, we confirmed that active caspase‐3 and ‐8 were detected in p27‐negative cells. These findings are the first to link the activation of caspases (caspase‐3 and caspase‐8) with cell cycle progression in asynchronously growing leukemia cells.
Materials and Methods
Cell lines and cell culture. The leukemia cell lines PEER and Jurkat were maintained in RPMI‐1640 (Sigma, St Louis, MO, USA) supplemented with 10% heat‐inactivated fetal bovine serum (Life Technologies, Tokyo, Japan) and incubated in a humidified incubator with 5% CO2 at 37°C. Cell concentration was maintained at 0.1–20 × 105 cells/mL by subculturing every 5 days. Viability was calculated using trypan blue dye exclusion. If viability was greater than 95%, cells could be harvested.
Antibodies and reagents. The following antibodies and reagents were used in the present study: agonistic anti‐Fas monoclonal antibody (clone CH‐11; MBL, Nagoya, Japan), anticaspase‐3 monoclonal antibody (clone 19; Immunotech, Marseille, France), anticaspase‐8 monoclonal antibody (clone 5F7; MBL), rabbit antiactin antibody (Sigma), anti‐FLICE inhibitory protein (FLIP) monoclonal antibody (clone G‐11; Santa Cruz Biotechnology, Santa Cruz, CA, USA), fluorescein isothiocyanate (FITC)‐conjugated antiactive caspase‐3 monoclonal antibody (clone C92‐605; BD PharMingen, San Diego, CA, USA), ribonuclease A (Sigma), phycoerythrin (PE)‐conjugated anticyclin A antibody reagent set (clone BF683; BD PharMingen), PE‐conjugated anticyclin B1 antibody reagent set (clone GNS‐1; BD PharMingen), anticyclin E monoclonal antibody (clone HE12; BD PharMingen), PE‐conjugated rat antimouse IgG1 monoclonal antibody (clone A85‐1; BD PharMingen), anti‐p27 polyclonal antibody (sc‐528; Santa Cruz Biotechnology), APO LOGIX carboxyfluorescein caspase‐8 detection kits (Cell Technology, Minneapolis, MN, USA), and phorbol 12‐myristate 13‐acetate (PMA) (Sigma).
DNA content and caspase‐3 double staining analysis. Cells (106/tube, cell density 5 × 105 cells/mL) were treated with agonistic anti‐Fas (clone CH‐11, final concentration 50 ng/mL) for the desired treatment time at 37°C before being collected and processed for cell cycle analysis. Briefly, cells were harvested and washed with 2 mL phosphate‐buffered saline (PBS). Cells were incubated with 20 µL FITC‐conjugated active caspase‐3 and 100 µL of 20 µg/mL propidium iodide (PI)‐containing RNase A at 37°C for 15 min, washed with 4 mL PBS and resuspended in 500 µL FACScan fluid. They were then analyzed immediately on a FACScan flow cytometer (Becton Dickinson, Tokyo, Japan) using CELL‐Quest software (Tokyo, Japan).
Cyclin/Ki‐67 and caspase‐3 double staining analysis. Cells were pretreated with anti‐Fas antibody using the desired method as mentioned above, and then collected and processed for cell cycle analysis. Before intracellular staining, cells were fixed and permeabilized according to the manufacturer's directions (IntraStain, Fixation and Permeabilization Kit, Dako Cytomation, Tokyo, Japan). Cells were stained as follows: (i) incubated with anticyclin A and antiactive caspase‐3 antibodies for 15 min at room temperature in the dark; (ii) incubated with anticyclin B1 and antiactive caspase‐3 antibodies for 15 min at room temperature in the dark; (iii) incubated with anti‐Ki‐67 and antiactive caspase‐3 antibodies for 15 min at room temperature in the dark; or (iv) cells were first incubated with anticyclin E antibody at room temperature for 15 min in the dark then washed with 2 mL PBS, followed by incubation with secondary antibody PE‐conjugated antimouse antibody and antiactive caspase‐3 antibody at room temperature for 15 min in the dark. All of these treatment groups were then washed with 2 mL PBS and resuspended in 500 µL FACScan fluid and analyzed immediately on a FACScan flow cytometer (Becton Dickinson, Tokyo, Japan), and analyzed using CELL‐Quest software. Control cells were stained with isotype‐matched control antibodies at room temperature or the secondary PE‐labeled antibody and analyzed. At least 50 000 cells were analyzed in all experiments.
DNA content and caspase‐8 double staining analysis. Cells were pretreated with anti‐Fas antibody as mentioned above. Cells (3 × 105 cells/tube, density of 106 cells/mL) were incubated for 1 h at 37°C under 5% CO2 with 10 µL 30× working dilution FAM‐Peptide‐FMK, and incubated with 100 µL of 20 µg/mL PI containing RNase A at 37°C for 15 min, then analyzed immediately on a FACScan flow cytometer (Becton Dickinson, Tokyo, Japan) using CELL‐Quest software.
Cyclin/Ki‐67 and caspase‐8 double staining analysis. Cells were pretreated with anti‐Fas antibody as mentioned above. Cells (3 × 105 cells/tube, density of 106 cells/mL) were incubated for 1 h at 37°C under 5% CO2 with 10 µL 30× working dilution FAM‐Peptide‐FMK, and washed with 2 mL 1× working dilution wash buffer. They were then fixed and permeabilized according to the manufacturer's directions (IntraStain, Fixation and Permeabilization Kit, Dako Cytomation). Cells were stained as follows: (i) incubated with anticyclin A antibody for 15 min at room temperature in the dark; (ii) incubated with anticyclin B1 antibody for 15 min at room temperature in the dark; (iii) incubated with anti‐Ki‐67 antibody for 15 min at room temperature in the dark; or (iv) cells were first incubated with anticyclin E antibody at room temperature for 15 min in the dark then washed with 2 mL PBS followed by incubation with 5 uL secondary antibody PE‐conjugated antimouse antibody at room temperature for 15 min in the dark. All of these treatment groups were then washed with 2 mL PBS and resuspended in 400 µL FACScan fluid and analyzed immediately on a flow cytometer (Becton Dickinson, Tokyo, Japan), and analyzed using CELL‐Quest software.
