

Abstract
Metastatic gastrointestinal stromal tumors (GIST) have an extremely poor prognosis; however, their immunohistochemical and genetic features have not been assessed satisfactorily and the mechanisms responsible for their high malignant potential remain unclear. We examined the immunohistochemical differences between gastric GIST and metastatic lesions in the liver of four patients who had undergone a postgastrectomy hepatectomy for metachronous liver metastases. We also carried out genetic analysis of the tumors in three of the four cases. In all cases, the immunoreactivity profiles, including KIT (CD117), CD34, smooth muscle actin (SMA), desmin, S‐100 and vimentin, were similar between the gastric and metastatic tumors, but the Ki67 labeling index in the metastatic GIST was higher than that of the primary GIST. Interestingly, in the case who had received neoadjuvant imatinib therapy before gastrectomy, its therapeutic effect was observed in most of the primary lesion, with the exception of a specific small area with high cellularity. Genetic analysis revealed no acquired mutations in the c‐kit or PDGFRA genes in the metastatic lesions in any of the patients, but loss of heterozygosity (LOH) of the c‐kit gene was observed mainly in the metastatic tumors in two of the three cases. Furthermore, in the case of neoadjuvant imatinib therapy, LOH of the c‐kit gene was shown in the high cellularity area in the primary lesion and metastatic liver GIST. It is suggested that LOH of the c‐kit gene is an important event that leads to imatinib resistance and metastatic progression of GIST. In conclusion, both gastric and metastatic GIST had almost the same immunohistochemical features, except for their proliferative activity, and LOH of the c‐kit gene played an important role in the process of liver metastasis. (Cancer Sci 2006; 97: 127 – 132)
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumors of the gastrointestinal tract, and are thought to originate from the interstitial cells of Cajal or its precursors.( 1 , 2 , 3 ) GIST express the KIT receptor, the product of the c‐kit protooncogene.( 1 ) KIT is a transmembrane growth factor receptor with tyrosine kinase activity. KIT receptor signaling is initiated by the binding of ligands (stem‐cell factor), receptor dimerization, and concomitant activation of tyrosine kinase. Most GIST have gain‐of‐function mutations in the c‐kit gene resulting in ligand‐independent KIT receptor activation.( 1 ) Most of the c‐kit mutations are located in exon 11, which encodes the KIT receptor juxtamembrane domain, whereas others are located in exons 9, 13 or 17.( 1 , 4 , 5 , 6 ) In the small subset of GIST without c‐kit mutations, alternative oncogenic activating mutations (e.g. in exons 12 or 18 of the PDGRFA gene) may be involved.( 7 , 8 ) Recently the relationship between c‐kit or PDGFRA mutations and clinical outcome has been assessed.( 9 , 10 , 11 , 12 , 13 , 14 , 15 ) However, only a few studies have examined the genetic difference between primary and metastatic liver GIST, and the mechanisms responsible for the metastatic potential of GIST remain unclear.
Imatinib mesylate (Glivec, Gleevec, STI571; Novartis, Basel, Switzerland) specifically inhibits KIT and PDGFR, resulting in high response rates in patients with GIST.( 16 , 17 , 18 , 19 , 20 ) According to the National Comprehensive Cancer Network (NCCN) practice guidelines, imatinib treatment should be recommended through the management strategies for recurrent GIST.( 21 ) However, many patients with GIST develop resistance to imatinib during therapy in spite of a good response to initial treatments. This secondary imatinib resistance will remain a matter of vital importance to recurrent GIST patients until new effective therapeutic agents are legalized. Nevertheless, meticulous analysis of both immunohistochemical and genetic features are essential in order to overcome imatinib resistance.
We investigated the immunohistochemical features of both primary and metastatic liver GIST in four patients with metachronous liver metastases of gastric GIST. We also analyzed genetic abnormalities of both lesions in three of these patients who had undergone a hepatectomy in 2004 to clarify the factors that are responsible for metastatic potential.
