

Abstract
Histone‐modified enzymes are involved in various cell functions, including proliferation, differentiation, cell death and carcinogenesis. The protein MOZ (monocytic leukemia zinc finger protein) is a Myst (MOZ, Ybf2 (Sas3), Sas2, Tip60)‐type histone acetyltranseferase (HAT) that generates fusion genes, such as MOZ–TIF2, MOZ–CBP and MOZ–p300, in acute myeloid leukemia (AML) by chromosomal translocation. MOZ associates with AML1 (RUNX1), PU.1, and p53, and cooperatively activates target gene transcription. Gene targeting in mice has revealed that MOZ is essential for the generation and maintenance of hematopoietic stem cells (HSC) and for the appropriate development of myeloid, erythroid and B‐lineage cell progenitors. In AML, MOZ fusion genes lead to repressed differentiation, hyper‐proliferation, and self‐renewal of myeloid progenitors through deregulation of MOZ‐regulated target gene expression. It is therefore necessary to analyze the roles of MOZ and MOZ fusion genes in normal and malignant hematopoiesis to elucidate the mechanisms underlying and develop therapies for MOZ‐related AML. (Cancer Sci 2008; 99: 1523–1527)
In acute myeloid leukemia (AML), fusion genes often form as the result of specific chromosomal translocations.( 1 , 2 ) Many of these fused genes, including MOZ, are transcription‐related factors involved in the development or self‐renewal of hematopoietic stem cells (HSC) and/or in hematopoietic cell differentiation.( 3 , 4 ) Monocytic leukemia zinc finger protein (MOZ) is a Myst (MOZ, Ybf2 (Sas3), Sas2, Tip60)‐type histone acetyltranseferase (HAT) that functions as a transcriptional coactivator for hematopoietic transcription factors such as AML1.( 5 , 6 ) Recently, we and others used gene‐targeted mice to reveal critical roles for MOZ in hematopoiesis, particularly in the self‐renewal of HSC.( 7 , 8 ) MOZ fusion genes can also transform HSC and myeloid progenitor cells into leukemia cells and confer unto them self‐renewal activity.( 9 , 10 ) Researching the functions of MOZ and MOZ fusion genes in normal and malignant hematopoiesis can aid in understanding the mechanisms of AML development and leukemic cell‐renewal activity. Such discoveries may allow the development of improved AML therapies.
Involvement of the MOZ gene in chromosomal translocations
Chromosomal translocations, which are frequently detected in patients with AML, include t(8;21), inv(16), t(15;17) and t(11; v) (v: variable) result in the gene fusions AML1–ETO, CBFβ–MYH11, PML–RARα and MLL‐fusions, respectively. MOZ was first identified as a gene involved in the translocation t(8;16)(p11;p13) resulting in the MOZ–CBP fusion gene.( 11 ) The MOZ–p300, MOZ–TIF2 and MOZ–NcoA3 fusion genes were later identified in AML from t(8;22)(p11;q13),( 12 , 13 ) inv(8)(p11;q13)( 14 , 15 ) and t(8; 20)(p11;q13),( 16 ) respectively (Fig. 1). MOZ is also involved in a patient of pediatric therapy‐related myelodysplastic syndrome with a novel chromosomal translocation t(2;8)(p23;p11).( 17 ) MOZ‐related factor (MORF; Myst4, Querkopf), a HAT that is highly homologous to MOZ,( 18 , 19 ) also generates fusion genes with CBP and probably also with Gcn5 in various disorders, including AML( 20 , 21 ) therapy‐related myelodysplastic syndrome( 22 ) and uterine leiomyomata.( 23 )
Figure 1.
