

Abstract
Dendritic cells (DC) begin maturation in response to complex stimuli consisting of antigens and pattern molecules (PAMP) for the activation of the immune system. Immune adjuvant usually contains PAMP. Infection represents one event that is capable of inducing such a complex set of stimuli. Recently, DC were subdivided into a number of subsets with distinct cell‐surface markers, with each subset displaying unique differential maturation in response to pattern molecules to induce various types of effector cells. In the present study, we review how pattern recognition molecules and adaptors in each DC subset drive immune effector cells and their effect in the stimulated DC. Although tumor cells harbor tumor‐associated antigens, they usually lack PAMP. Hence, we outline the properties of exogenously‐added PAMP in the modulation of raising tumor immunity. In addition, we describe the mechanism by which DC‐dependent natural killer activation is triggered for the induction of antitumor immunity. (Cancer Sci 2010; 101: 313–320)
Adjuvants are typically administered with target antigens in order to enhance the host immune response. Freund complete adjuvant (FCA), Freund incomplete adjuvant (FIA), and hydrated alumina (alum) are representative adjuvants that are used as antigen conjugates to potentiate immune responses and antibody production in animals. Although the mechanism by which these reagents enhance immunity was not completely understood, it appeared that the addition of adjuvants to antigens potentially induced immunity by “making it dirty”.( 1 ) However, more recently the agonistic features of adjuvants for pattern‐recognition receptors (PRR) have been highlighted based on elucidation of the ligand properties of Toll‐like receptors (TLR) and TLR‐mediated dendritic cell (DC) maturation. The accumulated evidence on TLR‐dependent DC maturation has solidified the current understanding that DC TLR confer the direction of the effector driving on the DC that present antigens. We hold that antigens determine the object toward which immune cells are proliferated, whereas adjuvants determine what effectors will be selected for immunological output.( 2 ) The fundamental concepts of the immune system should be re‐evaluated through the understanding of TLR‐mediated DC immune responses, which will also revolutionize the concepts related to antitumor immunity.
The two major arms of the innate immune signaling pathway, the MyD88 and toll‐interleukin 1 receptor domain (TIR)‐containing adoptor molecule (TICAM‐1) pathways (Fig. 1), have been identified through the investigation of TLR signaling.( 3 ) Although MyD88 is dominant in mammals living on land, most aquatic vertebrates preferentially use TICAM‐1 for TLR signaling.( 4 ) TLR employing MyD88 adaptors usually recognize bacterial patterns, whereas TLR taking TICAM‐1 recognize virus products, including nucleic acids. In addition to these PRR, the retinoic acid‐inducible protein I (RIG‐I)‐like receptor and nucleotide‐binding oligomerization domain‐containing protein (NOD)‐like receptor (NLR) systems are located in the cytoplasm( 5 , 6 ) and are inherent in most animals.( 7 ) PRR systems are also distributed across the cell membrane and cytoplasm. The mineral oil component of FIA, crystallized uric acid, and alum are able to activate the NLR‐inflammasome pathway,( 5 ) which yields interleukin (IL)‐1β and IL‐18. These cytokines in turn stimulate their respective receptors to activate the MyD88 pathway in myeloid DC (mDC).( 8 ) The activation of the MyD88 pathway in mDC is a common feature in bacterial stimulation.
Figure 1.
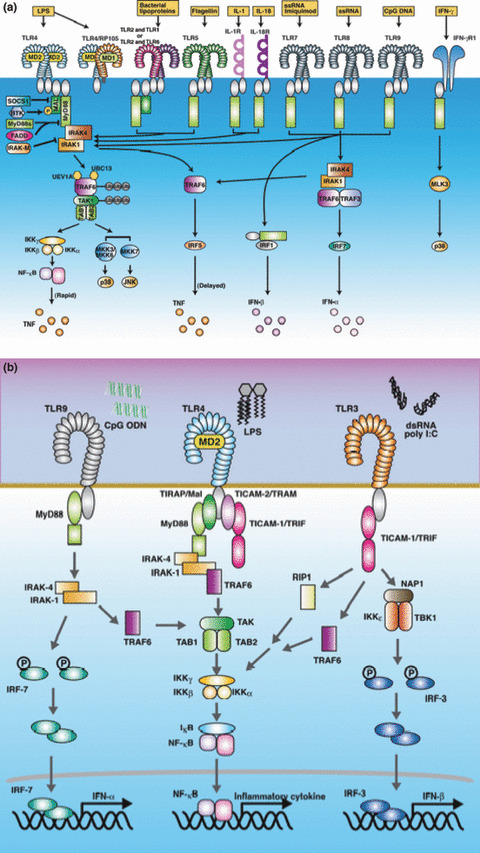
MyD88 and Toll‐interleukin I receptor‐domain (TIR)‐containing adoptor molecule (TICAM‐1) pathways. MyD88 is an adaptor for all Toll‐like receptors (TLR), except TLR3 (a). TLR2 and TLR4 recruit MyD88 via the bridging adaptor Toll‐interleukin I receptor‐domain (TIR)‐containing adoptor protein (TIRAP) (MAL) (b). Other TLR directly recruit MyD88. MyD88 activates nuclear factor‐kappa β (NF‐κB) in most cell types, except plasmacytoid dendritic cells (pDC), which activate the interferan‐regulatory factor (IRF‐7) transcription factor. MyD88 pathway is involved in the production of pro‐inflammatory cytokines in most cells. In contrast, the MyD88 pathway in pDC and the TICAM‐1 pathway in myeloid dendritic cells (DC) activate the type interferon (IFN) promoter via IRF‐3 or IRF‐7 (b). TLR4 can recruit both MyD88 and TICAM‐1, whereas other TLR recruit either of them. Each TLR responds to different agonistic stimuli, as shown in Table 1. DC, dendritic cells; IFN, interferon; IRF, interferon‐regulatory factor; pDC, plasmacytoid DC; TICAM‐1, Toll‐interleukin 1 receptor domain (TIR)‐containing adaptor molecule; TIRAP, Toll‐interleukin 1 receptor (TIR) domain‐containing adapter protein; TLR, toll like receptor.