Materials and Methods
Patients
Primary and metastatic tumors from four patients who had undergone a hepatectomy for metachronous liver metastases of gastric GIST at the Department of Surgery, Hamamatsu University School of Medicine (Hamamatsu, Japan) were evaluated. The subjects consisted of three men and one woman, with a mean age of 66.5 years (range 63–72 years) when the hepatectomy was carried out. The mean time from gastrectomy to detection of liver metastases was 74.3 months (range 10–159 months). Neither chemotherapy nor radiotherapy was received by any of the patients throughout the study. One of the four patients had received imatinib for multiple liver metastases before the hepatectomy (case 2) and another had received neoadjuvant imatinib therapy before the gastrectomy (case 3). Other clinical findings of these four patients are shown in Table 1.
Table 1.
Clinical findings in four cases of metastatic liver gastrointestinal stromal tumors (GIST)
| Case No. | Age † (years) | Sex | Primary tumor size (cm) | Therapy prior to operation 1 | Operation 1 ‡ | Time § (months) | Therapy prior to operation 2 | Operation 2 ¶ | Disease outcome |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 63 | Male | 2.6 × 2.4 × 2.2 | None | LE | 90 | None | LS + PH | DOD |
| 2 | 68 | Female | 3.8 × 2.7 × 2.2 | None | DG | 38 | IM (200 mg/day) | PH | NED |
| 3 | 63 | Male | 6.5 × 5 × 3 | IM (400 mg/day) | PG | 10 | None | RL | NED |
| 4 | 72 | Male | 18 × 16.5 × 8.8 | None | LE | 159 | None | RL | NED |
Age at hepatectomy.
‡ Gastrectomy.
§ Time from gastrectomy to detection of metastatic liver GIST.
¶ Hepatectomy. DG, distal gastrectomy; DOD, died of disease; IM, imatinib mesylate; LE, local excision; LS, lateral segmentectomy; NED, no evidence of disease; PG, proximal gastrectomy; PH, partial hepatectomy; RL, right lobectomy.
Immunohistochemistry
Formalin‐fixed paraffin‐embedded tissues were used for conventional H&E staining and for the immunohistochemical examination, as described previously.( 22 ) A rabbit polyclonal antibody against human KIT (CD117, 1/50; DakoCytomation, Kyoto, Japan) or against bovine S‐100 protein (1/300; DakoCytomation), and a mouse monoclonal antibody against human CD34 (QB end10, 1/50; DakoCytomation), human desmin (D33, 1/50; DakoCytomation), smooth muscle actin (SMA) (1A4, 1/50; DakoCytomation), vimentin (undiluted; DakoCytomation), or Ki‐67 (1/400; MIB‐1; DakoCytomation) were used as the primary antibodies at the indicated dilutions. Binding of the polymer‐conjugated secondary antibody and visualization were carried out using a Histofine MAX kit (Nichirei, Osaka, Japan).
DNA isolation
Genomic DNA from the primary GIST of the three patients who agreed to this genetic analysis was extracted from formalin‐fixed, paraffin‐embedded tissues using DEXPAT (Takara Bio, Shiga, Japan). Metastatic liver GIST specimens were flash‐frozen and stored at −80°C and genomic DNA was extracted following standard procedures. Briefly, the DNA was isolated by digestion in lysis buffer (10 mM Tris–HCl [pH 8.0], 5 mM ethylenediaminetetracetic acid, 0.5% sodium dodecylsulfate), containing proteinase K (Invitrogen, Carlsbad, CA, USA) at 37°C for 18 h, followed by phenol/chloroform extraction. After precipitation by the addition of two volumes of ethanol and 0.1 volumes of sodium acetate (3 M), the DNA was washed once with ethanol (70%[v/v]) and dissolved in water. Normal control genomic DNA was isolated from normal liver adjacent to the tumors.