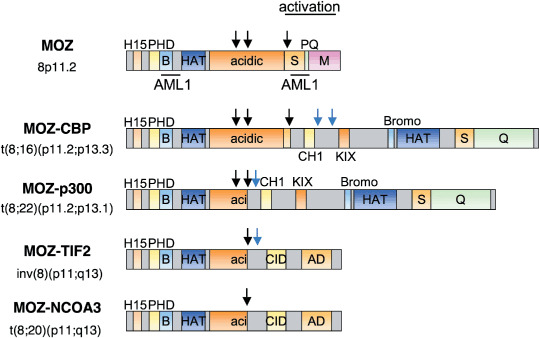
Structure of normal monocytic leukemia zinc finger protein (MOZ) and leukemia‐associated MOZ fusion proteins. MOZ has various functional domains including H1/5 (H15; histone H1/H5–like), PHD (plant homeobox–like domain) zinc finger, B basic (b), histone acetyltranseferase (HAT) and acidic domains. S, PQ and M indicate serine‐, proline‐ and glutamine‐, and methionine‐rich domains, respectively. MOZ fusions with four known partners are shown: CBP, p300, TIF2 and the recently identified NCOA3. MOZ fusion partners contained CH1 (cystein/histidine rich domain 1), KIX (kinase‐inducible) domain, Bromodomain (Bromo), CID (CBP interacting domain) and AD (activation domain). The breakpoints in the MOZ gene and its fusion partners are shown as black arrows and blue arrows, respectively.
All MOZ fusion partner genes are involved in histone modification and transcriptional regulation. CBP and p300 are major HAT( 24 ) that function as coactivators for various transcription factors. TIF2 (NcoA2/GRIP1) and NcoA3 (TRAM‐1/RAC3/pCIP/AIB‐1) are adaptor proteins that combine nuclear receptors with CBP.( 25 ) AML that express MOZ fusion genes are typically monocytic leukemias classified as M4/M5 among French–American–British (FAB) subtypes;( 18 ) MOZ‐related translocation is found in about 6.5% of such AML subtypes.( 16 )
In all MOZ fusion genes, breakpoints of MOZ are located in or around its acidic domain (Fig. 1). As a result, the N‐terminal region of MOZ is retained and the C‐terminal region is replaced with the fusion partners, such as CBP or p300. The N‐terminal region of MOZ contains a H15 (histone H1/H5) domain related to nuclear localization,( 5 ) a PHD (plant homeobox‐like domain) zinc finger involved in binding to methylated histone, a basic domain and a Myst‐type HAT domain. The HAT domain contains a C2HC zinc finger and helix–turn–helix motifs that bind to nucleosomes( 26 ) and DNA.( 27 ) Taken together, these structures suggest that MOZ fusion genes may affect the transcription of MOZ target genes with deregulated acetylation states.
HAT activity and transcriptional regulation
Histone acetylation is one of the major epigenetic mechanisms to regulate gene expression.( 28 ) MOZ is capable of acetylating histones H2A and H3 at K14, and H4 at K5, 8, 12, and 16 in vitro. ( 5 , 6 ) Five Myst‐type acetyltransferases are known in humans: MOF (Myst1); HBO1(Myst2); MOZ (Myst3); MORF (Myst4); and TIP60.( 29 ) With the exception of MOF, all of the Myst‐type acetyltransferases are involved in transcriptional complexes with inhibition of growth (ING) family proteins.( 30 , 31 ) MOZ and MORF form the yeast NuA3‐like transcriptional complex with ING5, BRPF1/2/3 and hEaf6. The yeast NuA3 complex aids the acetylation of histone H3 at K14, which is associated with transcription and replication.( 31 )
MOZ specifically interacts with transcription factors such as AML1,( 5 ) PU.1,( 8 ) p53, Runx2( 32 ) and NF‐κB,( 33 ) functioning as their transcriptional coactivator. AML1 forms large multiprotein complexes including CBFβ as a ‘core component’ as well as several classes of chromatin modulators such as p300 or CBP,( 34 ) MOZ,( 13 ) PML( 35 ) and HIPK2( 36 ) as a ‘regulatory complex’ (Fig. 2).( 37 ) MOZ cooperatively activates AML1‐dependent transcription of genes such as MPO (myeloperoxidase) ( 5 ) or macrophage inflammatory protein (MIP)‐1α.( 38 ) Interaction of MOZ with Runx232, p65/NF‐κB( 33 ) or p53 (S. R. and I. K., unpublished data) can activate their transcription factor‐dependent gene expression. The role of HAT activity in MOZ function remains unclear. For instance, the HAT domain of MOZ is required for NF‐κB‐dependent transcription( 33 ) but not for AML1‐mediated transcription,( 5 ) suggesting target‐dependent roles for HAT activity in transcriptional regulation. The leukemia‐associated fusion protein MOZ–CBP inhibits AML1‐dependent activation of the MPO promoter( 5 ) but activates NF‐κB‐dependent transcription.( 33 ) These results indicate that effects of MOZ fusion proteins on transcription are dependent on promoter contexts.