Cell cycle analysis. Cells were arrested in early G1 phase by treatment with PMA. Jurkat and PEER were preincubated with PMA (final concentration 10 nM) for 24 h, cells (2 × 105) were harvested and incubated with 370 µL PI–Triton X‐containing RNase A at room temperature for 15 min in dark, then analyzed immediately on a flow cytometer using ModFit LT software (Tokyo, Japan).
Western blot analysis. Before generating cytoplasmic extracts, cells (1 × 107) pretreated under various conditions were washed in ice‐cold PBS then lysed at 4°C by gentle pipetting after 5 min in hypotonic buffer (20 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM Na3VO4, 1 mM ethylene diamine tetra‐acetic acid, 10% glycerol, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 1 µg/mL aprotinin, 1 µg/mL pepstatin, 1 µg/mL leupeptin and 1 mM dithiothreitol) with 0.2% Nonidet P (NP)‐40. After centrifugation at 4°C (8000g in a microcentrifuge) for 10 s, supernatants were collected as cytoplasmic extracts. Protein concentrations were determined using the Total Protein Assay system according to the manufacturer's directions (Pierce, Rockford, IL, USA) and normalized for protein content (30 µg/lane). The proteins were subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis followed by semidry transfer to a nitrocellulose membrane (Millipore Corporation, Bedford, MA, USA). Transferred nitrocellulose blots were blocked with 5% non‐fat dry milk in PBS and incubated with primary antibody against caspase‐3 (1:1000), caspase‐8 (1:1000) or actin (1:1000) at 4°C overnight. The membranes were washed three times with 0.05% PBST at room temperature and incubated with horseradish peroxidase‐conjugated antimouse IgG or antirabbit secondary antibody (1:1000) for 1 h at room temperature. After three washes with 0.05% PBST, antibodies bound to protein blots were detected using Western Lightening Chemiluminescence Reagent Plus (Perkin Elmer Life Science, Boston, MA, USA).
Cell sorting. Cell sorting was carried out using FACSAria (Becton Dickinson). To sort active caspase‐3‐positive and ‐negative groups, the cells were pretreated with anti‐Fas antibody for 4 h, then fixed and permeabilized according to the manufacturer's directions (IntraStain, Fixation and Permeabilization Kit, Dako Cytomation) and stained with antiactive caspase‐3 antibody. To sort peptide caspase‐8‐positive and ‐negative groups, the cells were pretreated with anti‐Fas antibody for 6 h, then incubated at a density of 106 cells/mL for 1 h at 37°C under 5% CO2 with 30× working dilution FAM‐Peptide‐FMK. They were then washed with 1× working dilution wash buffer and fixed according to the manufacturer's directions. The labeled cells were sorted into tubes precoated with a buffer containing 0.5 mL PBS. The sorting windows were selected to include cells that were positive or negative for active caspase, based on the active caspase fluorescence and distribution. The sorted cells were then analyzed by western blotting.
Post‐sorting western blot analysis. Cells were lysed with Laemmli sample buffer (2% sodium dodecyl sulfate, 62.5 mM Tris HCl [pH 6.8], 2.5% sucrose, 5% 2‐mercaptoethanol, 0.02% bromophenol blue). Samples were boiled for 5 min and then supersonicated. Total cellular proteins from 5 × 105 cells per sample per lane were subjected to western blot analysis as mentioned above. The blots were incubated with primary rabbit antihuman p27 antibody (1:200), mouse antihuman cyclin E antibody (2 µg/mL), mouse anticyclin D1 antibody (2 µg/mL) or mouse anti‐FLIP antibody (1:100) at 4°C overnight then incubated with horseradish peroxidase‐conjugated antimouse IgG or antirabbit secondary antibody (1:1000) for 1 h at room temperature. The proteins were detected using Western Lightening Chemiluminescence Reagent Plus.
Results
Cell cycle dependency of caspase activation. We hypothesized that activation of caspases might be dependent on cell cycle progression. To investigate whether caspase‐3 or caspase‐8 would be activated in the specific phase of the cell cycle, we carried out simultaneous flow cytometric analysis of cell cycle markers and the active forms of caspases (1, 2, 3, 4, 5, 6). Leukemia cells (PEER and Jurkat) treated with anti‐Fas antibody were analyzed using dual analysis of DNA content and expression of active caspase‐3 (Fig. 1). As shown by the continuity of the G1 cluster and active caspase‐3‐expressing cell population, it is most likely that caspase‐3 would be initially activated in the G1 compartment of the cell cycle. In addition, we found that cells expressing active forms of caspase‐3 were negative for cyclin A and cyclin B1 (Fig. 2). Interestingly, the majority of cells expressing active forms of caspase‐3 were positive for cyclin E, although a small population of cyclin E‐negative cells expressed active forms of caspase‐3 (Fig. 2). Cells expressing the active form of caspase‐3 first appeared in the cyclin E‐positive fraction (data not shown). As expression of cyclins A, B1 and E were induced in S, G2/M and late G1/early S phase of the cell cycle, respectively, these results indicate that caspase‐3 is activated in cells entering late G1 and early S phase. To confirm this conclusion, we used the antibody Ki‐67 (widely used as a proliferation marker) whose expression in cycling cells is maximal during mitosis and decreases during G1, only to increase again during the S phase of cell cycle.( 21 ) We found that cells expressing active forms of caspase‐3 first appeared in the cell population negative for Ki‐67 (Fig. 3).