Polymerase chain reaction and direct sequencing for c‐kit and PDGFRA
Exons 9, 11, 13 and 17 of the c‐kit gene were amplified by polymerase chain reaction (PCR) using the following oligonucleotide primer pairs: for exon 9, 5′‐ATTTATTTTCCTAGAGTAAGCCAGGG‐3′ and 5′‐ATCATGACTGATATGGTAGACAGAGC‐3′; for exon 11, 5′‐CTCCAGAGTGCTCTAATGACTGAGAC‐3′ and 5′‐GTCACTGTTATGTGTACCCAAAAAGG‐3′; for exon 13, 5′‐GCTTGACATCAGTTTGCCAGTTGTGC‐3′ and 5′‐GACAGACAATAAAAGGCAGCTTGGAC‐3′; and for exon 17, 5′‐CTTTTCTCCTCCAACCTAATAGTG‐3′ and 5′‐TTGAAACTAAAAATCCTTTGCAGGAC‐3′. Exons 12 and 18 of the PDGFRA gene were amplified by PCR using the following primer pairs: for exon 12, 5′‐GTGCACTGGGACTTTGGTAATTC‐3′ and 5′‐GTGCAAGGGAAAAGGGAGTCTTG‐3′; and for exon 18, 5′‐GCTATTCAGCTACAGATGGCTTG‐3′ and 5′‐AAGTGAAGGAGGATGAGCCTGAC‐3′. PCR was carried out in a reaction volume of 50 µL containing DNA from 5 µL of DEXPAT extract described above or approximately 500 ng DNA obtained through standard procedures. The PCR products were electrophoresed through a 4.0% agarose gel with ethidium bromide. Each band was excised from the gel and extracted with the Geneclean Spin Kit (Q BIO Gene, Irvine, CA, USA). Direct sequencing of the DNA extract from the gel was carried out using the DYEnamic ET Terminator Cycle Sequencing Kit (Amersham Bioscience, Piscataway, NJ, USA) and 3100 DNA Analyzer (Applied Biosystems, Foster City, CA, USA). Direct sequencing was carried out at least twice using different PCR products obtained from the same samples.
Microsatellite analysis
The loss of heterozygosity (LOH) at microsatellite markers D4S2996, D4S2889, D4S3254, and at another CA repeat named HK8810 (located near the c‐kit gene) was evaluated with PCR of tumor and normal specimens using the following oligonucleotide primer pair for HK8810, 5′‐TCAAGAGACAGAGAGACAGAAAG‐3′ and 5′‐TTCCTGAGCACATATCTAACCAC‐3′. The PCR products were electrophoresed through a 5.0% polyacrylamide gel and stained with ethidium bromide.
Fluorescence in situ hybridization analysis
Dual‐color interphase fluorescence in situ hybridization (FISH) analysis was carried out on 4‐µm paraffin‐embedded tissue sections of primary and metastatic GIST according to a protocol published previously.( 23 , 24 , 25 , 26 , 27 , 28 , 29 ) Spectrum Orange‐labeled bacterial artificial chromosome (BAC) clones for KIT/4q12 (RP11‐568A2) were cohybridized with Spectrum Green‐labeled chromosome 4 centromeric probe (CEP4; Vysis, North Chicago, IL, USA). The samples were observed immediately using a fluorescence microscope (BX‐51; Olympus, Tokyo, Japan) equipped with epifluorescence filters and a photometric CCD camera (Sensicam; PCO Company, Kelheim, Germany).
Results
Immunohistochemical findings
The immunohistochemical findings of the four cases are summarized in Table 2. Liver metastatic lesions showed almost similar findings to the primary tumor, except that the intensity of Ki67 staining of liver GIST was higher than that of the primary GIST in all cases. In case 2, who received imatinib treatment for metastatic liver GIST before hepatectomy, degenerative change and hemorrhage were observed in the central area of liver GIST; however, many viable cells were still left in the most peripheral lesions (data not shown). Interestingly, in case 3, who underwent neoadjuvant imatinib therapy before gastrectomy, the cellularity of a particular small area was much higher (Fig. 1a,b) and KIT immunoreactivity was stronger than that of the major part in the primary GIST (Fig. 1c). Moreover, the intensity of Ki67 staining was also high in this area (Fig. 1d), and immunohistochemical features in the small area were also observed in metastatic liver GIST (Fig. 1e–g). These findings suggest that this small area with high metastatic potential was resistant to imatinib, although neoadjuvant imatinib was effective in the major part of primary GIST.
Table 2.