Figure 2.
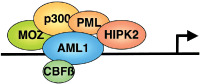
Role of monocytic zinc finger protein (MOZ) in acute myeloid leukemia (AML)1‐dependent transcription. AML1 acts as both a sequence‐specific transacting factor and a scaffold to intermediate the association of HIPK2 and histone acetyltranseferase (HAT). MOZ functions as coactivator for AML1 and activicates AML1‐mediated transcription.
During hepatocarcinogenesis, MOZ activates MafK and Nrf2‐dependent transcription of the GSTP gene.( 39 ) In moz mutant zebrafish, hoxa2b and hoxb2a expression is lost and the support skeleton of the second pharyngeal segment is transformed into a mirror‐image duplicated jaw skeleton.( 40 , 41 ) Defective Hox gene expression in moz mutants is partially recovered by the histone deacetylase inhibitor trichostatin A. Consistent with moz‐deficient zebrafish, HoxA9 expression is also decreased in hematopoietic cells in MOZ‐deficient mice.( 8 ) Additionally, expression of HoxA9, HoxA10 and Meis1 are increased in AML patients with MOZ fusion genes.( 42 ) These results indicate that MOZ‐regulated genes are involved in development, hematopoiesis and carcinogenesis.
Roles of MOZ in hematopoiesis
MOZ cooperatively activates target genes of AML1 and PU.1 that play important roles in hematopoiesis.( 5 , 8 ) We previously demonstrated that MOZ‐deficient mice are embryonic lethal at E15 with a pale appearance and a small liver, the major organ that produces hematopoietic cells at this period.( 8 ) These mice have significantly decreased production of mature erythrocytes; the embryonic lethal state likely results from defective erythrocyte maturation around E15, because primitive erythrocytes are exchanged with definitive erythrocytes during this period.
Cell numbers in MOZ‐deficient fetal livers are about half those of wild‐type animals. MOZ‐deficient hematopoietic progenitor cells produce colonies on methylcellulose medium with appropriate cytokines with cell numbers of one‐fifth to one‐tenth those of wild‐type cells. HSC and their progenitors are severely decreased in MOZ‐deficient fetal livers, and CD19+ B‐lineage cells are also significantly decreased. In contrast, populations of granulocytes and monocytes, as well as myeloid progenitor KLSF (c‐Kit+, Lin−, Sca‐1−, FcRγ+), the population of which is small in normal fetal liver and bone marrow, are increased in MOZ‐deficient fetal liver. KLSF cells in normal and MOZ‐deficient fetal liver can form myeloid colonies in methylcellulose medium and differentiate into either granulocytes or monocytes in vitro. Increases in the committed myeloid cells suggest that myeloid differentiation is inhibited around the KLSF stage in MOZ(–/–) mice, because MOZ has important roles in the regulation of this stage. Taken together, these results indicate that MOZ plays key roles for appropriate development and differentiation of lymphoid and myeloid cells (Fig. 3).
Figure 3.
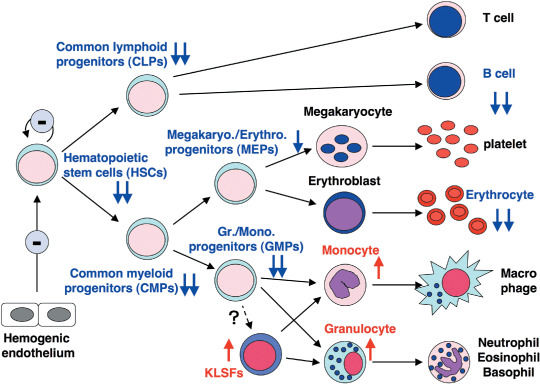
Schematic for fetal hematopoiesis phenotypes of monocytic zinc finger protein (MOZ)‐deficient mice. The MOZ‐deficient fetal liver contains reduced hematopoietic progenitor and hematopoietic stem cells (HSC) have lost their self‐renewal activity. Levels of CD19+ B‐lineage cells and mature erythrocytes are also severely reduced. In contrast, myeloid cell and KLSF (c‐Kit+, Lin−, Sca‐1−, FcRγ+) populations are increased in the MOZ‐deficient fetal liver.