Figure 1.
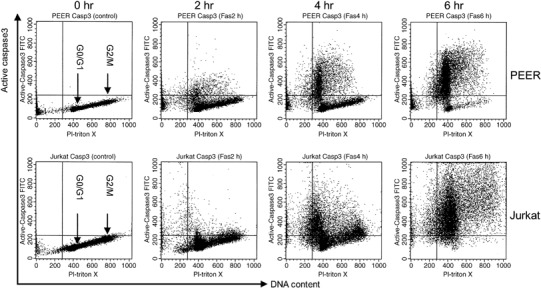
DNA content and active caspase‐3 double staining analysis of leukemia cells treated with anti‐Fas antibody. Cells (PEER and Jurkat) were pretreated with anti‐Fas antibody (50 ng/mL) for 0, 2, 4 and 6 h. After incubation, cells were harvested and stained with fluorescein isothiocyanate‐conjugated antiactive caspase‐3 antibody and propidium iodide (PI)–Triton X for DNA content, then analyzed by flow cytometry. Similar results were obtained in three repeated experiments.
Figure 2.
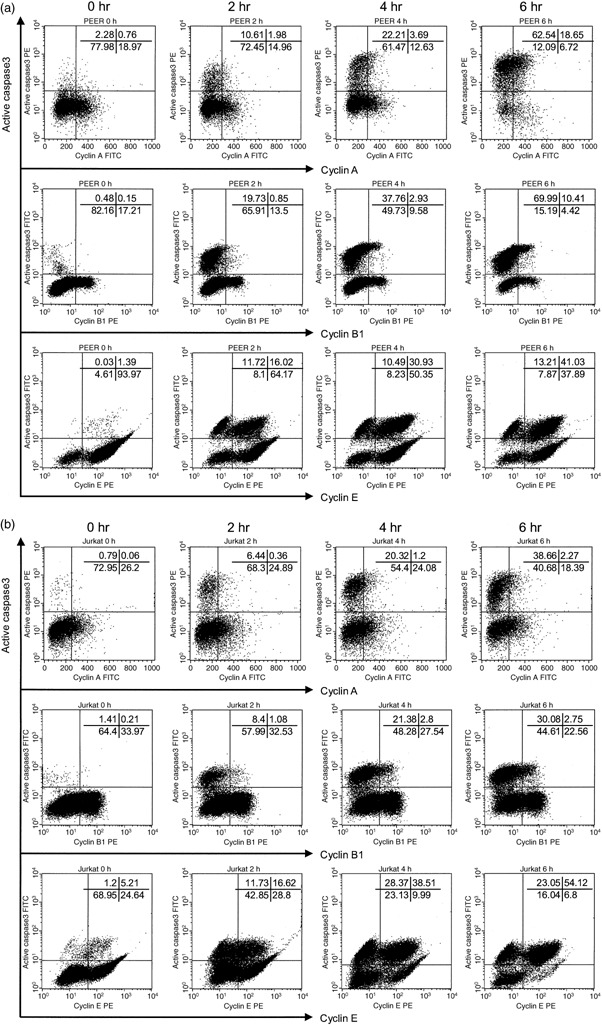
Cyclins and active caspase‐3 double staining analysis of leukemia cells treated with anti‐Fas antibody. (a) PEER and (b) Jurkat cells were pretreated with anti‐Fas antibody (50 ng/mL) for 0, 2, 4 and 6 h. For intracellular staining, cells were first fixed and permeabilized, then stained with fluorescein isothiocyanate‐conjugated antiactive caspase‐3 antibody and phycoerythrin (PE)‐conjugated anticyclin A/B1/E, then analyzed by flow cytometry. Similar results were obtained in three repeated experiments.
Figure 3.
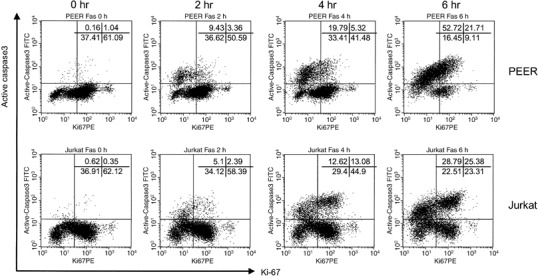
Ki‐67 and active caspase‐3 double staining analysis of leukemia cells treated with anti‐Fas antibody. Cells (PEER and Jurkat) were pretreated with anti‐Fas (50 ng/mL) antibody for 0, 2, 4 and 6 h. For intracellular staining, cells were first fixed and permeabilized, then stained with fluorescein isothiocyanate‐conjugated antiactive caspase‐3 antibody and phycoerythrin (PE)‐conjugated anti‐Ki‐67, then analyzed by flow cytometry. Similar results were obtained in three repeated experiments.
Figure 4.
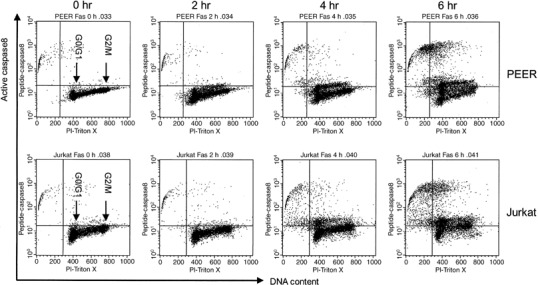
DNA content and active caspase‐8 double staining analysis of leukemia cells treated with anti‐Fas antibody. Cells (PEER and Jurkat) were incubated with anti‐Fas antibody (50 ng/mL) for 0, 2, 4 and 6 h. After incubation, cells were harvested and stained with FAM‐peptide‐FMK (peptide caspase‐8) and propidium iodide (PI)–Triton X for DNA content, then analyzed by flow cytometry.