The MyD88 pathway of plasmacytoid DC (pDC) is unique, as TLR7 and TLR9 predominantly activate interferon‐regulatory factor (IRF)‐7 and induce interferon (IFN)‐α.( 9 ) Human mDC lack TLR7 and TLR9 and the IFN‐inducing MyD88 pathway, although mouse mDC harbor the TLR7 and TLR9 MyD88 pathway, which are inducible by RNA and CpG DNA respectively.( 10 ) In contrast, TICAM‐1 links the type I IFN‐inducing pathways in the mDC of both humans and mice,( 11 ) while TLR3 represents the sensor of dsRNA of viral origin.( 12 ) In addition, viral products, double‐stranded (ds) RNA, and 5’‐triphosphate RNA stimulate the intracytoplasmic helicases melanoma‐differentiation‐associated gene 5 (MDA5) and RIG‐I, which in turn activate the IRF‐3‐ and IRF‐7‐activating kinases (TANK‐binding kinase (TBK1)/I kappa B kinase (IKK)ε.( 5 , 11 ) The adaptor of this pathway is IPS‐1,( 5 ) and it is therefore known as the interferon‐beta promoter stimulator 1 (IPS‐1) pathway. The IPS‐1 pathway shares the downstream signaling components, including the kinases, with the TICAM‐1 pathway to activate the IFN‐inducing pathway.( 13 ) Thus, the representative inflammatory responses in pattern recognition are rooted in the properties of the adaptors in the case of TLR, MyD88, and TICAM‐1. In DC, these pathways play a significant role in differential maturation.
Bacterial and viral pattern molecules revisited
It is known that FCA contains heat‐killed mycobacteria (the causative agent of tuberculosis), which functions as a ligand of TLR.( 14 ) These are MyD88‐dependent properties and the features of the DC maturation profiles with these TLR ligands have been examined (Table 1). Although the toxicity of the TLR agonists is not removed, their role in triggering antitumor immunity, including cytokine‐ and effector‐inducing abilities, are being examined with respect to their practical use for patients with cancer. Alum (aluminum hydroxide) acts as an NLR agonist involving the secondary activation of MyD88( 15 ) and is currently used as a standard adjuvant in humans. However, a sufficient immune potential may not be accomplished with a single stimulation of the NLR system. The adjuvant BCG– cell wall skeleton (CWS), which contains mycolic acid, arabinogalactan, and peptidoglycan (PGN), has been used for patients with cancer, and a good prognosis was reported after BCG–CWS treatment.( 16 ) This adjuvant contains muramyl dipeptide (MDP) as a center for the activation of TLR2 and TLR4 and also involves MyD88 activation.( 17 ) The DC maturation profile induced by BCG‐CWS is comparable to that induced by Pam2 peptides that activates TLR2 (4) BCG–CWS does not contain DNA, which excludes the possibility of activating TLR9. Only rare examples of fatal shock and interstitial pneumonia have been reported with BCG–CWS that stimulates TLR2 and TLR4.( 18 )
Table 1.
Human TLR and pattern molecules with MyD88‐ or Toll‐interleukin I receptor domain (TIR)‐containing adoptor molecule (TICAM‐1)‐activating properties
| Human TLR | Ligands |
|---|---|
| TLR1 | Pam3 |
| TLR2 | Pam2, Pam3, PGN |
| TLR3 | dsRNA |
| TLR4 | LPS, virus fusion units |
| TLR5 | Flagellin |
| TLR6 | Pam2 |
| TLR7 | ssRNA |
| TLR8 | ssRNA |
| TLR9 | CpG DNA |
| TLR10 | – |
| MyD88 activators (Lipoproteins, PGN) | Reference |
|---|---|
| M161Ag (MALP‐2) | ( 62 ) |
| TAN33 | ( 63 ) |
| OM‐174 | ( 64 ) |
| BCG‐CWS (Azuma lot) | ( 22 ) |
| SMP105 | ( 65 ) |
| TICAM‐1 activators (RNA, lipid A) | |
| DI RNA (stem loop) | ( 66 ) |
| Poly(A:U) | ( 67 ) |
| Poly(I:C12U) | ( 68 ) |
| PolyI:C(LC) | ( 20 ) |
| MPLA | ( 21 ) |
| Anti‐human TLR monoclonal antibodies | ||
|---|---|---|
| TLR1 | TLR1.136 | ( 58 ) |
| TLR2 | TLR2.45 | ( 59 ) |
| TLR3 | TLR3.7 | ( 60 ) |
| TLR4 | HTA125 | ( 61 ) |
| TLR6 | TLR6.127 | ( 58 ) |
dsRNA, double‐stranded RNA; TLR, Toll‐like receptor. SMP105 is a lot of BCG‐CWS that activates only TLR2.
In contrast, viral products, including dsRNA (and its analog polyI:C), and the lipopolysaccharide (LPS) of Gram‐negative bacteria were identified as TLR ligands with TICAM‐1 agonistic function.( 3 ) dsRNA and LPS stimulate TLR3 and TLR4, respectively, both of which link the adaptor TICAM‐1.( 3 , 11 ) As they activate nuclear factor (NF)‐κB and IRF‐3, cytokine storm (hypercytokinemia) or endotoxin‐like shock tends to occur in vivo.( 19 ) It is therefore mandatory to reduce their toxic properties before they are applied to human patients. Importantly, polyI:CLC (TLR3‐complexed poly inosinic: polycytidylic (IC) with carboxymethylcellulose and poly‐L‐lysine to improve resistance to ribonucleases (i.e. TLR3),( 20 ) and monophosphoryl lipid A (i.e. TLR4)( 21 ) have been considered promising candidates for immunotherapy. These TLR agonists mainly stimulate the TICAM‐1 pathway without the robust activation of the MyD88 pathway( 20 , 21 ) and rarely induce side‐effects, such as cytokine storms, skin festering, and the symptoms of inflammation during preclinical trials. It is important that the differential view of the MyD88 and TICAM‐1 adjuvants in terms of their DC maturation and effector‐driving properties be examined. The development of TLR agonists with properties superior to those of alum can be expected to be revealed through these studies. In this review, the molecular mechanisms of effector activation by DC TLR are outlined and discussed.