MOZ is also required for self‐renewal of hematopoietic stem cells. When MOZ‐deficient fetal liver cells are transplanted into irradiated mice, recipient mice do not develop a reconstituted hematopoietic system, even when excess numbers of fetal liver cells are transplanted. Gene expression analysis reveals that the expression of c‐Kit, c‐Mpl and HoxA9 is reduced in MOZ‐deficient compared with wild‐type mice. Thomas et al. reported the lethality at birth of mice with a MOZ C‐terminal deletion (MOZ‐ΔC mice)( 7 ) a mouse model that is slightly different from the MOZ‐deficient mice discussed above. Hematopoietic deficiency in MOZ‐ΔC mice was observed in terms of decreased numbers of blood cells, significantly reduced hematopoietic progenitors (blasts) and HSC and an increase in nucleated erythrocytes. The HSC from these mice also have reduced CFU–S12 and hematopoietic reconstitution activities. Thymus T‐cell development is normal, although thymocyte numbers are substantially reduced. On the other hand, the spleens of the MOZ‐ΔC mice are critically affected.
As described above, MOZ interacts with AML1 and PU.1 transcription factors that have various functional roles in hematopoiesis. AML1‐deficient mice display deficiencies in their ability to generate HSC, CLP (common lymphoid progenitors) and B cells and have an increased number of myeloid lineage cells.( 43 , 44 , 45 , 46 ) PU.1‐deficient mice display defects in CLP and most myeloid progenitor cells except MEP (megakaryo/erythroid progenitors).( 47 , 48 , 49 ) MOZ‐deficient mice show less severe phenotypes than either AML1‐ and PU.1‐deficient mice, which is presumably because these transcription factors are not completely inactive in the absence of MOZ.( 8 ) It is interesting that although the number of myeloid lineage cells is decreased in PU.1‐null mice,( 47 ) they are increased in PU.1 knockdown mice, as observed in MOZ‐null mice.( 50 ) These data suggest that part of the MOZ‐deficient phenotype is due to the reduced activity of transcription factors.
Numerous studies have shown that HSC self‐renewal activity is controlled by transcription modulating factors, signal transduction molecules or cell cycle regulators.( 51 , 52 ) MOZ‐deficient mice, in which HSC lose their self‐renewal activity, display reduced expression of HoxA9,( 53 ) c‐Kit( 54 ) and c‐Mpl,( 55 , 56 ) which are known to be involved in HSC self‐renewal activity. Therefore, HSC dysfunction in MOZ‐deficient mice results from the reduced expression of MOZ‐target genes including HoxA9, c‐Kit, and c‐Mpl.
MOZ‐associated leukemia and leukemic stem cells
The MOZ gene is involved in specific chromosomal translocations in monocytic leukemia. These translocations result in the expression of MOZ fusion proteins such as MOZ–CBP, MOZ–p300, MOZ–TIF2 and MOZ–NcoA3. It has been reported that MOZ–TIF2 can efficiently induce AML in mice after transplantation of HSC or myeloid progenitors in which MOZ–TIF2 is introduced by retroviral vectors.( 9 ) It has also been suggested that MOZ–TIF2 confers self‐renewal activity to committed progenitor cells.( 57 ) Several regions of the MOZ–TIF2 fusion gene are necessary to give rise to AML, including the C2HC zinc finger motif that is associated with nucleosome binding and the Myst domain (both of the MOZ‐derived region), and the LXXLL motif that is related to CBP recruitment (of the TIF2 region).( 9 ) The MOZ PHD finger and the acetyl‐coenzyme A binding site that is indispensable for HAT activity contribute to, but are not essential for, AML development. In the MOZ–TIF2‐induced AML mouse model, levels of myeloid progenitor populations, including GMP (granulocyte/monocyte progenitors) and KLSF, are substantially increased.( 57 ) Myeloid progenitor KLSF are also elevated in MOZ‐deficient mice.( 8 ) These results suggest that MOZ dysfunction is associated with AML induction by MOZ–TIF2.