Summary of immunohistochemical findings
| Case no. | Lesion | KIT | CD34 | SMA | Desmin | S‐100 | Vimentin | Ki67 labeling index (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | Primary | +++ | +++ | – | – | – | ++ | 1 |
| Liver meta | +++ | +++ | – | – | – | ++ | 20 | |
| 2 | Primary | +++ | +++ | – | – | – | +++ | <1 |
| Liver meta | +++ | +++ | – | – | – | +++ | 10 | |
| 3 | Primary‐L † | + | + | + | – | – | ++ | 2 |
| Primary‐H ‡ | +++ | +++ | ++ | – | – | ++ | 14 | |
| Liver meta | +++ | +++ | ++ | – | – | ++ | 23 | |
| 4 | Primary | +++ | +++ | + | – | – | – | 1.2 |
| Liver meta | +++ | +++ | + | – | – | – | 4.3 |
Low cellularity area in the primary lesion.
High cellularity area in the primary lesion. +++, Strong staining intensity; +, weak staining intensity; –, negative staining.
Figure 1.
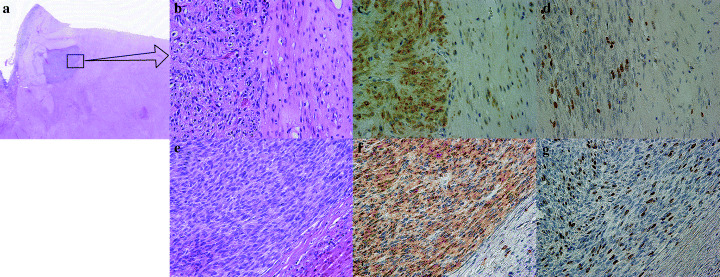
Pathological findings of case 3. (a) Primary lesion resected after neoadjuvant imatinib treatment. Higher cellularity was observed in the small area than in the peripheral large portions (10×, H&E). (b) High‐power view of Fig. 1a. Stromal hyalinization was seen in the peripheral large portions (right side). The viable cellular part is in the left side (200×, H&E). (c) KIT immunostaining of the primary lesion. KIT immunoreactivity was much stronger in the small area (200×, KIT). (d) Ki67 immunostaining of the primary lesion. The Ki67 labeling index was much stronger in the small area (14%) than in the peripheral lesions (2%) (200×, MIB1). (e) Metastatic liver gastrointestinal stromal tumor. Higher cellularity was observed in a similar manner to the small area of the primary lesion (200×, H&E). (f) KIT immunostaining of liver metastasis. Immunoreactivity was similar to the small area of the primary lesion (200×, KIT). (g) Ki67 immunostaining of liver metastasis. The Ki67 labeling index was higher (23%) than the primary lesion (200×, MIB1).
Genetic findings
We carried out mutation analysis for exons 9, 11, 13 and 17 of the c‐kit gene and exons 12 and 18 of the PDGFRA gene of both primary and metastatic liver GIST to clarify whether acquired mutations in these genes were responsible for liver metastasis. The same mutation was observed only in exon 11 of the c‐kit gene between both primary and metastatic liver GIST, and no acquired mutations were seen in any of the patients. Details of the mutation at exon 11 in the c‐kit gene and the predicted amino acid sequence of each patient are shown in Fig. 2.
Figure 2.
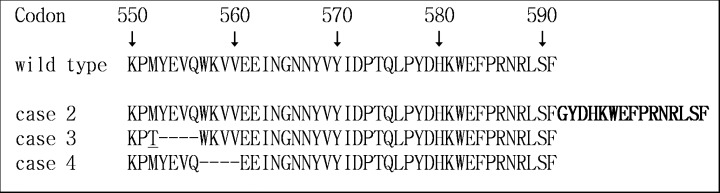
c‐kit mutation at exon 11 and predicted amino acid sequence. Point mutation is underlined, bold letters represent an insertion and ‘‐‐‐‐’ represents a deletion.
We evaluated microsatellite marker analysis near the c‐kit gene with PCR of tumors and normal specimens to investigate whether the LOH of the c‐kit gene contributed to the progression of GIST. First, the microsatellite markers D4S2996, D4S2889 and D4S3254, which are located near the c‐kit gene, were amplified with by PCR using genomic DNA extracted from normal tissues of three patients. However, these markers revealed much less polymorphisms (data not shown). We then used another CA repeat marker, named HK8810, which is located between D4S2889 and D4S3254, and it showed polymorphisms among these three patients (Fig. 3). Microsatellite analysis revealed LOH of the c‐kit gene at both primary and metastatic lesions of case 2 (Fig. 3). Interestingly in case 3, LOH was observed in the small area that showed higher cellularity and a high Ki67 labeling index immunohistochemically, whereas it was not seen in the majority of the primary lesion tissue with lower cellularity (Fig. 3). Furthermore, the metastatic liver GIST in case 3 showed a LOH similar to the higher cellularity area of the primary lesion (Fig. 3). These findings suggest that the cell population with highly malignant potential in the primary GIST induced by LOH of the c‐kit gene had metastasized to the liver.