Adjuvants stimulate tumor‐associated myeloid cells and DC
We have speculated from in vitro studies that immature mDC are matured to antigen‐presenting mDC by BCG–CWS, a TLR2 agonist,( 22 ) which also induces a variety of immune effector cells, including CD8+ T cells (CTL)( 23 ) and NK cells.( 24 ) These effector cells can damage tumor cells under high effector target (E/T) ratios in vitro.( 23 , 24 ) Indeed, tumor B16 mutanoma growth is retarded in tumor‐bearing mice (C57BL/6) when BCG–CWS‐matured mDC or secondary‐induced CTL are injected in the area surrounding the tumor. It is the CTL, but not NK, cells that are the main effector responsible for tumor regression in vivo.( 23 ) Unexpectedly, however, the immune cells which infiltrate into the tumor largely consist of macrophages and not lymphocytes or mDC in mouse models (Shime H and Seya T, unpublished observation, 2009). The properties of these macrophages remain experimentally undetermined. As the tumor‐infiltrating macrophages contain many subsets, and some of them often possess immune suppressing properties,( 25 ) these macrophages could be related to myeloid‐derived suppressor cells (MDSC) and act as inflammation inducers to sustain tumor growth. Thus, BCG‐CWS‐mediated functional modification of these macrophages and their effect on tumor growth in mice remains to be determined. Specific questions also remain concerning adjuvant administration to patients. How myeloid cells mature to DC after they are phagocytozing tumor‐associated antigens, how mature mDC are located by effector cells, and how tumors regress in such situations still remain unanswered.
Treg, a regulatory population of CD4 T cells, has an inhibitory activity against antitumor immunity( 26 ) and has been shown to inhibit CD8 CTL tumoricidal activity in vitro.( 26 ) Several reports indicate that Treg cells infiltrate into tumors and support tumor progression.( 27 , 28 ) However, mDC are present at very low levels in the tumor masses where Treg cells invade. Again, the functional modulation of Treg cells in the local tumor environment by adjuvants or mDC is unclearly illustrated.
Recently, several myeloid cell populations have been discovered that are associated with tumor cell progression, including interferon‐producing killer DC (IKDC),( 29 ) MDSC,( 25 , 30 ) and tumor‐associated macrophages (TAM).( 31 ) Although the maturation or activation of these myeloid cells is likely crucial for tumor progression, only a few reports have investigated their maturation mechanism and effect on tumors by adjuvant treatment. Early‐acting pattern molecules can act on tumor cells to release late‐acting substances. In fact, damage‐associated molecular patterns (DAMP), such as high‐modify group box protein (HMGB1), uric acids, heat‐shock protein (HSP), and DNA complexes,( 32 ) are secondary liberated from tumors, and stimulate the TAM. Whether these stimuli alter the tumor‐progressing ability of the macrophages should be a point of consideration for adjuvant therapy. The types of TLR present in these myeloid cells and the effect of administered adjuvants are topics that need to be investigated.
Many studies on TLR knockout mice allowed us to describe the properties of mouse bone marrow‐derived DC (BMDC) treated with a variety of adjuvants( 33 ) and to show the points for induction of immune effector cells through the adjuvant immunotherapy of cancer. Ambivalent functions between mDC and MDSC in a tumor environment can affect the conformation of antitumor immunity.
MyD88‐ and TICAM‐1‐mediated DC maturation
Soon after the discovery of the TLR,( 34 ) it was shown that TLR agonists have a DC maturation activity.( 35 ) DC maturation is characterized by TLR adaptors, which have common features, including the upregulation of major histocompatibility complex (MHC), costimulators and NK‐activating ligands, and the following features which are unique to each adaptor in mDC.( 36 ) MyD88‐dependent DC maturation has two modes, with NK activation and CTL induction occurring concomitantly with the activation of NF‐κB, followed by the induction of inflammatory cytokines.( 37 ) Using BCG–CWS as an adjuvant for the TLR2 agonist, we examined how the TLR2 agonist acts on mDC and tumor cells.( 23 ) While NK activation by MyD88 is feasible in vitro, TLR2 agonists exhibit minimal NK‐mediated tumor‐suppression activity in tumor‐implant mice.( 24 ) The TLR2‐dependent antitumor NK activity is abrogated in MyD88−/− mice, suggesting the presence of a NK‐activation pathway via MyD88.( 24 ) However, following an in vitro analysis, it was revealed that TLR2–MyD88 in NK cells, but not in mDC, is rather dominant in this mode of NK activation, and that activated NK cells barely enter the tumor mass. For this reason, the subcutaneous administration of BCG–CWS marginally retards tumor growth in mice via the activation of NK cells.
In contrast, mDC maturation is accompanied with potent antigen presentation secondary to cross‐priming in TLR2‐primed mDC.( 23 ) Tumor antigen‐specific CTL induction is facilitated in mice with an implant tumor burden, concomitant with the retardation of tumor growth. This CTL induction is MyD88 dependent, since TLR2‐mediated cross‐priming does not occur in MyD88−/− mDC. Neither CTL induction nor the retardation of tumor growth significantly occurs in MyD88‐deficient mice. Thus, MyD88 in mDC preferentially participates in cross‐priming and driving CTL in vivo. The downstream molecules of MyD88 associated with mDC CTL driving are unknown.