Figure 5.
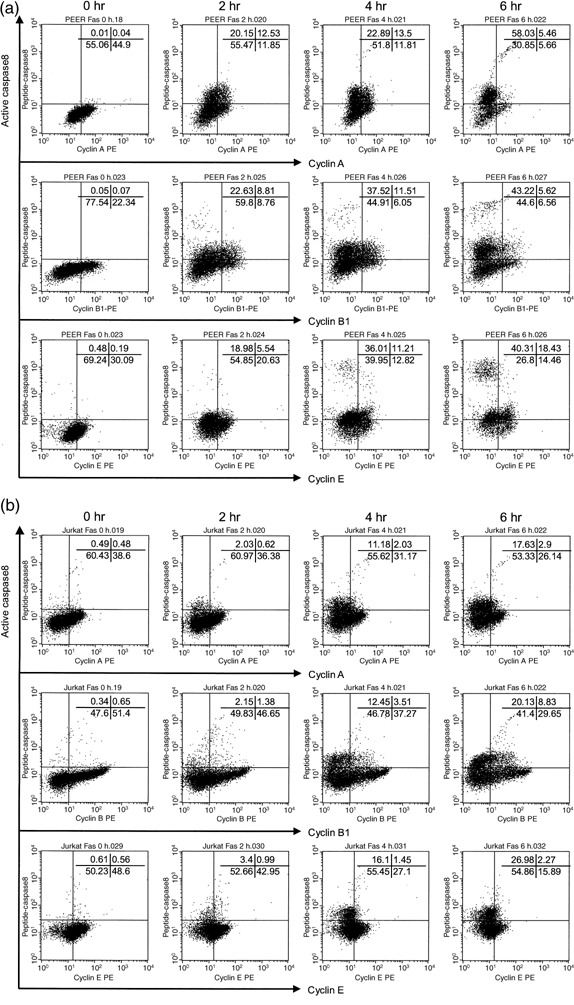
Cyclins and active caspase‐8 double staining analysis of leukemia cells treated with anti‐Fas antibody. (a) PEER and (b) Jurkat cells were incubated with anti‐Fas (50 ng/mL) antibody for 0, 2, 4 and 6 h. Before intracellular staining, cells were preincubated with FAM‐peptide‐FMK (peptide caspase‐8), then fixed and permeabilized and stained with phycoerythrin (PE)‐conjugated cyclin A/B1 and analyzed by flow cytometry. For cyclin E staining, cells were additionally stained with PE‐conjugated secondary antibody. Similar results were obtained in three repeated experiments.
Figure 6.
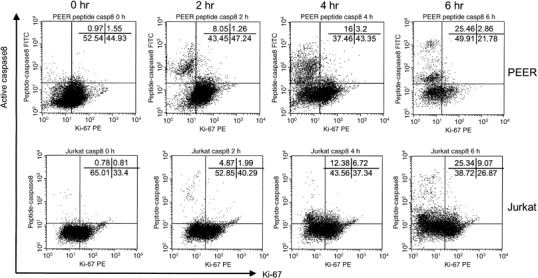
Ki‐67 and active caspase‐8 double staining analysis of leukemia cells treated with anti‐Fas antibody. Cells (PEER and Jurkat) were incubated with anti‐Fas antibody (50 ng/mL) for 0, 2, 4 and 6 h. Before intracellular staining, cells were preincubated with FAM‐peptide‐FMK (peptide caspase‐8), then fixed and permeabilized and stained with phycoerythrin (PE)‐conjugated Ki‐67 and analyzed by flow cytometry. Similar results were obtained in three repeated experiments.
Similarly, we analyzed the expression of active forms of caspase‐8 in leukemia cells treated with anti‐Fas antibody. The continuity of the G1 cluster and active caspase‐8‐expressing cell population suggested that activation of caspase‐8 would be started in cells in the G1 compartment (Fig. 4). We also found that caspase‐8 was activated in cells negative for cyclins A, B1 and E, and Ki‐67 (5, 6). These data indicate that caspase‐8 would be initially activated in early or middle G1 phase prior to the activation of caspase‐3 in late G1 and early S phase of the cell cycle.
Activation of caspases is abolished when cells are arrested in early G1 phase. We previously demonstrated that PMA can stop cell cycle progression of MML‐1 cells in early G1 phase and completely inhibits Fas‐mediated apoptosis.( 19 ) To further confirm that caspases are activated in middle or late G1 phase of the cell cycle, PEER and Jurkat cells were arrested in early G1 phase by treatment with PMA. Cells were preincubated with 10 nM PMA for 24 h; more than 70% of cells were arrested in early G1 phase (Fig. 7a). When PMA‐treated cells were further incubated with anti‐Fas antibody the activation of caspase‐3 or caspase‐8 was almost completely abolished (Fig. 7b). The inhibition of caspase activation in PMA‐treated cells was also confirmed by western blot analysis (Fig. 7c). Both caspase‐3 and caspase‐8 were cleaved rapidly in control cells but not at all in PMA‐treated cells. It is of note that the amounts of membrane Fas receptor, procaspase‐3 and procaspase‐8 remained unchanged by treatment with PMA alone (data not shown). These data suggest that neither caspase‐3 nor caspase‐8 can be activated in early G1 phase of the cell cycle.