Figure 3.
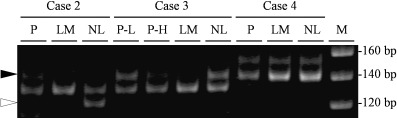
Microsatellite marker analysis near the c‐kit gene. White and black arrowheads indicate the location of the missing alleles of cases 2 and 3, respectively. LM, liver metastasis; M, marker; NL, normal liver; P, primary lesion; P‐H, primary lesion with high cellularity; P‐L, primary lesion with low cellularity.
We used a FISH assay to confirm the results of the microsatellite analysis. BAC clones for KIT/4q12 and chromosome 4 centromeric probes were cohybridized to paraffin‐embedded tissue sections of primary and metastatic GIST. Only a single KIT and chromosome 4 centromere signal was observed in the area where LOH of the c‐kit gene was indicated by microsatellite marker analysis in cases 2 and 3 (data not shown and Fig. 4). These findings verified that the LOH of the c‐kit gene occurred as a result of allelic loss of chromosome 4.
Figure 4.
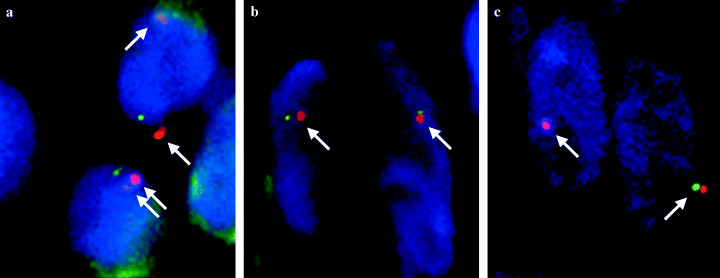
Dual‐color interphase fluorescence in situ hybridization on the primary lesion and the liver metastasis of case 3. Spectrum green‐labeled (green signal) and spectrum orange‐labeled (red signal) DNA probes containing sequence specific for KIT/4q12 and CEP4, respectively, were used. (a) The majority of cells in the primary lesion with low cellularity have two signals. (b) The majority of cells in a small area of the primary lesion with high cellularity have one or no signals. (c) In the cells in the liver metastasis, only one or no signals were observed. The arrows indicate pairs of signals of KIT/4q12 and CEP4.
Discussion
Recurrent GIST have an extremely poor prognosis. Postoperative recurrence or metastasis has been observed after surgical resection in 40–90% of patients.( 30 , 31 , 32 ) Imatinib mesylate is revolutionary in the treatment of metastatic GIST. Imatinib is a selective, potent, small molecule inhibitor of a family of tyrosine kinase signaling enzymes, including KIT and PDGFR, that inhibits proliferation and promotes apoptosis in GIST cells.( 18 ) More than 80% of patients with metastatic or unresectable GIST have achieved an objective confirmed partial response or maintained stable disease following imatinib therapy.( 17 , 19 , 20 ) It is quite reasonable that imatinib is administered as a first‐line treatment for metastatic GIST, which is recommended in the NCCN practice guidelines.( 21 ) However, we must also bear in mind the fact that imatinib was reported to become ineffective at a certain period after initial treatment for metastatic or recurrent GIST, even when it was effective initially.( 33 , 34 , 35 , 36 , 37 , 38 ) In order to clarify the mechanism of imatinib resistance, genetic analysis of both primary and metastatic lesions is essential. However, pathological and genetic features of recurrent GIST have not been satisfactorily assessed. Only a few studies have assessed the features of both primary and metastatic GIST.