In the present study, we used polyI:C for evaluating the TICAM‐1 potential in mDC maturation and antitumor immunity.( 38 ) The TICAM‐1 pathway allows mDC to activate IRF‐1 and IRF‐3, which in turn activate the IFN‐β promoter, as well as unidentified antitumor factors (Fig. 1). The data imply that cross‐priming and the NK‐driving signal are also dependent upon TICAM‐1, but the transcription factors utilized by TICAM‐1 are wholly distinct from those of MyD88. The search for the molecules that participate in the TICAM‐1 CTL driving is underway, and a molecule downstream of IRF‐1, but not IRF‐3, has been shown to be crucial for in vivo CTL induction. In contrast, TICAM‐1‐mediated antitumor NK activation largely relies on the IRF‐3‐derived NK‐activating molecule (INAM), in addition to the reported cytokines IL‐15, IFN‐α, and IL‐12p70.( 39 )
MyD88 and TICAM‐1 activate different signaling platforms for the recruitment of second adaptors.( 3 ) In mDC, TLR2 and TLR4 recruit the combined adaptor Mal/Toll‐interleukin I receptor domain‐containing adoptor protein (TIRAP)–MyD88 to signal the transcription factor NF‐κB.( 4 ) In contrast, TLR3 and TLR4 can utilize TICAM‐1 as the adaptor.( 3 ) TLR4 recruits the combined adaptor Toll‐IL‐IR domain‐containing adoptor inducing IFN‐beta‐related adaptor molecule (TRAM) (TICAM‐2)–TICAM‐1 while TLR3 directly recruits TICAM‐1 for signaling.( 3 ) TLR4 is unique in that it uses both MyD88 and TICAM‐1 adaptors (Fig. 1). The classic example in which both routes are activated is during LPS‐induced endotoxic shock.( 40 ) Like BCG‐CWS and PolyI:C, activation of either one route would be required for a condition of less toxic adjurants. Studies of the TICAM‐1 signalosome suggest that upon TLR3 activation, TICAM‐1 recruits a variety of molecules as secondary adaptors, including NAK‐associated protein I (NAP1),( 41 ) receptor‐interacting protein I (RIP1),( 42 ) similar to NAP I TBK adoptor (SINTBAD),( 43 ) adenovirus 5 E1A‐binding protein (BS69),( 44 ) and TNF receptor‐associated factor (TRAF) family proteins.( 45 ) Whether or not these molecules are associated with antitumor CTL or NK induction remains to be determined.
The mode by which mDC are matured differs in the MyD88 and the TICAM‐1 pathways. The TICAM‐1 pathway preferentially induces IL‐12 and type I IFN in mDC and drives NK activation.( 38 ) Type I IFN induction by MyD88 has been observed only in pDC.( 9 , 10 ) In contrast, mDC MyD88 strongly induces pro‐inflammatory cytokines, such as tumor necrosis factor‐α, IL‐1β, and IL‐6.( 9 , 46 ) The molecular mechanism that facilitates the cross‐presentation ability in mDC is currently unknown.
DC subsets and TLR expression
BMDC (representative of mDC) and pDC can be prepared from mouse bone marrow cells by using granulocyte‐macrophage colony‐stimulating factor (GM–CSF) and the Flt3 ligand( 46 ) while Langerhans cells can be generated by the addition of transforming growth factor‐β to GM–CSF and IL‐4.( 47 ) The DC subsets in the spleen and the intestinal tract can be separated using a flow cytometry (FACS) sorter. The characteristics of these mouse DC subsets have been described previously.( 36 ) In humans, monocyte‐derived DC can be used as mDC, but their characteristics are somewhat different from mDC prepared in the peripheral blood using the mDC marker plasmocytoid DC antigen (PDCA1). Human peripheral blood pDC can be isolated from whole blood using PDCA4.
The distribution of TLR of the DC subset were examined by using human TLR‐specific monoclonal antibodies generated in our laboratory, and the TLR repertoires of monocyte‐derived DC and pDC were determined (Table 2). The TLR distribution roughly resembles mouse DC, although a clear result could not be obtained with mouse BMDC and pDC because of a lack of appropriate specific antibodies against mouse TLR.( 36 ) The discrepancy of appropriate TLR7 levels in mouse BMDC and human mDC could be a result of differences in the inducible nature of mouse, but not human, TLR7. It was also shown that human mDC express TLR8, while mouse mDC do not.( 36 ) The failure of CpG DNA to raise effective antitumor immunity can be attributable to the low or absent induction of TLR9 in human mDC, unlike the situation in mouse mDC.
Table 2.
TLR expression profiles in human DC subsets
| Freshly isolated | In vitro‐differentiated | ||||
|---|---|---|---|---|---|
| Monocytes | mDC* | pDC** | DCs | Macrophages | |
| TLR1 | ++ | + | − | + | ++ |
| TLR2 | ++ | ++ | − | ++ | ++ |
| TLR3 | − | ++ | − | ++ | + |
| TLR4 | ++ | + | − | + | + |
| TLR6 | ++ | + | − | + | + |
| TLR7 | − | − | + | − | − |
| TLR8 | + | + | − | + | + |
| TLR9 | − | − | + | − | − |
Positive and negative symbols denote the results of the flow cytometry (FACS) analyses using monoclonal antibodies, except TLR7, TLF8, and TLR9. Results were determined by reverse transcription–polymerase chain reaction. TLR3, TLR7, TLR8, and TLR9 reside in the endosome to recognize nucleotide derivatives. (*) PDCA1+ cells; (**) PDCA4+ cells. PDCA, plasmacytoid dendritic cell antigen; DC, dendritic cell; mDC, myeloid dendritic cells; pDC, plasmacytoid dendritic cells; TLR, Toll‐like receptor.
DC subsets and effector induction
CTL and NK cells can be induced by mature mDC, with or without the presentation of MHC class I antigens, while CD4 T‐cell subsets are induced by the presentation of class II antigens. In addition to CTL and NK cells, the tumor‐modulating functions of Th1, Th2, Th17, and Treg were evaluated (Fig. 2). NK activation is a result of the balance between NK‐activating and inhibitory ligands on mDC. NK cells can also be activated with cytokines, such as IL‐2, IL‐15, IFN‐α/β, and IL‐12.( 48 ) CTL is a result of the activation of the CD8+ T cell by the presentation of class I antigens on mDC. Other effectors are the result of the activation of CD4+ T cells by MHC class II antigen presentation on mDC. A master transcription factor in addition to T‐bet, GATA‐3, RORgT, and Foxp3 are known to exist for Th1, Th2, Th17, and each Treg on the CD4 lymphocyte side.( 49 ) However, there is little information concerning the mDC properties driving these effector cells.
Figure 2.