It is also worthwhile to note that a graded reduction of PU.1 induces myeloproliferative disorder (MPD) or AML in mice.( 50 ) MOZ–CBP inhibits AML1‐dependent activation of the MPO promoter( 5 ) but activates NF‐κB‐dependent transcription.( 33 ) MOZ–TIF2 also represses retinoic‐acid‐receptor‐ and p53‐dependent transcription.( 58 ) Therefore, MOZ fusion proteins differently regulate transcription of their targets, with transcription effects being largely dependent on the promoter context (Fig. 4).
Figure 4.
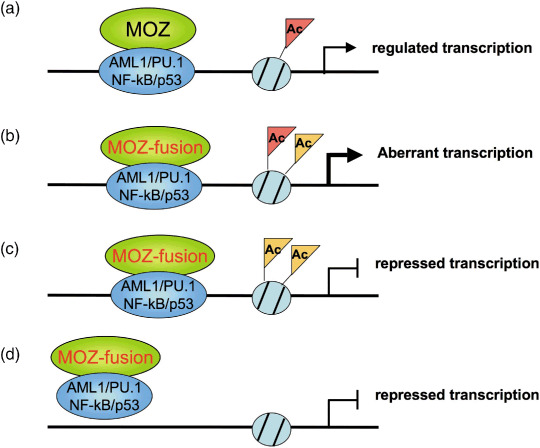
Models of aberrant transcription in acute myeloid leukemia (AML) with monocytic leukemia zinc finger protein (MOZ) fusion genes. (a) MOZ maintains appropriate chromatin status to regulate target gene expression and hematopoiesis. (b) Constitutively active transcription is induced by MOZ fusion proteins through unusual chromatin status such as hyper‐acetylation in leukemia. (c) MOZ fusions induce unusual chromatin status and then down‐regulate target gene expression. (d) MOZ fusions deplete transcription factors and/or their coactovators, and then down‐regulate target gene expression. Ac, acetylation.
Camos et al. previously reported the gene expression profiles of AML patients with a t(8; 16) MOZ‐CBP translocation compared with those of AML patients carrying other translocations.( 42 ) Prolactin and the proto‐oncogene RET are specifically over‐expressed in MOZ–CBP patients, as are the homeobox genes HoxA9, A10 and Meis1. Overexpression of Hox genes are often observed in leukemia patients including those with MLL gene rearrangements.( 59 ) However, other hox genes, such as HoxA3, A7, or HoxB5 are not elevated in MOZ–CBP patients. One can conclude that HoxA9 is a target gene for both normal MOZ and leukemia‐associated MOZ fusion proteins, because its expression is impaired in MOZ‐deficient hematopoietic cells. It will be necessary to analyze the transcriptional regulation of HoxA9 by MOZ or MOZ fusion genes to reveal the molecular mechanisms underlying the development of MOZ‐related AML.
Conclusions
MOZ is a Myst‐type acetyltransferase that coordinately activates the target genes of hematopoiesis‐essential transcription factors such as AML1 and PU.1. MOZ is also indispensable for hematopoietic cell development and HSC self‐renewal. In acute monocytic leukemia, MOZ fuses with CBP, p300 or TIF2, all of which are involved in histone modification. MOZ fusion proteins positively and negatively affect transcription of MOZ target genes to induce leukemia. Further investigation is needed into the functions of MOZ and MOZ fusion genes in hematopoiesis and leukemogenesis in order to develop improved AML therapeutics.
Acknowledgments
This work was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Health, Labor and Welfare and from the Ministry of Education, Culture, Sports, Science, and Technology, and by the Program for Promotion of Fundamental Studies from the National Institute of Biomedical Innovation of Japan.