This is the first report in which immunohistochemical and genetic differences, including LOH of the c‐kit gene, were assessed between gastric and metastatic liver GIST. In all cases, the immunohistochemical and genetic features of both lesions were almost the same except for the Ki67 labeling index. The high proliferative activity of metastatic lesions support clinical findings that metastatic liver GIST often grow much more rapidly than primary tumors. As such, what is the mechanism of high proliferative potential in metastatic GIST? It is possible that the rapid growth could be induced by some genetic changes. Some studies have suggested that an acquired mutation at the c‐kit or PDGFRA gene could contribute to the gain in malignant potential.( 33 , 34 , 35 , 36 , 37 , 38 ) We also assessed the genetic differences between primary and metastatic liver GIST, but no acquired mutations of either the c‐kit or PDGFRA genes were detected in any of the cases.
Several mechanisms responsible for the acquired resistance to imatinib have been described. These have been categorized as being either KIT‐dependent or KIT‐independent. Kit‐dependent mechanisms have been the best studied, due to secondary mutations resulting in substitution of some residues at critical binding sites for imatinib.( 33 , 34 , 35 , 36 , 37 , 38 ) In the imatinib‐treated patients in our study, LOH of the c‐kit gene was detected instead of acquired mutations. Considering these findings, LOH of the c‐kit gene might be responsible for liver metastases because of acquired high metastatic potential and, presumably, resistance to imatinib. The other changes described previously, such as loss of 1p, 14q or 22q or gene amplifications on 8q or 17q in accordance with c‐kit alteration, might play more definitive roles in the metastatic process.( 39 , 40 ) More extensive study covering the whole genome would perhaps answer this question. Debiec‐Rychter et al. showed by FISH analysis that the KIT, PDFGRA and CEP4 loci were lost in only six of 26 progressive tumors of patients treated with imatinib.( 36 ) Considering that not all imatinib‐resistant tumors have LOH, our observation may be more consistent with the alternate (but not mutually exclusive) idea that loss of KIT, PDFGRA and CEP4 loci is responsible for resistance to imatinib. Furthermore, they explained that while in three of the tumors this hemizygosity was already observed in the primary tumor biopsy specimens, in the three other specimens it was only present in the progressive lesions.( 36 ) In case 3 in our study, who had undergone neoadjuvant imatinib therapy, hemizygosity of chromosome 4 was observed only in the small area of the primary lesion, which seemed pathologically imatinib resistant. In contrast, neoadjuvant imatinib therapy made a contribution to the appearance of this resistant clone. Therefore LOH of the c‐kit gene was, probably, undetectable due to this heterozygosity without imatinib treatment. These findings suggest difficulty in the pathological and genetic analysis of biopsy specimens. This pitfall due to heterozygosity from biopsy specimens should be taken into account in future studies.
The NCCN guidelines point out that surgery does not cure recurrent GIST, and they recommend imatinib treatment for recurrence because the results of a randomized clinical trial revealed that recurrence was observed within several months after imatinib had been stopped.( 21 ) In our study, however, only one of the four cases recurred after hepatectomy, and in case 2, imatinib treatment for metastatic liver GIST was not able to be continued because of adverse effects. Furthermore, LOH of the c‐kit gene was demonstrated in two cases whose lesions were clinically resistant to imatinib. Considering these result, surgery should be considered for both primary and metastatic GIST as the first‐line treatment. Resection or ablation for metastatic liver GIST is beneficial even when imatinib treatment becomes ineffective.
In conclusion, LOH of the c‐kit gene could be responsible for the gain in high proliferative activity, resulting in enhanced metastatic potential, which also plays an important role in imatinib resistance. Genetic heterogeneity of primary GIST caused difficulties in reaching an accurate diagnosis, even postoperatively. Meticulous assessment of resected specimens may pave the way to overcoming imatinib resistance and to improve the patient's prognosis.
Acknowledgments
The work was supported in part by the 21st century COE program, Ministry of Education, Culture, Sports, Science and Technology (15390125, 16591307, 17015017, 17790910), Ministry of Health, Labour and Welfare (15–22), Smoking Research Foundation and Foundation for Promotion of Cancer Research. We thank Drs Katsutoshi Miura, Hisaki Igarashi, Yayoi Kawabata and Kiyoko Nagura for excellent technical assistance.