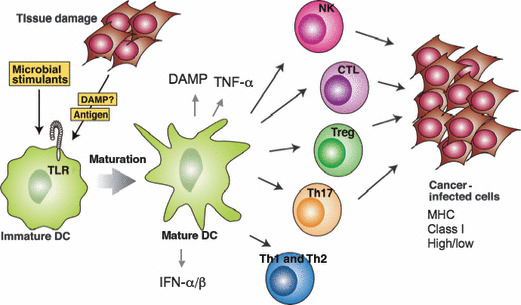
Selective induction of immune effector lymphocytes by different agonistic stimuli. Each pattern molecule (PAMP) has its own uniqueness in myeloid dendritic cell (mDC) maturation. Differential maturation of mDC results in different effector driving as shown. CD8 T, various CD4 T, and B cells are proliferated by myeloid DC (mDC) with different properties. Tumor regression is a marker for evaluating which lymphocytes are activated in response to pathogen‐associated molecular patterns (PAMP).
Each DC subset seems to correspond to a specific effector, although the selection mechanism by which DC induce various effectors is not clear in most instances. However, it is known that CD8+ DC induce Treg( 50 ) and NK cells( 51 ) in the mouse spleen, and lamina propria pDC in the mouse enteric canal promotes immunoglobulin A production.( 52 ) In addition, CD70+/CD11c+ DC induce Th17 cells by the adenosine tri phosphate (ATP) of enterobacteria,( 53 ) and BMDC activate NK cells via the TICAM‐1 pathway.( 54 ) Further examples of DC subsets that preferentially function with specific effectors will likely be demonstrated through practical experiments.
Mechanism of DC‐mediated antitumor NK activation
It has been reported that BMDC drive antitumor NK activation in a TICAM‐1‐dependent manner.( 38 , 54 ) This NK activation does not rely upon a soluble factor, such as a cytokine, but instead was generated by BMDC–NK cell–cell contact.( 39 ) Therefore, there must be an NK‐activating molecule that is induced on the BMDC surface in response to TICAM‐1 signaling (Fig. 3a). We focused our attention on this key molecule, which is crucial for antitumor NK immunity and found that DC‐mediated NK activation occurred normally in IRF‐7 −/− BMDC stimulated with polyI:C, but this response was absent in IRF‐3 −/− BMDC.( 39 ) Therefore, the putative NK‐driving signal in mDC involves transcription factor IRF‐3 downstream of the activated TICAM‐1. Ultimately, the NK activation molecule was identified using a screening method in which candidate molecules were expressed in IRF‐3 −/− BMDC using a lentiviral vector.( 39 ) We named this molecule INAM. When INAM was expressed in mDC, it promoted NK activation in the mixture of mDC (expressing INAM) and NK cells; however, INAM did not exhibit an NK‐activating function on BaF3 cells. INAM is an NK‐activating molecule peculiar to BMDC whose TICAM‐1 has been activated, and there have been no reports suggesting the presence of this kind of molecule until recently (Fig. 3b). In BMDC, INAM receives a sugar chain modification by a similar membrane protein to tetraspanin with a molecular weight of 45 kDa. INAM is distributed in the spleen and lymph nodes, and is actually expressed by a variety of lymphocyte subsets present in the lymph nodes. It has been predicted to make a loop card structure on the surface of the cell in two portions based on the amino acid sequence.( 39 )
Figure 3.
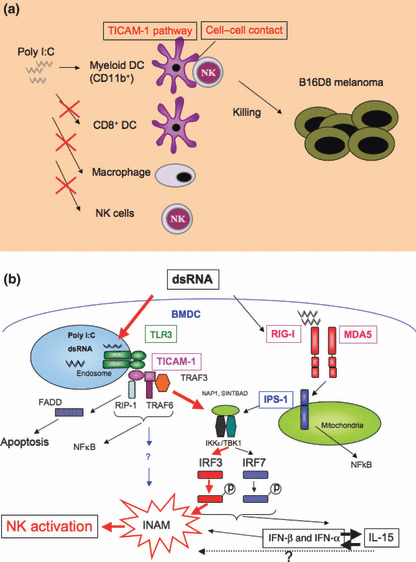
A molecular mechanism of myeloid dendritic cell (mDC)‐mediated natural killer (NK) activation. (a) CD11b+ bone marrow‐derived dendritic cells (BMDC) act for natural killer (NK) activation by double‐stranded (ds) RNA. NK cells express tumoricidal activity against major histocompatibility complex (MHC) low implant tumors if they are primed by polyI:C plus bone marrow‐derived dendritic cells (BMDC), but not other myeloid cells. Dendritic cell–NK cell–cell contact is essential for the induction of polyI:C‐mediated antitumor NK cells. (b) Route for mDC maturation for the induction of NK activation.( 39 )
It is predicted that INAM is related to the composition of immune synapses in the BMDC–NK contact. When BMDC, which forcibly express INAM, are prepared and adoptively transferred around the tumors of tumor‐bearing mice, the tumor is efficiently regressed. These results suggest that INAM is the factor directly responsible for driving antitumor NK activation. Humans have an ortholog of INAM, although its distribution profile appears to be somewhat different than to that of mice.
Points to trigger antitumor immune potential
Effector tumor cell–cell contact is essential for tumor damage by immune effector cells. The material liberated from cancer cells on one side generates the modulators of the PRR of mDC and influences the trigger of effector induction. The host molecules that modulate PRR are the previously‐mentioned DAMP.( 32 ) For effective tumor damage, the effector must reach the tumor mass. A suitable strategy is needed for determining the basic factor(s) of the immune response involved in cancer, and can be achieved by using immunomodulatory reagents and gene‐disrupted mice with abrogated TLR pathways.
We have analyzed how BMDC acquire effector‐driving functions by focusing on the innate immune response. The results suggest that PRR stimuli become a trigger that leads to the alteration of precancerous cells to the malignant form. However, PRR are indispensable to the activation of antitumor immunity. In both cases, myeloid cells are intimately involved in the process of tumor–immune cell interaction. Indeed, BCG has high therapeutic potential for patients with bladder transitional epithelial cancer,( 55 ) but it has less of an effect on a variety of other solid cancers. This discrepancy can be rooted in the fact that myeloid cells interact with tumor cells with ambivalent reaction profiles (Fig. 4). An effective strategy for tackling the issue of immune abnormality has yet to be proposed, and even the fundamental immune aberrance present in the microenvironment of tumors is not generally recognized by researchers. It has been speculated that tumor cells produce cytokines that modulate the inflammatory environment as tumor develops. When tumor is surgically excised, many constitutional accidents are often diminished,( 56 ) which can reflect the fact that tumors develop concomitantly with immune modulation. It has become clear that some modulating factors of the innate immune system, such as DAMP, cause cancer‐mediated idiosyncrasies (Fig. 2).