References
- 1. Look AT. Oncogenic transcription factors in the human acute leukemias. Science 1997; 278: 1059–64. [DOI] [PubMed] [Google Scholar]
- 2. Mrozek K, Heinonen K, Bloomfield CD. Clinical importance of cytogenetics in acute myeloid leukaemia. Best Pract Res Clin Haematol 2001; 14: 19–47. [DOI] [PubMed] [Google Scholar]
- 3. Scandura JM, Boccuni P, Cammenga J, Nimer SD. Transcription factor fusions in acute leukemia: variations on a theme. Oncogene 2002; 21: 3422–44. [DOI] [PubMed] [Google Scholar]
- 4. Di Croce L. Chromatin modifying activity of leukaemia associated fusion proteins. Hum Mol Genet 2005; 14 (Spec 1): R77–84. [DOI] [PubMed] [Google Scholar]
- 5. Kitabayashi I, Aikawa Y, Nguyen LA, Yokoyama A, Ohki M. Activation of AML1‐mediated transcription by MOZ and inhibition by the MOZ–CBP fusion protein. Embo J 2001; 20: 7184–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Champagne N, Pelletier N, Yang XJ. The monocytic leukemia zinc finger protein MOZ is a histone acetyltransferase. Oncogene 2001; 20: 404–9. [DOI] [PubMed] [Google Scholar]
- 7. Thomas T, Corcoran LM, Gugasyan R et al . Monocytic leukemia zinc finger protein is essential for the development of long‐term reconstituting hematopoietic stem cells. Genes Dev 2006; 20: 1175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Katsumoto T, Aikawa Y, Iwama A et al . MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev 2006; 20: 1321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deguchi K, Ayton PM, Carapeti M et al . MOZ–TIF2‐induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2‐mediated recruitment of CBP. Cancer Cell 2003; 3: 259–71. [DOI] [PubMed] [Google Scholar]
- 10. Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer‐stem‐cell research. Nat Rev Cancer 2005; 5: 311–21. [DOI] [PubMed] [Google Scholar]
- 11. Borrow J, Stanton VP Jr, Andresen JM et al . The translocation t(8;16) (p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB–binding protein. Nat Genet 1996; 14: 33–41. [DOI] [PubMed] [Google Scholar]
- 12. Chaffanet M, Gressin L, Preudhomme C, Soenen‐Cornu V, Birnbaum D, Pebusque MJ. MOZ is fused to p300 in an acute monocytic leukemia with t(8;22). Genes Chromosomes Cancer 2000; 28: 138–44. [DOI] [PubMed] [Google Scholar]
- 13. Kitabayashi I, Aikawa Y, Yokoyama A et al . Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22) (p11;q13) chromosome translocation. Leukemia 2001; 15: 89–94. [DOI] [PubMed] [Google Scholar]
- 14. Carapeti M, Aguiar RC, Goldman JM, Cross NC. A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood 1998; 91: 3127–33. [PubMed] [Google Scholar]
- 15. Liang J, Prouty L, Williams BJ, Dayton MA, Blanchard KL. Acute mixed lineage leukemia with an inv (8) (p11q13) resulting in fusion of the genes for MOZ and TIF2. Blood 1998; 92: 2118–22. [PubMed] [Google Scholar]
- 16. Esteyries S, Perot C, Adelaide J et al . NCOA3, a new fusion partner for MOZ/MYST3 in M5 acute myeloid leukemia. Leukemia 2007; 22: 663–5. [DOI] [PubMed] [Google Scholar]
- 17. Imamura T, Kakazu N, Hibi S et al . Rearrangement of the MOZ gene in pediatric therapy‐related myelodysplastic syndrome with a novel chromosomal translocation t(2;8) (p23;p11). Genes Chromosomes Cancer 2003; 36: 413–19. [DOI] [PubMed] [Google Scholar]
- 18. Yang XJ, Ullah M. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene 2007; 26: 5408–19. [DOI] [PubMed] [Google Scholar]
- 19. Thomas T, Voss AK, Chowdhury K, Gruss P. Querkopf, a MYST family histone acetyltransferase, is required for normal cerebral cortex development. Development 2000; 127: 2537–48. [DOI] [PubMed] [Google Scholar]