References
- 1. Hirota S, Isozaki K, Moriyama Y et al. Gain‐of‐function mutations of c‐kit in human gastrointestinal stromal tumors. Science 1998; 279: 577–80. [DOI] [PubMed] [Google Scholar]
- 2. O'Leary T, Berman JJ. Gastrointestinal stromal tumors: answers and questions. Hum Pathol 2002; 33: 456–8. [DOI] [PubMed] [Google Scholar]
- 3. Kindblom LG, Remotti HE, Aldenborg F, Meis‐Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT) gastrointestinal stromal tumors show phenotypic characteristics of the intestinal cells of Cajal. Am J Pathol 1998; 152: 1259–69. [PMC free article] [PubMed] [Google Scholar]
- 4. Lux ML, Rubin BP, Biase TL et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000; 156: 791–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rubin BP, Singer S, Tsao C et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001; 61: 8118–21. [PubMed] [Google Scholar]
- 6. Hirota S, Nishida T, Isozaki K et al. Gain‐of‐function mutation at the extracellular domain of KIT in gastrointestinal stromal tumours. J Pathol 2001; 193: 505–10. [DOI] [PubMed] [Google Scholar]
- 7. Hirota S, Ohashi A, Nishida T et al. Gain‐of‐function mutations of platelet‐derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003; 125: 660–7. [DOI] [PubMed] [Google Scholar]
- 8. Heinrich MC, Corless CL, Duensing A et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003; 299: 708–10. [DOI] [PubMed] [Google Scholar]
- 9. Taniguchi M, Nishida T, Hirota S et al. Effect of c‐kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999; 59: 4297–300. [PubMed] [Google Scholar]
- 10. Sakurai S, Oguni S, Hironaka M, Fukayama M, Morinaga S, Saito K. Mutations in c‐kit gene exons 9 and 13 in gastrointestinal stromal tumors among Japanese. Jpn J Cancer Res 2001; 92: 494–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Antonescu CR, Sommer G, Sarran L et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res 2003; 9: 3329–37. [PubMed] [Google Scholar]
- 12. Ernst SI, Hubbs AE, Przygodzki RM, Emory TS, Sobin LH, O'Leary TJ. KIT mutation portends poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest 1998; 78: 1633–6. [PubMed] [Google Scholar]
- 13. Lasota J, Wozniak A, Sarlomo‐Rikala M et al. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of 200 cases. Am J Pathol 2000; 157: 1091–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lasota J, Jasinski M, Sarlomo‐Rikala M, Miettinen M. Mutations in exon 11 of c‐Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999; 154: 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miettinen M, El‐Rifai WHL, Sobin L, Lasota J. Evaluation of malignancy and prognosis of gastrointestinal stromal tumors: a review. Hum Pathol 2002; 33: 478–83. [DOI] [PubMed] [Google Scholar]
- 16. Joensuu H, Roberts PJ, Sarlomo‐Rikala M et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001; 344: 1052–6. [DOI] [PubMed] [Google Scholar]
- 17. van Osteroom AT, Judson I, Verweij J et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumors: a phase I study. Lancet 2001; 358: 1421–3. [DOI] [PubMed] [Google Scholar]
- 18. Buchdunger E, Cioffi CL, Law N et al. Abl protein‐tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c‐kit and platelet‐derived growth factor receptors. J Pharmacol Exp Ther 2000; 295: 139–45. [PubMed] [Google Scholar]
- 19. Dagher R, Cohen M, Williams G et al. Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res 2002; 8: 3034–8. [PubMed] [Google Scholar]
- 20. Demetri GD, von Mehren M, Blanke CD et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 2000; 231: 51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. George DD, Robert B, Charles DB et al. NCCN task force report: optimal management of patients with gastrointestinal stromal tumor (GIST) − expansion and update of NCCN clinical practice guidelines. J NCCN 2004; 2 (Suppl.): S1–S28 . [PubMed] [Google Scholar]
- 22. Igarashi H, Sugimura H, Maruyama K et al. Alteration of immunoreactivity by hydrated autoclaving, microwave treatment, and simple heating of paraffin‐embedded tissue sections. APMIS 1994; 102: 295–307. [DOI] [PubMed] [Google Scholar]