Figure 7.
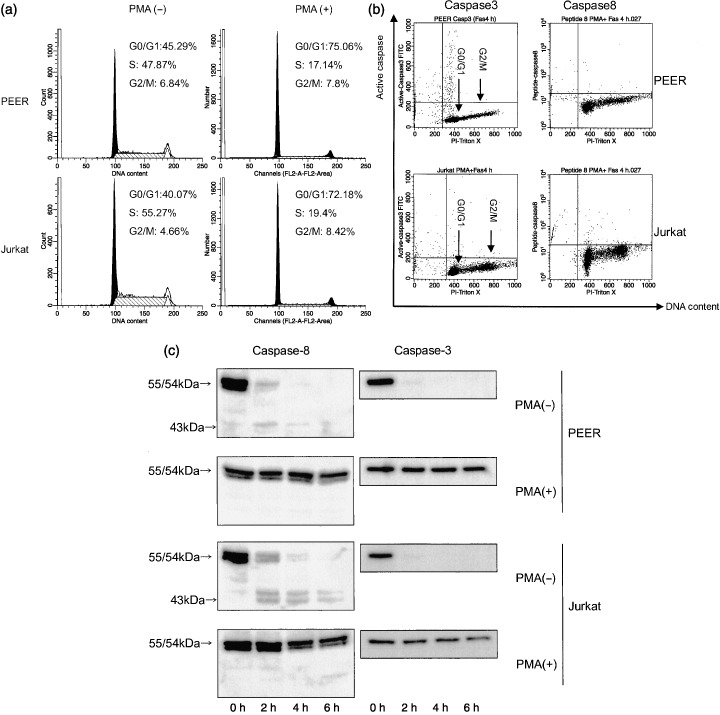
Activation of caspases was abolished when cells were arrested in early G1 phase. (a) Cells (PEER and Jurkat) were incubated with 10 nM PMA for 24 h then analyzed for DNA histogram. (b) Cells (PEER and Jurkat) were pretreated with phorbol 12‐myristate 13‐acetate (PMA) for 24 h then incubated with anti‐Fas antibody (50 ng/mL) for 0 and 4 h. After incubation, cells were harvested and analyzed for expression of active forms of caspase‐3 and caspase‐8, and DNA content. Similar results were obtained in three repeated experiments. (c) PEER and Jurkat cells were pretreated with 10 nM PMA for 24 h then apoptosis was induced with anti‐Fas antibody (50 ng/mL) for 0, 2, 4 and 6 h. Caspase expression was analyzed by western blotting.
Caspase activation in p27‐negative cells. To further confirm that caspases are activated in middle or late G1 phase of the cell cycle, we sorted cells based on active caspase‐3 and caspase‐8 fluorescence and distribution after cells were pretreated with anti‐Fas antibody for 4 h or 6 h, respectively. The active caspase‐3‐positive and ‐negative cells were sorted (Fig. 8a), and p27(Kip1) levels were analyzed by western blotting. As shown in Fig. 8b, the level of p27(Kip1) in active caspase‐3‐ and caspase‐8‐positive cells was markedly decreased compared with that in active caspase‐3‐negative cells. Because p27(Kip1) levels are high in resting early G1 cells but are rapidly reduced in activated cells in late G1 phase,( 22 ) caspases are most likely to be activated in cells entering into late G1 phase of the cell cycle. Cyclin D1/E or FLIP was equal between active caspase‐3‐ and caspase‐8‐positive and ‐negative cells.
Figure 8.
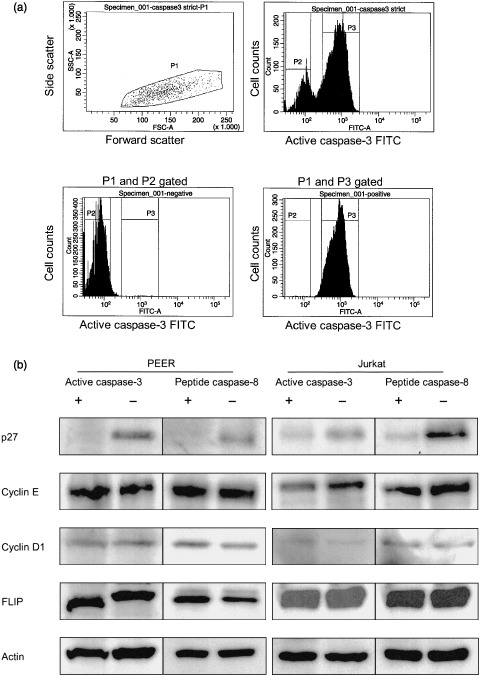
Caspases activated in p27‐negative cells. (a) Cells (PEER and Jurkat) was treated with anti‐Fas antibody (50 ng/mL) for 4 or 6 h then fixed, permeabilized and stained with fluorescein isothiocyanate‐conjugated (FITC) antiactive caspase‐3 antibody or peptide caspase‐8, respectively. The labeled cells were gated and sorted based on active caspase‐3 and caspase‐8 fluorescence. Diagrams show the regions used for PEER cell active caspase‐3 sorting. P1 and P2 represent cells negative for active caspase‐3, P1 and P3 are cells positive for active caspase‐3. Active caspase‐3‐ and caspase‐8‐positive or ‐negative Jurkat cells were sorted as described above. P27, cyclin D1/E, FLIP and actin levels in active caspase‐3‐positive and ‐negative cells, and peptide caspase‐8‐positive and ‐negative cells were analyzed by western blotting. The representative results are shown. Similar results were obtained in two independent experiments.