- 20. Panagopoulos I, Fioretos T, Isaksson M et al . Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16) (q22;p13). Hum Mol Genet 2001; 10: 395–404. [DOI] [PubMed] [Google Scholar]
- 21. Vizmanos JL, Larrayoz MJ, Lahortiga I et al . t(10;16) (q22;p13) and MORF–CREBBP fusion is a recurrent event in acute myeloid leukemia. Genes Chromosomes Cancer 2003; 36: 402–5. [DOI] [PubMed] [Google Scholar]
- 22. Kojima K, Kaneda K, Yoshida C et al . A novel fusion variant of the MORF and CBP genes detected in therapy‐related myelodysplastic syndrome with t(10;16) (q22;p13). Br J Haematol 2003; 120: 271–3. [DOI] [PubMed] [Google Scholar]
- 23. Moore SD, Herrick SR, Ince TA et al . Uterine leiomyomata with t(10;17) disrupt the histone acetyltransferase MORF. Cancer Res 2004; 64: 5570–7. [DOI] [PubMed] [Google Scholar]
- 24. Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene 2004; 23: 4225–31. [DOI] [PubMed] [Google Scholar]
- 25. Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol 2003; 17: 1681–92. [DOI] [PubMed] [Google Scholar]
- 26. Akhtar A, Becker PB. The histone H4 acetyltransferase MOF uses a C2HC zinc finger for substrate recognition. EMBO Rep 2001; 2: 113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holbert MA, Sikorski T, Carten J, Snowflack D, Hodawadekar S, Marmorstein R. The human monocytic leukemia zinc finger histone acetyltransferase domain contains DNA‐binding activity implicated in chromatin targeting. J Biol Chem 2007; 282: 36603–13. [DOI] [PubMed] [Google Scholar]
- 28. Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 1998; 12: 599–606. [DOI] [PubMed] [Google Scholar]
- 29. Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucl Acids Res 2004; 32: 959–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doyon Y, Cayrou C, Ullah M et al . ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell 2006; 21: 51–64. [DOI] [PubMed] [Google Scholar]
- 31. Avvakumov N, Cote J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007; 26: 5395–407. [DOI] [PubMed] [Google Scholar]
- 32. Pelletier N, Champagne N, Stifani S, Yang XJ. MOZ and MORF histone acetyltransferases interact with the Runt‐domain transcription factor Runx2. Oncogene 2002; 21: 2729–40. [DOI] [PubMed] [Google Scholar]
- 33. Chan EM, Chan RJ, Comer EM et al . MOZ and MOZ–CBP cooperate with NF‐kappaB to activate transcription from NF‐kappaB‐dependent promoters. Exp Hematol 2007; 35: 1782–92. [DOI] [PubMed] [Google Scholar]
- 34. Kitabayashi I, Yokoyama A, Shimizu K, Ohki M. Interaction and functional cooperation of the leukemia‐associated factors AML1 and p300 in myeloid cell differentiation. Embo J 1998; 17: 2994–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nguyen LA, Pandolfi PP, Aikawa Y, Tagata Y, Ohki M, Kitabayashi I. Physical and functional link of the leukemia‐associated factors AML1 and PML. Blood 2005; 105: 292–300. [DOI] [PubMed] [Google Scholar]
- 36. Aikawa Y, Nguyen LA, Isono K et al . Roles of HIPK1 and HIPK2 in AML1‐ and p300‐dependent transcription, hematopoiesis and blood vessel formation. Embo J 2006; 25: 3955–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoshida H, Kitabayashi I. Chromatin regulation by AML1 complex. Int J Hematol 2008; 87: 19–24. [DOI] [PubMed] [Google Scholar]
- 38. Bristow CA, Shore P. Transcriptional regulation of the human MIP‐1alpha promoter by RUNX1 and MOZ. Nucl Acids Res 2003; 31: 2735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ohta K, Ohigashi M, Naganawa A et al . Histone acetyltransferase MOZ acts as a co‐activator of Nrf2–MafK and induces tumour marker gene expression during hepatocarcinogenesis. Biochem J 2007; 402: 559–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miller CT, Maves L, Kimmel CB. moz regulates Hox expression and pharyngeal segmental identity in zebrafish. Development 2004; 131: 2443–61. [DOI] [PubMed] [Google Scholar]