Figure 4.
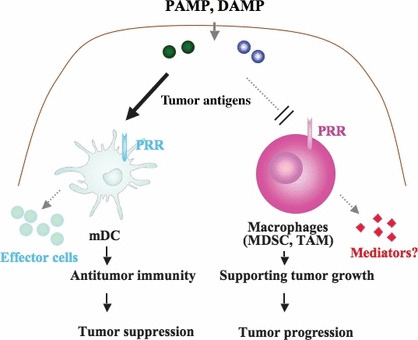
Diverged functions of myeloid cells in tumor mass. A variety of myeloid subsets reside in tumor masses. Some of the subsets exhibit an immune suppressive feature that facilitates escape of tumor cells from immune effectors. Since pattern molecules (PAMP) act on both myeloid dendritic cells (mDC) and myeloid‐derived immune suppressing cells, complicated immune responses occur in tumors. Selective maturation of mDC circumventing the exacerbation of tumor progression by myeloid suppressor cells should be considered as adjuvant therapy of cancer. DAMP, damage‐associated molecular patterns; MDSC, myeloid‐derived suppressor cells; PRR, pattern‐recognition receptors; TAM, tumor‐associated macrophages.
Up until now, the effectiveness of cancer immunotherapy has been primarily evaluated based on tumor regression and the survival prognosis of patients. A representative study involved the evaluation of peptide vaccine therapy for cancer treatment. According to the report by Rosenberg,( 57 ) the peptide vaccine administered to melanoma patients had an effective rating of approximately 2.6%. For future studies, it is necessary to determine the potential of peptide‐conjugating materials, including adjuvants and inflammation‐inducing reagents.( 20 ) A number of reports have suggested that adjuvants can greatly increase the efficiency rate of treatment, although the criteria is prerequistie to fairly evaluate the function of adjuvants in cancer patients.
The method for stimulating DC needs to be carefully selected, as the systemic administration of inflammation‐inducing material can also lead to the acceleration or invasion of developing malignancies at the same time (Fig. 4). The adoptive transfer of adjuvant‐treated mDC to patients is a promising choice; however, it might be difficult for this treatment to be adapted by the Japanese health insurance system. The molecular manipulation of a specific PRR in DC that is involved in effector driving can lead to effective treatment with minimal side‐effects. In this case, the route and molecule that selectively raises the degree of DC maturation without enhancing MDSC should be clarified. If the inflammatory signals that promote carcinogenesis are properly controlled using adjuvants, the design of DC maturation can be manipulated without helping tumor progression. The search for the functional molecule of antitumor effector induction in mDC will help establish an effective treatment of cancer and facilitate the evaluation of the efficacy of peptide vaccines. In the future, we hope that through continued research, cancer patients will have access to convenient and highly effective immunotherapy.
Acknowledgments
This work was supported in part by Grants‐in‐Aid from the Ministry of Education, Science, and Culture (Specified Project for Advanced Research) and the Ministry of Health, Labor, and Welfare of Japan by the Akiyama Foundation and Yakulto Foundation. Financial support from the Sapporo Biocluster “Bio‐S” Knowledge Cluster Initiative of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) is gratefully acknowledged. We are grateful to our laboratory members for their critical discussions, and Ms R. Hatsugai and Ms H. Sato in our laboratory for secretarial assistance.
References
- 1. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology Cold Spring Harb Symp Quant Biol 1989; 54: 1–13. [DOI] [PubMed] [Google Scholar]
- 2. Seya T, Matsumoto M. The extrinsic RNA‐sensing pathway for adjuvant immunotherapy of cancer. Cancer Immunol Immunother 2009; 58: 1175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Akira S. Toll‐like receptor signaling. J Biol Chem 2003; 278: 38105–8. [DOI] [PubMed] [Google Scholar]
- 4. Seya T, Akazawa T, Tsujita T, Matsumoto M. Role of Toll‐like receptors in adjuvant‐augmented immune therapies. Evid Based Complement Alternat Med. 2006; 3: 31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoneyama M, Onomoto K, Fujita T. Cytoplasmic recognition of RNA. Adv Drug Deliv Rev 2008; 60: 841–6. [DOI] [PubMed] [Google Scholar]
- 6. Martinon F, Gaide O, Pétrilli V, Mayor A, Tschopp J. NALP inflammasomes: a central role in innate immunity. Semin Immunopathol 2007; 29: 213–29. [DOI] [PubMed] [Google Scholar]
- 7. Ishii A, Kawasaki M, Matsumoto M, Tochinai S, Seya T. Phylogenetic and expression analysis of amphibian Xenopus Toll‐like receptors. Immunogenetics 2007; 59: 281–93. [DOI] [PubMed] [Google Scholar]
- 8. De Gregorio E, D’Oro U, Wack A. Immunology of TLR‐independent vaccine adjuvants. Curr Opin Immunol 2009; 21: 339–45. [DOI] [PubMed] [Google Scholar]
- 9. Honda K, Taniguchi T. Toll‐like receptor signaling and IRF transcription factors. IUBMB Life 2006; 58: 290–5. [DOI] [PubMed] [Google Scholar]
- 10. Hoshino K, Kaisho T. Nucleic acid sensing Toll‐like receptors in dendritic cells. Curr Opin Immunol 2008; 20: 408–13. [DOI] [PubMed] [Google Scholar]
- 11. Matsumoto M, Seya T. TLR3: interferon induction by double‐stranded RNA including poly(I:C). Adv Drug Deliv Rev 2008; 60: 805–12. [DOI] [PubMed] [Google Scholar]
- 12. Matsnmoto M, Funami K, Oshiumi H, Seya T. Toll‐like receptor 3: a link between toll‐ like receptor, interferon and viruses. Microbiol Immunol. 2004; 48: 147–54. [DOI] [PubMed] [Google Scholar]
- 13. Sasai M, Shingai M, Funami K et al. NAK‐associated protein 1 participates in both the TLR3 and the cytoplasmic pathways in type I IFN induction. J Immunol 2006; 177: 8676–83. [DOI] [PubMed] [Google Scholar]