Discussion
Caspases have been recognized as cysteine proteases that play a major role in the execution of the death signal mediated by Fas receptor (CD95). We hypothesized that caspase‐3 and caspase‐8 might be activated when cells enter into a specific phase of the cell cycle. Numerous studies have been done to clarify the relationship between the apoptotic process and cell cycle progression. Sensitivity to Fas‐mediated apoptosis has been reported to differ among different compartments of cell cycle phases,( 18 , 19 , 20 , 23 , 24 , 25 , 26 ) whereas one group has shown no variation in cellular sensitivity to Fas‐mediated apoptosis during cell cycle progression.( 27 ) In the present study, dual analysis of DNA content and expression of active forms of caspases demonstrated the continuity of the G1 cluster and cells expressing active forms of caspases (1, 4). These results suggest that activation of both caspase‐3 and caspase‐8 could be initiated in G1 phase of the cell cycle. To further identify the specific phase of the cell cycle for caspase activation, we analyzed the expression of cyclin molecules in cells expressing active forms of caspases. It was clearly demonstrated that cells expressing active caspase‐3 and caspase‐8 were negative for cyclin A and cyclin B1 (2, 5), indicating that caspase‐3 and caspase‐8 could not be activated in S or G2/M phase. More importantly, cells expressing active forms of caspase‐8 were negative for cyclin E, whereas cells expressing active forms of caspase‐3 were positive for cyclin E (2, 5). Because cyclin E is expressed from late G1 phase to early S phase,( 7 ) we concluded that caspase‐3 was activated in late G1 phase and early S phase, and caspase‐8 was activated before cells started to express cyclin E in late G1 phase. Taken together, it is most likely that caspase‐8 was activated in cells entering middle G1 phase before cyclin E started to be expressed in late G1 phase prior to the activation of caspase‐3 in late G1 phase and early S phase.
The expression of Ki‐67 in cycling cells reaches a peak during mitosis then decreases in G1 and increases again in S phase; some cells with a long G1 phase can become negative for Ki‐67 at the end of G1 phase.( 21 ) We also found that cells expressing active forms of caspase‐3 and caspase‐8 were negative for Ki‐67 (3, 6). In addition, cells arrested in early G1 phase became resistant to Fas‐mediated apoptosis and activation of both caspase‐3 and caspase‐8 were completely abolished (Fig. 7). Although PMA was reported to disrupt the recruitment of FADD to the DD of the Fas receptor,( 28 ) the present study clearly demonstrates that PMA arrested cells at early G1 phase and completely inhibited Fas‐mediated apoptosis.
To further confirm our conclusion, we carried out post‐sorting western blot experiments and showed that cells expressing active caspase‐3 were clearly negative for the cyclin E–cdk2 complex inhibitor p27(Kip1), which is a hallmark of the negative regulation of G1 progression.( 29 , 30 ) A previous study has shown that p27(Kip1) is expressed in resting early G1 cells, whereas its expression is rapidly reduced when cells are activated and enter into late G1/S phase.( 31 ) It is also reported that thymocytes undergoing apoptosis degrade the cdk inhibitor p27(Kip1) and upregulate cdk2 kinase activity.( 32 , 33 ) Our results showed that cells expressing active forms of caspase‐3 and caspase‐8 were negative for p27(Kip1), which means that caspase‐3 is most likely activated in late G1 phase of the cell cycle. This result is in agreement with others and with our own previous results.( 34 ) We also found that the expression level of p27(Kip1) distinctly increased when MML‐1 cells were arrested at early G1 phase with PMA treatment (data not shown). Our results also clearly showed that cells expressing the active form of caspase‐8 were negative for p27(Kip1). Therefore it can be concluded that caspase‐3 and caspase‐8 were activated in cells undergoing Fas‐mediated apoptosis when p27(Kip1) expression was downregulated in late G1 phase of the cell cycle.
Why are caspases activated mainly in middle/late G1 phase or early S phase of the cell cycle? It is assumed that the Fas‐mediated death signal can be transduced effectively when the expression of antiapoptotic molecules is downregulated or when the expression of proapoptotic molecules is upregulated. Recently, the expression of FLIP, an inhibitory molecule of caspase‐8, has been shown to be cell cycle dependent. The distinct decrease in FLIP protein levels occurs during late G1‐to‐early S phase transition. Using cell cycle‐blocking agents, it has been reported that activated T cells in G1 phase contain high levels of FLIP protein, whereas the amount of FLIP protein is decreased significantly in activated T cells entering into S phase.( 24 , 35 ) In addition, Bcl‐2, the another antiapoptotic molecule, which can inhibit the activation of both caspase‐3 and caspase‐8, is highly expressed in memory T cells, whereas Bcl‐2 expression is downregulated in effector T cells, implying a role for this molecule in modulating the susceptibility of T cells to undergo AICD.( 36 , 37 ) Furthermore, it has been shown that Bcl‐2 retards S phase entry by increasing p27 and p130 expression and by negatively regulating E2F1, which promotes the transcription of Bid and caspase‐8.( 38 , 39 , 40 ) However, we could not find any decrease in FLIP or Bcl‐2 expression in caspase‐3‐activated cells (Fig. 8b and data not shown). Interestingly, it was demonstrated that caspase‐3 is capable of cleaving p16, p21cip‐1/WAF‐1 and retinoblastoma (RB),( 41 ) to promote cell cycle progression in late G1/S phase transition. In early G1 phase, procaspase‐3 is bound to p21cip‐1/WAF‐1, which inhibits the function of cdk, leading to dephosphorylation of the pocket proteins p107 and p110Rb.( 42 ) Caspase‐3 inactivation in early G1 phase could be induced by the formation of a procaspase‐3–p21 complex,( 43 , 44 ) resulting in resistance to Fas‐mediated apoptosis. These results suggest the close relationship between caspase activation and G1/S phase cell cycle progression.