- 41. Crump JG, Swartz ME, Eberhart JK, Kimmel CB. Moz‐dependent Hox expression controls segment‐specific fate maps of skeletal precursors in the face. Development 2006; 133: 2661–9. [DOI] [PubMed] [Google Scholar]
- 42. Camos M, Esteve J, Jares P et al . Gene expression profiling of acute myeloid leukemia with translocation t(8;16) (p11;p13) and MYST3–CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res 2006; 66: 6947–54. [DOI] [PubMed] [Google Scholar]
- 43. Okuda T, Van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996; 84: 321–30. [DOI] [PubMed] [Google Scholar]
- 44. Ichikawa M, Asai T, Saito T et al . AML‐1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med 2004; 10: 299–304. [DOI] [PubMed] [Google Scholar]
- 45. Growney JD, Shigematsu H, Li Z et al . Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 2005; 106: 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Putz G, Rosner A, Nuesslein I, Schmitz N, Buchholz F. AML1 deletion in adult mice causes splenomegaly and lymphomas. Oncogene 2006; 25: 929–39. [DOI] [PubMed] [Google Scholar]
- 47. Kim HG, De Guzman CG, Swindle CS et al . The ETS family transcription factor PU.1 is necessary for the maintenance of fetal liver hematopoietic stem cells. Blood 2004; 104: 3894–900. [DOI] [PubMed] [Google Scholar]
- 48. Iwasaki H, Somoza C, Shigematsu H et al . Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood 2005; 106: 1590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nutt SL, Metcalf D, D’Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J Exp Med 2005; 201: 221–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rosenbauer F, Wagner K, Kutok JL et al . Acute myeloid leukemia induced by graded reduction of a lineage‐specific transcription factor, PU. 1 Nat Genet 2004; 36: 624–30. [DOI] [PubMed] [Google Scholar]
- 51. Stein MI, Zhu J, Emerson SG. Molecular pathways regulating the self‐renewal of hematopoietic stem cells. Exp Hematol 2004; 32: 1129–36. [DOI] [PubMed] [Google Scholar]
- 52. Huang X, Cho S, Spangrude GJ. Hematopoietic stem cells: generation and self‐renewal. Cell Death Differ 2007; 14: 1851–9. [DOI] [PubMed] [Google Scholar]
- 53. Lawrence HJ, Christensen J, Fong S et al . Loss of expression of the HOXA‐9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood 2005; 106: 3988–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Waskow C, Paul S, Haller C, Gassmann M, Rodewald HR. Viable c‐Kit (W/W) mutants reveal pivotal role for c‐kit in the maintenance of lymphopoiesis. Immunity 2002; 17: 277–88. [DOI] [PubMed] [Google Scholar]
- 55. Kimura S, Roberts AW, Metcalf D, Alexander WS. Hematopoietic stem cell deficiencies in mice lacking c‐Mpl, the receptor for thrombopoietin. Proc Natl Acad Sci USA 1998; 95: 1195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Antonchuk J, Hyland CD, Hilton DJ, Alexander WS. Synergistic effects on erythropoiesis, thrombopoiesis, and stem cell competitiveness in mice deficient in thrombopoietin and steel factor receptors. Blood 2004; 104: 1306–13. [DOI] [PubMed] [Google Scholar]
- 57. Huntly BJ, Shigematsu H, Deguchi K et al . MOZ–TIF2, but not BCR–ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004; 6: 587–96. [DOI] [PubMed] [Google Scholar]
- 58. Kindle KB, Troke PJ, Collins HM et al . MOZ–TIF2 inhibits transcription by nuclear receptors and p53 by impairment of CBP function. Mol Cell Biol 2005; 25: 988–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eklund EA. The role of HOX genes in malignant myeloid disease. Curr Opin Hematol 2007; 14: 85–9. [DOI] [PubMed] [Google Scholar]