- 14. Freund J, Stone SH. The effectiveness of tuberculo‐glycolipid as an adjuvant in eliciting allergic encephalomyelitis and aspermatogenesis. J Immunol 1959; 82: 560–7. [PubMed] [Google Scholar]
- 15. Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 2008; 453: 1122–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yasumoto K, Manabe H, Yanagawa E et al. Nonspecific adjuvant immunotherapy of lung cancer with cell wall skeleton of Mycobacterium bovis Bacillus Calmette‐Guérin. Cancer Res 1979; 39: 3262–7. [PubMed] [Google Scholar]
- 17. Uehori J, Fukase K, Akazawa T et al. Dendritic cell maturation induced by muramyl dipeptide (MDP) derivatives: monoacylated MDP confers TLR2/TLR4 activation. J Immunol 2005; 174: 7096–103. [DOI] [PubMed] [Google Scholar]
- 18. Diner EK, Verghese M. Interstitial pneumonitis secondary to intravesical bacillus calmette‐guerin for carcinoma in‐situ of the bladder. Int Braz J Urol 2004; 30: 400–2. [DOI] [PubMed] [Google Scholar]
- 19. Absher M, Stinebring WR. Toxic properties of a synthetic doublestranded RNA. Endotoxin‐like properties of poly I. C, an interferon stimulator. Nature 1969; 223: 715–7. [DOI] [PubMed] [Google Scholar]
- 20. Longhi MP, Trumpfheller C, Idoyaga J et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med 2009; 206: 1589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mata‐Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF‐biased agonist of TLR4. Science 2007; 316: 1628–32. [DOI] [PubMed] [Google Scholar]
- 22. Tsuji S, Matsumoto M, Takeuchi O et al. Maturation of human dendritic cells by cell wall skeleton of Mycobacterium bovis bacillus Calmette‐Guérin: involvement of toll‐like receptors. Infect Immun 2000; 68: 6883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akazawa T, Masuda H, Saeki Y et al. Adjuvant‐mediated tumor regression and tumor‐specific cytotoxic response are impaired in MyD88‐deficient mice. Cancer Res 2004; 64: 757–64. [DOI] [PubMed] [Google Scholar]
- 24. Fujimoto Y, Hashimoto M, Furuyashiki M, Katsumoto M, Seya T, Suda Y, Fukase K. Lipopeptides from Staphylococcus aureus as Tlr2 Ligands: prediction with mrna expression, chemical synthesis and immunostimulatory activities. Chembiochem 2009; 10: 2311–5. [DOI] [PubMed] [Google Scholar]
- 25. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid‐derived suppressor cells in tumor‐bearing mice. J Immunol 2008; 181: 5791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cao X. Regulatory T cells and immune tolerance to tumors. Immunol Res. 2009; Sep 10. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 27. Chaput N, Conforti R, Viaud S, Spatz A, Zitvogel L. The Janus face of dendritic cells in cancer. Oncogene 2008; 27: 5920–31. [DOI] [PubMed] [Google Scholar]
- 28. Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T‐cell recognition: molecular mechanisms and functional significance. Adv Immunol 2000; 74: 181–273. [DOI] [PubMed] [Google Scholar]
- 29. Ullrich E, Bonmort M, Mignot G et al. Therapy‐induced tumor immunosurveillance involves IFN‐producing killer dendritic cells. Cancer Res 2007; 67: 851–3. [DOI] [PubMed] [Google Scholar]
- 30. Nagaraj S, Gabrilovich DI. Tumor Escape Mechanism Governed by Myeloid‐Derived Suppressor Cells. Cancer Res 2008; 68: 2561–3. [DOI] [PubMed] [Google Scholar]
- 31. Gallina G, Dolcetti L, Serafini P et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest 2006; 116: 2777–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 2008; 8: 279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Akira S, Takeda K. Functions of toll‐like receptors: lessons from KO mice. C R Biol 2004; 327: 581–9. [DOI] [PubMed] [Google Scholar]
- 34. Medzhitov R, Preston‐Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997; 388: 394–7. [DOI] [PubMed] [Google Scholar]
- 35. Reise Sousa C. Toll‐like receptors and dendritic cells: for whom the bug tolls. Semin Immunol 2004; 16: 27–34. [DOI] [PubMed] [Google Scholar]
- 36. Iwasaki A, Medzhitov R. Toll‐like receptor control of the adaptive immune responses. Nat Immunol 2004; 5: 987–95. [DOI] [PubMed] [Google Scholar]
- 37. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol 2006; 311: 17–58. [DOI] [PubMed] [Google Scholar]
- 38. Akazawa T, Ebihara T, Okuno M et al. Antitumor NK activation induced by the Toll‐like receptor 3‐TICAM‐1 (TRIF) pathway in myeloid dendritic cells. Proc Natl Acad Sci U S A 2007; 104: 252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ebihara T, Azuma M, Oshiumi H, Taniguchi T, Matsumoto M, Seya T Identification of INAM, a polyI:C‐inducible membrane protein, that participates in dendritic cell‐mediated natural killer cell activation. J Exp Med. 2010; (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88‐deficient mice to endotoxin. Immunity 1999; 11: 115–22. [DOI] [PubMed] [Google Scholar]
- 41. Sasai M, Oshiumi H, Matsumoto M et al. Cutting Edge: NF‐kB‐activating kinase‐associated protein 1 participates in TLR3/Toll‐IL‐1 homology domain‐containing adapter molecule‐1‐mediated IFN Regulatory Factor 3 activation. J Immunol 2005; 174: 27–30. [DOI] [PubMed] [Google Scholar]
- 42. Meylan E, Burns K, Hofmann K et al. RIP1 is an essential mediator of Toll‐like receptor 3‐induced NF‐kappa B activation. Nat Immunol 2004; 5: 503–7. [DOI] [PubMed] [Google Scholar]