- 23. Kitayama Y, Igarashi H, Sugimura H. Amplification of FISH signals using intermittent microwave irradiation for analysis of chromosomal instability in gastric cancer. Mol Pathol 1999; 52: 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kitayama Y, Igarashi H, Sugimura H. Different vulnerability among chromosomes to numerical instability in gastric carcinogenesis: stage‐dependent analysis by FISH with the use of microwave irradiation. Clin Cancer Res 2000; 6: 3139–46. [PubMed] [Google Scholar]
- 25. Kitayama Y, Igarashi H, Sugimura H. Initial intermittent microwave irradiation for fluorescence in situ hybridization analysis in paraffin‐embedded tissue sections of gastrointestinal neoplasia. Lab Invest 2000; 80: 779–81. [DOI] [PubMed] [Google Scholar]
- 26. Kitayama Y, Igarashi H, Watanabe F, Maruyama Y, Kanamori M, Sugimura H. Nonrandom chromosomal numerical abnormality predicting prognosis of gastric cancer. a retrospective study of 51 cases using pathology archives. Lab Invest 2003; 83: 1311–20. [DOI] [PubMed] [Google Scholar]
- 27. Kobayashi K, Kitayama Y, Igarashi H et al. Intratumor heterogeneity of centromere numerical abnormality in multiple primary gastric cancers: application of fluorescence in situ hybridization with intermittent microwave irradiation on paraffin‐embedded tissue. Jpn J Cancer Res 2000; 91: 1134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakamura R, Song JP, Isogaki J, Kitayama Y, Sugimura H. Multiple (multicentric and multifocal) cancers in the ipsilateral breast with different histologies: profiles of chromosomal numerical abnormality. Jpn J Clin Oncol 2003; 33: 463–9. [DOI] [PubMed] [Google Scholar]
- 29. Song JP, Kitayama Y, Igarashi H et al. Centromere numerical abnormality in the papillary, papillotubular type of early gastric cancer, a further characterization of a subset of gastric cancer. Int J Oncol 2002; 21: 1205–11. [PubMed] [Google Scholar]
- 30. DeMatteo RP, Lewis JJ, Leung D et al. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 2000; 231: 51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roberts PJ, Eisenberg B. Clinical presentation of gastrointestinal stromal tumors and treatment of operable disease. Eur J Cancer 2002; 38: S37–8. [DOI] [PubMed] [Google Scholar]
- 32. Sturgeon C, Chejfec G, Espat NJ. Gastrointestinal stromal tumors: a spectrum of disease. Surg Oncol 2003; 12: 21–6. [DOI] [PubMed] [Google Scholar]
- 33. Tamborini E, Bonadiman L, Greco A et al. A new mutation in the KIT ATP pocket cause acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology 2004; 127: 294–9. [DOI] [PubMed] [Google Scholar]
- 34. Wakai T, Kanda T, Hirota S, Ohashi A, Shirai Y, Hatakeyama K. Late resistance to imatinib therapy in a metastatic gastrointestinal stromal tumor is associated with a second KIT mutation. Br J Cancer 2004; 90: 2059–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen LL, Trent JC, Wu EF et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res 2004; 64: 5913–19. [DOI] [PubMed] [Google Scholar]
- 36. Debiec‐Rychter M, Cools J, Dumez H et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of PKC412 inhibitor against imatinib‐resistant mutants. Gastroenterology 2005; 128: 270–9. [DOI] [PubMed] [Google Scholar]
- 37. Wardelmann E, Thomas N, Merkelbach‐Bruse S et al. Acquired resistance to imatinib in gastrointestinal stromal tumors caused by multiple KIT mutations. Lancet Oncol 2005; 6: 249–51. [DOI] [PubMed] [Google Scholar]
- 38. Antonescu CR, Besmer P, Guo T et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 2005; 11: 4182–90. [DOI] [PubMed] [Google Scholar]
- 39. El‐Rifai W, Sarlomo‐Rikala M, Andersson LC, Knuutila S, Miettinen M. DNA sequence copy number changes in gastrointestinal stromal tumors: tumor progression and prognostic significance. Cancer Res 2000; 60: 3899–903. [PubMed] [Google Scholar]
- 40. Fukasawa T, Chong JM, Sakurai S et al. Allelic loss of 14q and 22q, NF2 mutation, and genetic instability occur independently of c‐kit mutation in gastrointestinal stromal tumor. Jpn J Cancer Res 2000; 12: 1241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]