The data presented here clearly demonstrate that caspase‐3 is activated in late G1 phase or early S phase and caspase‐8 is activated in middle or late G1 phase. It would be worthwhile to further clarify the molecular mechanisms for caspase‐3 and caspase‐8 activation in the G1 and early S phases of the cell cycle. More importantly, the cell cycle dependency of caspase activation might be influential in the speed of caspase activation and induction of Fas‐mediated apoptosis.
Conclusion
We have investigated the relationship between cell cycle progression and the activation of caspases in Fas‐mediated apoptosis in leukemia cells. The data presented clearly demonstrate that caspase‐3 is activated in late G1 phase or early S phase and caspase‐8 is activated in middle or late G1 phase. The speed of cell cycle progression from G1 to S phase might be influential in the speed of caspase activation and induction of Fas‐mediated apoptosis.
Acknowledgments
This study was supported in part by Grants from the Ministry of Education, Science, Sports and Culture, Japan. We thank Dr Masahiro Masuya and Dr Yuka Sugimoto for expert assistance with the cell sorting.
References
- 1. Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain‐containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995; 81: 505–12. [DOI] [PubMed] [Google Scholar]
- 2. Muzio M, Chinnaiyan AM, Kischkel FC et al . FLICE, a novel FADD‐homologous ICE/CED‐3‐like protease, is recruited to the CD95 (Fas/APO‐1) death‐inducing signaling complex. Cell 1996; 85: 817–27. [DOI] [PubMed] [Google Scholar]
- 3. Scaffidi C, Fulda S, Srinivasan A et al . Two CD95 (APO‐1/Fas) signaling pathways. EMBO J 1998; 17: 1675–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491–501. [DOI] [PubMed] [Google Scholar]
- 5. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479–89. [DOI] [PubMed] [Google Scholar]
- 6. Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase‐3 to trigger DNA fragmentation during apoptosis. Cell 1997; 89: 175–84. [DOI] [PubMed] [Google Scholar]
- 7. Gong J, Traganos F, Darzynkiewicz Z. Threshold expression of cyclin E but not D type cyclins characterizes normal and tumour cells entering S phase. Cell Prolif 1995; 28: 337–46. [DOI] [PubMed] [Google Scholar]
- 8. Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin E, a nuclear protein essential for the G1‐to‐S phase transition. Mol Cell Biol 1995; 15: 2612–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dulic V, Lees E, Reed SI. Association of human cyclin E with a periodic G1–S phase protein kinase. Science 1992; 257: 1958–61. [DOI] [PubMed] [Google Scholar]
- 10. Yam CH, Fung TK, Poon RY. Cyclin A in cell cycle control and cancer. Cell Mol Life Sci 2002; 59: 1317–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hwang A, Maity A, McKenna WG, Muschel RJ. Cell cycle‐dependent regulation of the cyclin B1 promoter. J Biol Chem 1995; 270: 28 419–24. [DOI] [PubMed] [Google Scholar]
- 12. Maity A, McKenna WG, Muschel RJ. Evidence for post‐transcriptional regulation of cyclin B1 mRNA in the cell cycle and following irradiation in HeLa cells. EMBO J 1995; 14: 603–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Darzynkiewicz Z, Gong J, Juan G, Ardelt B, Traganos F. Cytometry of cyclin proteins. Cytometry 1996; 25: 1–13. [DOI] [PubMed] [Google Scholar]
- 14. Gong J, Traganos F, Darzynkiewicz Z. Discrimination of G2 and mitotic cells by flow cytometry based on different expression of cyclins A and B1. Exp Cell Res 1995; 220: 226–31. [DOI] [PubMed] [Google Scholar]
- 15. Meikrantz W, Schlegel R. Apoptosis and the cell cycle. J Cell Biochem 1995; 58: 160–74. [DOI] [PubMed] [Google Scholar]
- 16. Russell JH. Activation‐induced death of mature T cells in the regulation of immune responses. Curr Opin Immunol 1995; 7: 382–8. [DOI] [PubMed] [Google Scholar]
- 17. Janssen O, Sanzenbacher R, Kabelitz D. Regulation of activation‐induced cell death of mature T‐lymphocyte populations. Cell Tissue Res 2000; 301: 85–99. [DOI] [PubMed] [Google Scholar]
- 18. Li QS, Tanaka S, Kisenge RR, Toyoda H, Azuma E, Komada Y. Activation‐induced T cell death occurs at G1A phase of the cell cycle. Eur J Immunol 2000; 30: 3329–37. [DOI] [PubMed] [Google Scholar]
- 19. Komada Y, Zhou YW, Zhang XL et al . Fas receptor (CD95)‐mediated apoptosis is induced in leukemic cells entering G1B compartment of the cell cycle. Blood 1995; 86: 3848–60. [PubMed] [Google Scholar]
- 20. Komada Y, Sakurai M. Fas receptor (CD95)‐mediated apoptosis in leukemic cells. Leuk Lymphoma 1997; 25: 9–21. [DOI] [PubMed] [Google Scholar]
- 21. Lopez F, Belloc F, Lacombe F et al . Modalities of synthesis of Ki67 antigen during the stimulation of lymphocytes. Cytometry 1991; 12: 42–9. [DOI] [PubMed] [Google Scholar]