- 43. Ryzhakov G, Randow F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1‐binding domain with NAP1 and TANK. EMBO J 2007; 26: 3180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takaki H, Oshiumi H, Sasai M, Kawanishi T, Matsumoto M, Seya T. Oligomerized TICAM‐1(TRIF) in the cytoplasm recruits nuclear Bs69 to enhance NF‐kappaB activation and type I IFN induction. Eur J Immunol. 2009; 39: 3469–76. [DOI] [PubMed] [Google Scholar]
- 45. Sasai M, Oshiumi H, Funami K, Matsumoto M, Seya T Direct binding of TRAF2 and TRAF6 to TICAM‐1/TRIF adaptor of the Toll‐like receptor 3/4 pathway. Molec. Immunol 2010; (in press). [DOI] [PubMed] [Google Scholar]
- 46. Honda K, Ohba Y, Yanai H et al. Spatiotemporal regulation of MyD88‐IRF‐7 signalling for robust type‐I interferon induction. Nature 2005; 434: 1035–40. [DOI] [PubMed] [Google Scholar]
- 47. Geissmann F, Prost C, Monnet JP, Dy M, Brousse N, Hermine O. Transforming growth factor beta1, in the presence of granulocyte/macrophage colony‐stimulating factor and interleukin 4, induces differentiation of human peripheral blood monocytes into dendritic Langerhans cells. J Exp Med 1998; 187: 961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu J, Lanier LL. Natural killer cells and cancer. Adv Cancer Res 2003; 90: 127–56. [DOI] [PubMed] [Google Scholar]
- 49. Chen Z, O’Shea JJ. Th17 cells: a new fate for differentiating helper T cells. Immunol Res 2008; 41(2): 87–102. [DOI] [PubMed] [Google Scholar]
- 50. Yamazaki S, Dudziak D, Heidkamp GF et al. CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol 2008; 181: 6923–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Miyake T, Kumagai Y, Kato H et al. Poly I:C‐induced activation of NK cells by CD8 alpha+ dendritic cells via the IPS‐1 and TRIF‐dependent pathways. J Immunol 2009; 183: 2522–8. [DOI] [PubMed] [Google Scholar]
- 52. Tezuka H, Abe Y, Iwata M et al. Regulation of IgA production by naturally occurring TNF/iNOS‐producing dendritic cells. Nature 2007; 448: 929–33. [DOI] [PubMed] [Google Scholar]
- 53. Atarashi K, Nishimura J, Shima T et al. ATP drives lamina propria T(H)17 cell differentiation. Nature 2008; 455: 808–12. [DOI] [PubMed] [Google Scholar]
- 54. Ebihara T, Azuma M, Oshiumi H et al. Hepatitis C virus‐infected hepatocytes extrinsically modulate dendritic cell maturation to activate T cells and natural killer cells. Hepatology 2008; 48: 48–58. [DOI] [PubMed] [Google Scholar]
- 55. Alexandroff AB, Jackson AM, O’Donnell MA, James K. BCG immunotherapy of bladder cancer: 20 years on. Lancet 1999; 353: 1689–94. [DOI] [PubMed] [Google Scholar]
- 56. Kodama K, Higashiyama M, Takami K et al. Innate immune therapy with a Bacillus Calmette‐Guérin cell wall skeleton after radical surgery for non‐small cell lung cancer: a case‐control study. Surg Today 2009; 39: 194–200. [DOI] [PubMed] [Google Scholar]
- 57. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10: 909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nakao Y, Funami K, Kikkawa S et al. Surface‐expressed TLR 6 Participates in the Recognition of Diacylated Lipopeptide and Peptidoglycan in Human Cells. J Immunol 2005; 174: 1566–73. [DOI] [PubMed] [Google Scholar]
- 59. Uehori J, Matsumoto M, Tsuji S et al. Simultaneous blocking of human Toll‐like receptor 2 and 4 suppresses myeloid dendritic cell activation induced by Mycobacterium bovis bacillus Calmette‐Guérin (BCG)‐peptidoglycan (PGN). Infect Immun 2003; 71: 4238–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Matsumoto M, Kikkawa S, Kohase M, Miyake K, Seya T. Establishment of a monoclonal antibody against human Toll‐like receptor 3 that blocks double‐stranded RNA‐mediated signaling. Biochem Biophys Res Commun 2002; 293: 1364–9. [DOI] [PubMed] [Google Scholar]
- 61. Bosisio D, Polentarutti N, Sironi M et al. Stimulation of toll‐like receptor 4 expression in human mononuclear phagocytes by interferon‐gamma: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood 2002; 99: 3427–31. [DOI] [PubMed] [Google Scholar]
- 62. Nishiguchi M, Matsumoto M, Takao T et al. Mycoplasma fermentans lipoprotein M161Ag‐induced cell activation is mediated by Toll‐like receptor 2: role of N‐terminal hydrophobic portion in its multiple functions. J Immunol 2001; 166: 2610–6. [DOI] [PubMed] [Google Scholar]
- 63. Matsuo‐Tanabe M, Kawamoto T, Tanida S, Matsumoto M, Seya T Toll‐like receptor 2 agonists with unique properties synthesized with reference to a product of Streptosporangium . Immunology 2004: 235–41. [Google Scholar]
- 64. Brandenburg K, Lindner B, Schromm A et al. Physicochemical characteristics of triacyl lipid A partial structure OM‐174 in relation to biological activity. Eur J Biochem 2000; 267: 3370–7. [DOI] [PubMed] [Google Scholar]
- 65. Murata M. Activation of Toll‐like receptor 2 by a novel preparation of cell wall skeleton from Mycobacterium bovis BCG Tokyo (SMP‐105) sufficiently enhances immune responses against tumors. Cancer Sci 2008; 99: 1435–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shingai M, Ebihara T, Begum NA et al. Differential type I IFN‐inducing abilities of wild‐type versus vaccine strains of measles virus. J Immunol 2007; 179: 6123–33. [DOI] [PubMed] [Google Scholar]
- 67. Sugiyama T, Hoshino K, Saito M et al. Immunoadjuvant effects of polyadenylic:polyuridylic acids through TLR3 and TLR7. Int Immunol 2008; 20: 1–9. [DOI] [PubMed] [Google Scholar]
- 68. Navabi H, Jasani B, Reece A et al. A clinical grade poly I:C‐analogue (Ampligen) promotes optimal DC maturation and Th1‐type T cell responses of healthy donors and cancer patients in vitro. Vaccine 2009; 27: 107–15. [DOI] [PubMed] [Google Scholar]