- 22. Agrawal D, Hauser P, McPherson F, Dong F, Garcia A, Pledger WJ. Repression of p27kip1 synthesis by platelet‐derived growth factor in BALB/c 3T3 cells. Mol Cell Biol 1996; 16: 4327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beletskaya IV, Nikonova LV, Beletsky IP. Cell cycle specificity of Fas‐mediated apoptosis in WIL‐2 cells. FEBS Lett 1997; 412: 91–3. [DOI] [PubMed] [Google Scholar]
- 24. Algeciras‐Schimnich A, Griffith TS, Lynch DH, Paya CV. Cell cycle‐dependent regulation of FLIP levels and susceptibility to Fas‐mediated apoptosis. J Immunol 1999; 162: 5205–11. [PubMed] [Google Scholar]
- 25. Kuntzen C, Sonuc N, De Toni EN et al . Inhibition of c‐Jun‐N‐terminal‐kinase sensitizes tumor cells to CD95‐induced apoptosis and induces G2/M cell cycle arrest. Cancer Res 2005; 65: 6780–8. [DOI] [PubMed] [Google Scholar]
- 26. Dao T, Huleatt JW, Hingorani R, Crispe IN. Specific resistance of T cells to CD95‐induced apoptosis during S phase of the cell cycle. J Immunol 1997; 159: 4261–7. [PubMed] [Google Scholar]
- 27. Hueber A, Durka S, Weller M. CD95‐mediated apoptosis: no variation in cellular sensitivity during cell cycle progression. FEBS Lett 1998; 432: 155–7. [DOI] [PubMed] [Google Scholar]
- 28. Meng XW, Heldebrant MP, Kaufmann SH. Phorbol 12‐myristate 13‐acetate inhibits death receptor‐mediated apoptosis in Jurkat cells by disrupting recruitment of Fas‐associated polypeptide with death domain. J Biol Chem 2002; 277: 3776–83. [DOI] [PubMed] [Google Scholar]
- 29. Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin‐Cdk protein kinase activity, is related to p21. Cell 1994; 78: 67–74. [DOI] [PubMed] [Google Scholar]
- 30. Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 1996; 272: 877–80. [DOI] [PubMed] [Google Scholar]
- 31. Decker T, Schneller F, Hipp S et al . Cell cycle progression of chronic lymphocytic leukemia cells is controlled by cyclin D2, cyclin D3, cyclin‐dependent kinase (cdk) 4 and the cdk inhibitor p27. Leukemia 2002; 16: 327–34. [DOI] [PubMed] [Google Scholar]
- 32. Mohapatra S, Agrawal D, Pledger WJ. p27Kip1 regulates T cell proliferation. J Biol Chem 2001; 276: 21 976–83. [DOI] [PubMed] [Google Scholar]
- 33. Tsukiyama T, Ishida N, Shirane M et al . Down‐regulation of p27Kip1 expression is required for development and function of T cells. J Immunol 2001; 166: 304–12. [DOI] [PubMed] [Google Scholar]
- 34. Fournel S, Genestier L, Robinet E, Flacher M, Revillard JP. Human T cells require IL‐2 but not G1/S transition to acquire susceptibility to Fas‐mediated apoptosis. J Immunol 1996; 157: 4309–15. [PubMed] [Google Scholar]
- 35. Irmler M, Thome M, Hahne M et al . Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388: 190–5. [DOI] [PubMed] [Google Scholar]
- 36. Grayson JM, Zajac AJ, Altman JD, Ahmed R. Cutting edge: increased expression of Bcl‐2 in antigen‐specific memory CD8+ T cells. J Immunol 2000; 164: 3950–4. [DOI] [PubMed] [Google Scholar]
- 37. Grayson JM, Harrington LE, Lanier JG, Wherry EJ, Ahmed R. Differential sensitivity of naive and memory CD8+ T cells to apoptosis in vivo . J Immunol 2002; 169: 3760–70. [DOI] [PubMed] [Google Scholar]
- 38. Belanger S, Cote M, Lane D, L’Esperance S, Rancourt C, Piche A. Bcl‐2 decreases cell proliferation and promotes accumulation of cells in S phase without affecting the rate of apoptosis in human ovarian carcinoma cells. Gynecol Oncol 2005; 97: 796–806. [DOI] [PubMed] [Google Scholar]
- 39. Cheng N, Janumyan YM, Didion L, Van Hofwegen C, Yang E, Knudson CM. Bcl‐2 inhibition of T‐cell proliferation is related to prolonged T‐cell survival. Oncogene 2004; 23: 3770–80. [DOI] [PubMed] [Google Scholar]
- 40. Cao Q, Xia Y, Azadniv M, Crispe IN. The E2F‐1 transcription factor promotes caspase‐8 and bid expression, and enhances Fas signaling in T cells. J Immunol 2004; 173: 1111–17. [DOI] [PubMed] [Google Scholar]
- 41. Alenzi FQ. Links between apoptosis, proliferation and the cell cycle. Br J Biomed Sci 2004; 61: 99–102. [DOI] [PubMed] [Google Scholar]
- 42. Hingorani R, Bi B, Dao T, Bae Y, Matsuzawa A, Crispe IN. CD95/Fas signaling in T lymphocytes induces the cell cycle control protein p21cip‐1/WAF‐1, which promotes apoptosis. J Immunol 2000; 164: 4032–6. [DOI] [PubMed] [Google Scholar]
- 43. Suzuki A, Tsutomi Y, Akahane K, Araki T, Miura M. Resistance to Fas‐mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene 1998; 17: 931–9. [DOI] [PubMed] [Google Scholar]
- 44. Suzuki A, Tsutomi Y, Miura M, Akahane K. Caspase 3 inactivation to suppress Fas‐mediated apoptosis: identification of binding domain with p21 and ILP and inactivation machinery by p21. Oncogene 1999; 18: 1239–44. [DOI] [PubMed] [Google Scholar]